1. Introduction
Vinylcyclobutanes undergo ring expansion to cyclohexenes. Woodward and Hoffmann, in their
Conservation of Orbital Symmetry treatise, formally classified this type of skeletal rearrangement as a [1,3]-sigmatropic carbon migration [
1]. For a given vinylcyclobutane leading from a migrating carbon to a migration terminus, four discrete products could be formed by
si,
sr,
ai, and
ar routes. These designations refer to the potential for inversion (
i) or retention (
r) of configuration at the migrating carbon and suprafacial (
s) or antarafacial (
a) sigma bond formation at the migration terminus relative to the disposition of the original sigma bond with respect to the π bond framework. According to the Woodward-Hoffmann selection rules, the two symmetry-allowed products are
si and
ar, and the two symmetry-forbidden products are
sr and
ai. Given the geometric prohibition of antarafacial migration in bicyclic vinylcyclobutanes, only
si and
sr products can form without effecting excessive distortions of the molecular carbon skeleton. Although a one-step concerted process may have been assumed under orbital symmetry control of the vinylcyclobutane-to-cyclohexene rearrangement, recent experimental and computational studies converge toward a stepwise diradical mechanistic analysis [
2].
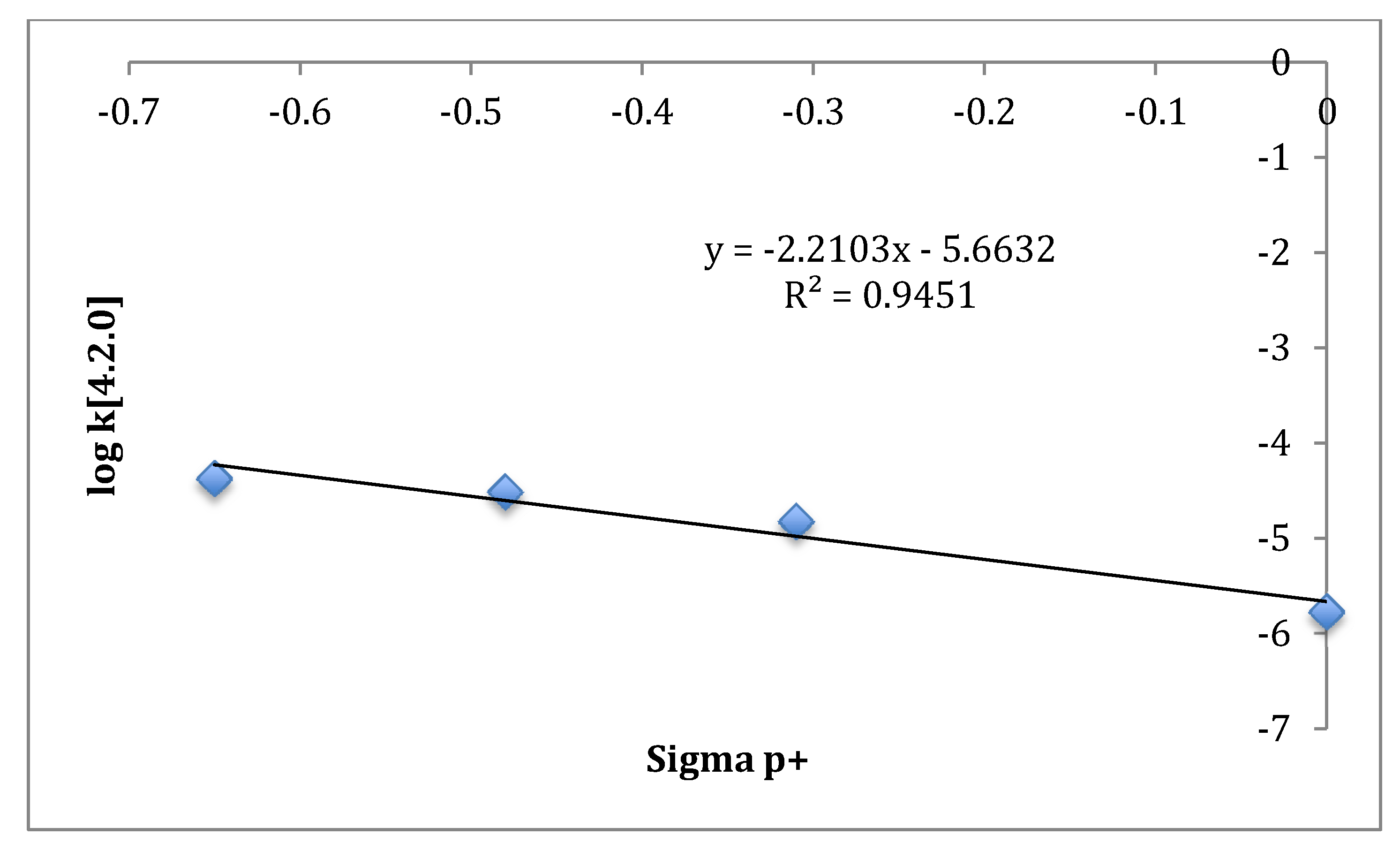
Thermal reactions of bicyclo[3.2.0]hept-2-enes and bicyclo[4.2.0]oct-2-enes, despite their homologous relationship, afford different product stereoselectivities as well as preferred exit channels. The
si/
sr ratios for the more conformationally labile bicyclo[4.2.0]oct-2-enes are lower than those reported for bicyclo[3.2.0]hept-2-enes [
3]. The relative kinetic importance of the observed exit channels for bicyclo[4.2.0]oct-2-enes labeled with a deuterium [
4], methyl [
5], or methoxy [
3] at a migrating carbon is
kep >
kf ≥
k13. The abbreviation
kep represents the rate of epimerization or one-centered stereomutation at C8;
kf, the rate of direct fragmentation;
k13, the total rate of [1,3]- sigmatropic migration including both
si and
sr products. This order is markedly different from the observation that [1,3]-carbon shifts afford the dominant products in the corresponding bicyclo[3.2.0]hept-2-enes [
3,
6,
7]. An early review of [1,3]-carbon rearrangements offered a significant prediction of the role of exit channels such as fragmentation and epimerization in the mechanistic assessment process: “The nature of exit channels such as fragmentations and stereomutations, which are undoubtedly mediated by diradical transition structures, are important in the formulation of a consistent mechanistic framework” [
2].
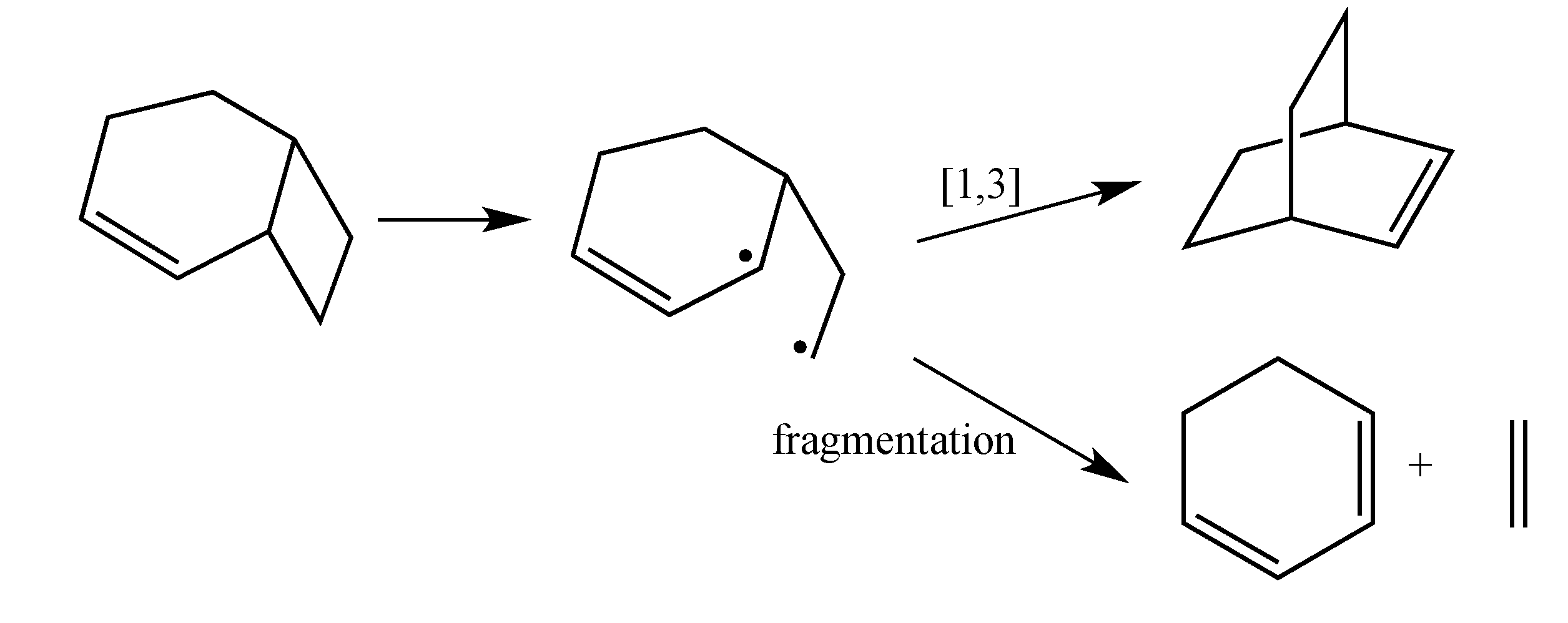
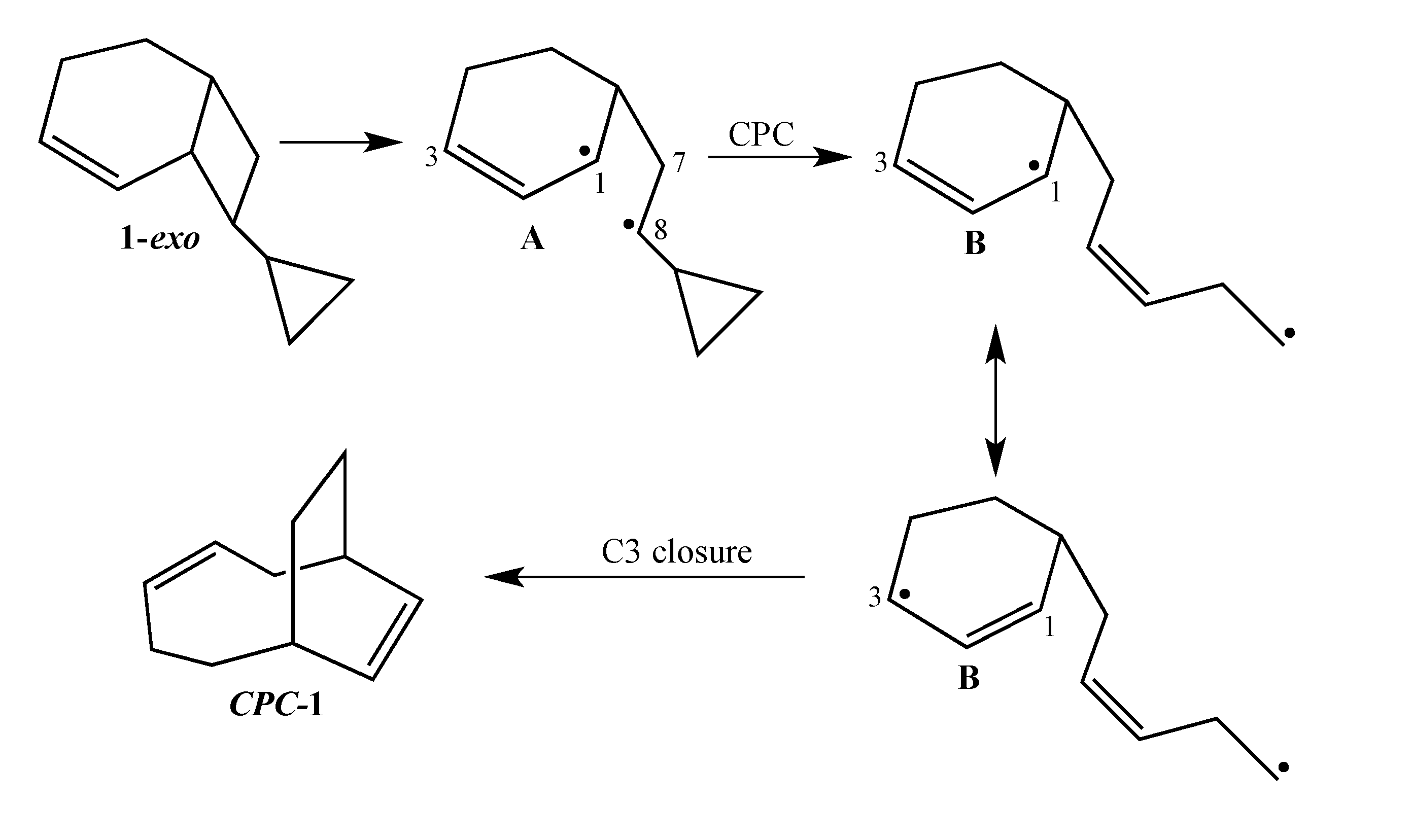
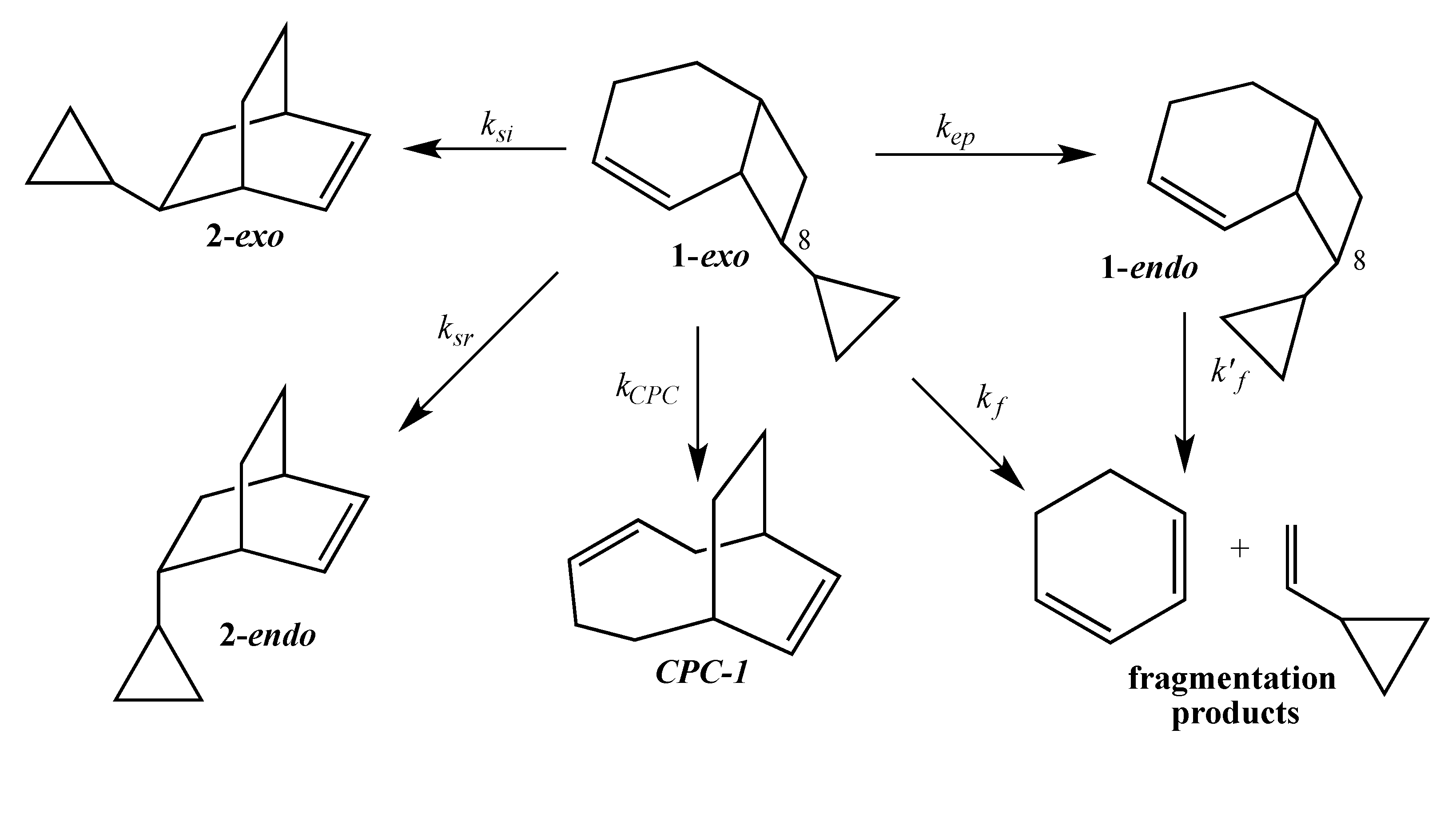
The current mechanistic formulation for the vinylcyclobutane–to–cyclohexene rearrangement is a stepwise diradical process. Representations of this mechanism for the parent compound bicyclo[4.2.0]oct-2-ene are provided in
Scheme 1, which shows that bicyclo[4.2.0]oct-2-ene can either isomerize via a [1,3]-shift to bicyclo[2.2.2]oct-2-ene or fragment to 1,3-cyclohexadiene and ethylene [
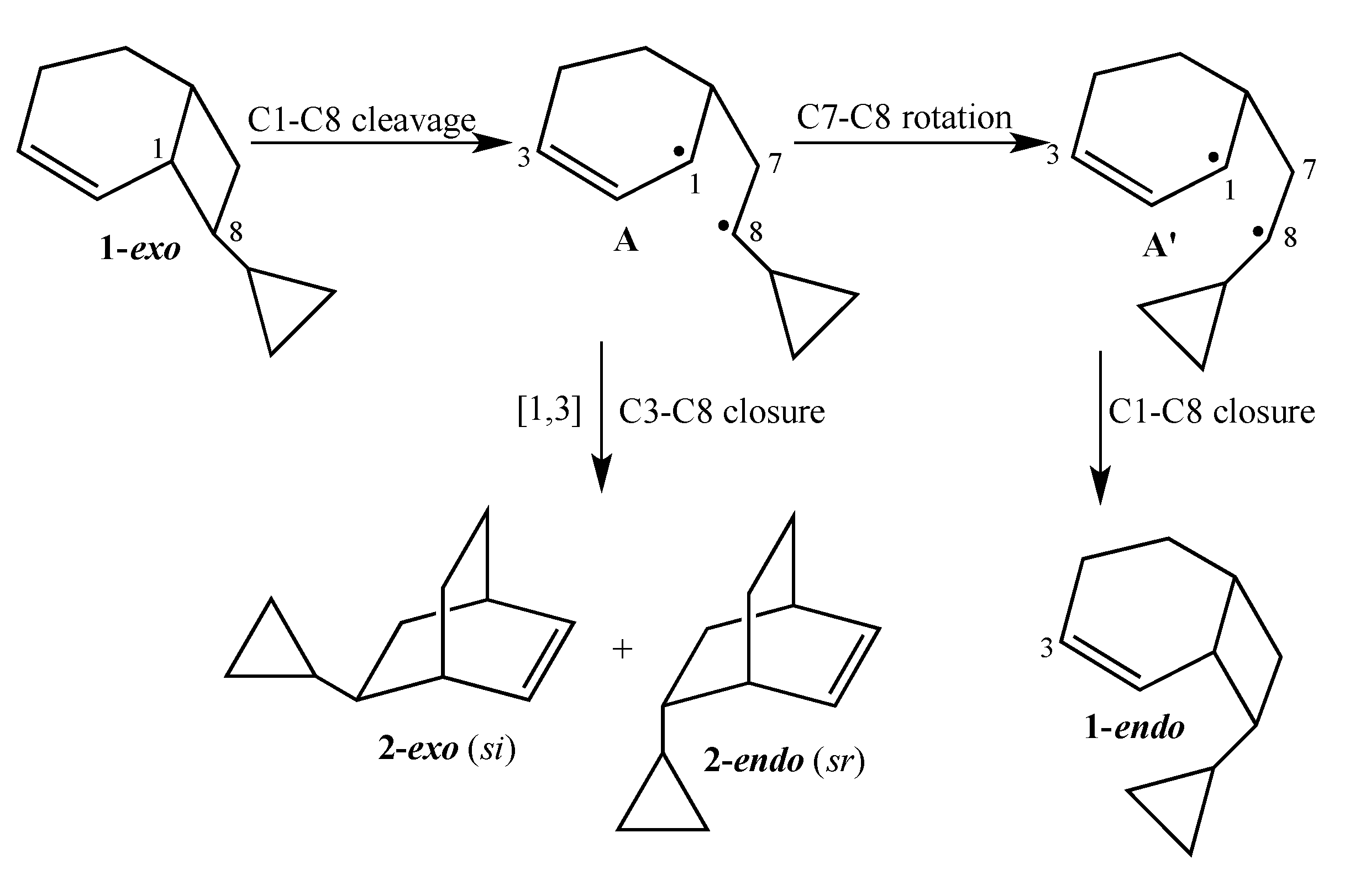
4]. Due to the presence of a stereochemical marker at C8, the analog
exo-8-cyclopropylbicyclo[4.2.0]oct-2-ene (
1-exo) can undergo [1,3]-sigmatropic migration to the
si (
2-exo) and
sr (
2-endo) products or C8 epimerization to
1-endo (
Scheme 2). The presence of a cyclopropyl substituent at C8 in
1-exo affords a unique potential for cyclopropylcarbinyl (CPC)-to-homoallylic radical rearrangement of diradical
A to diradical
B (
Scheme 3). Whereas key aspects of what transpires in a cyclopropylcarbinyl (CPC)-to-homoallylic radical rearrangement have been established for decades, this phenomenon has not yet been recognized in diradical species. Although no CPC rearrangement products were observed in the thermal reaction of
exo-7-cyclopropylbicyclo[3.2.0]hept-2-ene, the argument that bicyclo[4.2.0]oct-2-enes might yield diradical transition structures with more “weakly interacting radical centers” [
3] suggests the potential for
1-exo to form a CPC product such as bicyclo[5.2.2]undeca-3,8-diene,
CPC-1 (
Scheme 3).
Scheme 1.
Gas Phase Reaction of Bicyclo[4.2.0]oct-2-ene.
Scheme 1.
Gas Phase Reaction of Bicyclo[4.2.0]oct-2-ene.
Scheme 2.
Gas Phase Reactions of 1-exo.
Scheme 2.
Gas Phase Reactions of 1-exo.
Scheme 3.
Potential CPC Ring Closure Product CPC-1.
Scheme 3.
Potential CPC Ring Closure Product CPC-1.
3. Experimental
3.2. Thermal Reactions
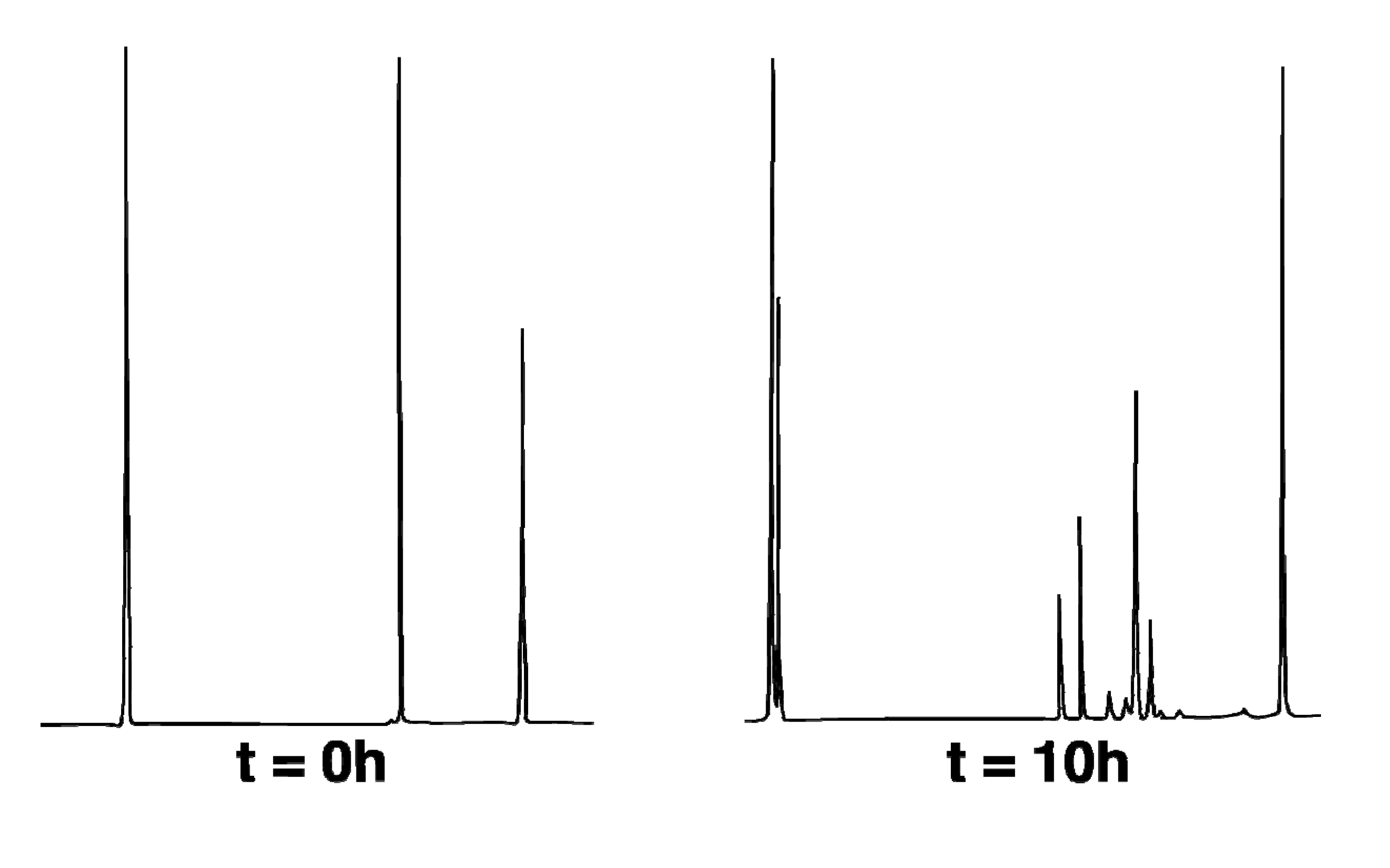
Thermal reactions of hydrocarbon 1-exo were carried out at 275.0 °C (with temperature control to ± 0.1 °C provided by a Bayley Precision Temperature Controller Model 124) in based-treated capillary tubes immersed in a molten salt bath (composed of a eutectic mixture of NaNO2 and KNO3). Temperatures were measured with an Omega DP11 thermocouple with a digital readout to ± 0.1 °C. Run times were measured to ± 0.01 min with a Precision Solid State Time-it. The internal standard (ISTD) was dodecane. Thermolysis samples were analyzed on an HP 5890A GC equipped with an HP cross-lined methyl silicone column (50 m × 0.2 mm i.d. × 0.10 µm film thickness) operating at an initial temperature of 100 °C held for 1 min followed by a temperature ramp of 0.1 °C/min to a maximum temperature of 150 °C. Retention times (min) were as follows: 11.4 (2-endo), 11.8 (2-exo), 12.3 (CPC-1), 12.8 (1-exo), 13.0 (1-endo), 15.5 (ISTD).
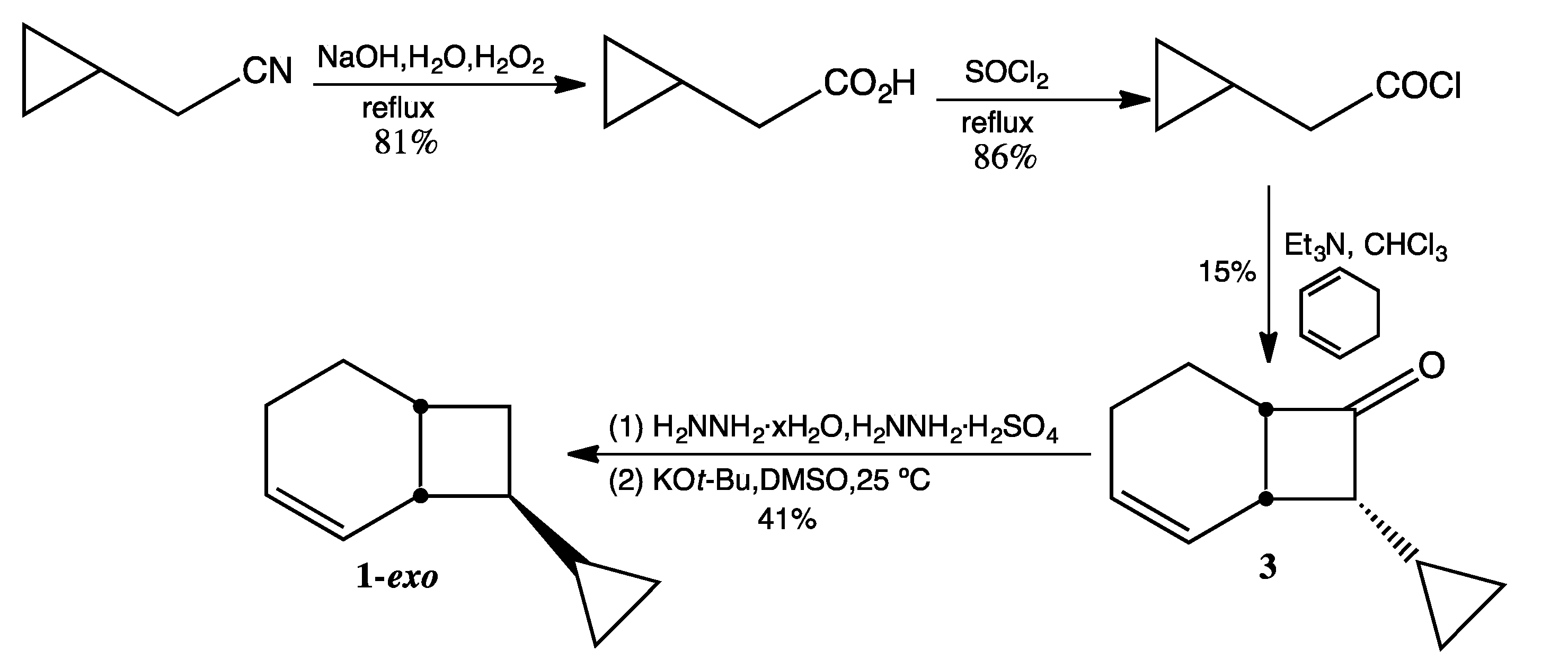
3.3. Preparation of 1-exo (Scheme 4)
Cyclopropylacetic Acid. The methodology of Fenick and Falvey [
8] was employed for the hydrolysis of commercially available cyclopropylacetonitrile.
1H-NMR (500 MHz,
d6-DMSO) δ 11.99 (br s, 1H), 2.09 (d, 2H), 0.92 (m, 1H), 0.44 (d, 2H), 0.11 (d, 2H).
13C-NMR (125 MHz,
d6-DMSO) δ 174.2 (C=O), 38.9 (CH
2), 7.0 (CH), 4.2 (2 CH
2). FTIR (neat) ν
max 3007, 3010, 1704, 1222, 828 cm
−1.
Cyclopropylacetyl chloride. Cyclopropylacetic acid (10.0 g, 100 mmol) and thionyl chloride (14.6 mL, 200 mmol) were combined and refluxed overnight at 40 °C under argon. Short-path distillation at atmospheric pressure afforded two fractions: fraction 1 (bp ~75 °C, unreacted thionyl chloride) and fraction 2 (bp 130–135 °C, cyclopropylacetyl chloride, 10.2 g, 86%). 1H-NMR (500 MHz, CDCl3) δ 2.77 (d, 2H), 1.13 (m, 1H), 0.65 (d, 2H), 0.24 (d, 2H). 13C-NMR (125 MHz, CDCl3) δ 173.4 (C=O), 51.9 (CH2), 7.1 (CH), 4.6 (2 CH2). FTIR (neat) νmax 3086, 3010, 1795, 1024, 925, 830, 707 cm−1.
endo-8-Cyclopropylbicyclo[4.2.0]oct-2-en-6-one (3). Triethylamine (14.0 mL, 100 mmol), freshly distilled from CaH2, was dissolved in chloroform (60 mL, purified by washing with conc. H2SO4 and distilled from CaH2) and then added dropwise to a solution of cyclopropylacetyl chloride (12.0 g, 100 mmol) in 1,3-cyclohexadiene (80 mL, 850 mmol). After stirring at rt for 24 h, the chloroform was removed via simple distillation. After addition of 150 mL of ether, the suspended solid was removed by vacuum filtration. The ether layer was washed with water and then brine, dried over MgSO4 (anhydrous), and concentrated under reduced pressure. After removal of hydrocarbon fractions on a silica gel column with pentane as the eluting solvent, the ketone product was eluted with 95:5 pentane:ether. Removal of solvent by rotary evaporation from five fractions yielded relatively pure ketone (2.5 g, 15%). 1H-NMR (500 MHz, CDCl3) δ 5.95 (d, 2H), 3.49 (m, 1H), 3.01 (dt, 1H), 2.74 (dt, 1H), 2.04 (m, 2H), 1.99 (m, 1H), 1.52 (m, 1H), 0.77 (m, 1H), 0.61 (m, 1H), 0.42 (m, 1H), 0.29 (m, 1H), 0.14 (m, 1H). 13C-NMR (125 MHz, CDCl3) δ 212.4 (C=O), 129.6 (=CH), 126.3 (=CH), 66.5 (CH), 54.5 (CH), 27.7 (CH), 21.3 (CH2), 18.4 (CH2), 6.5 (CH), 4.5 (CH2), 2.2 (CH2). FTIR (neat) νmax 3080, 3006, 1767, 1646, 685 cm−1. LRMS (EI) m/z 162 (M+, 1), 134 (5), 119 (5), 107 (8), 105 (10), 91 (32), 82 (27), 80 (100), 79 (61), 77 (25); HRMS (EI) calcd for C11H14O (M+) 162.1045, found 162.1043.
8-Cyclopropylbicyclo[4.2.0]oct-2-ene (1). A mixture of hydrazine sulfate (2.1 g, 15.9 mmol) dissolved in hydrazine hydrate (10 mL) and compound 3 (2.5 g, 15.4 mmol) was refluxed overnight at 65 °C. The reaction mixture was extracted several times with ether, and the combined organic layers were washed with distilled water and brine, dried over MgSO4 (anhydrous), and concentrated under reduced pressure to afford crude hydrazone (1.3 g, 48%). FTIR (neat) νmax 3368, 3202, 3075, 3017, 1682, 1645, 1018, 739 cm−1. To a solution of potassium tert-butoxide (1.08 g, 9.0 mmol), sublimed under high vacuum at 190 °C and dissolved in 25 mL anhydrous DMSO, was added 8-cyclopropylbicyclo[4.2.0]oct-2-en-7-one hydrazone (1.3 g, 8.0 mmol) dropwise over 6 h. After stirring overnight, the reaction mixture was quenched with 5 mL of cold water and extracted four times with pentane. The combined pentane extracts were washed ten times with water to remove DMSO and dried over MgSO4 (anhydrous). The pentane was removed via simple distillation to yield crude product (1.02 g, 85%) in an exo:endo ratio of ca. 9:1. Preparative GC (on a 12' × ¼'' DC710 packed column at 130 °C) afforded 1-exo in >99% purity by GC. 1H-NMR (500 MHz, CDCl3) δ 5.74 (m, 2H), 2.39 (br m, 2H), 1.99 (m, 1H), 1.93 (m, 1H), 1.70 (m, 2H), 1.64 (m, 2H), 1.48 (m, 1H), 0.87 (m, 1H), 0.37 (dd, 2H), 0.05 (dd, 2H). 13C-NMR (125 MHz, CDCl3) δ 130.3 (=CH), 127.0 (=CH), 46.2 (CH), 38.4 (CH), 29.2 (CH), 28.2 (CH2), 25.7 (CH2), 22.3 (CH2), 15.5 (CH), 2.9 (CH2), 2.8 (CH2). FTIR (neat) νmax 3076, 3015, 1644, 1015, 697 cm−1. LRMS (EI) m/z 148 (M+, 1), 105 (3), 91 (8), 81 (9), 80 (100), 79 (45), 77 (11); HRMS (EI) calcd for C11H16 (M+) 148.1252, found 148.1255.
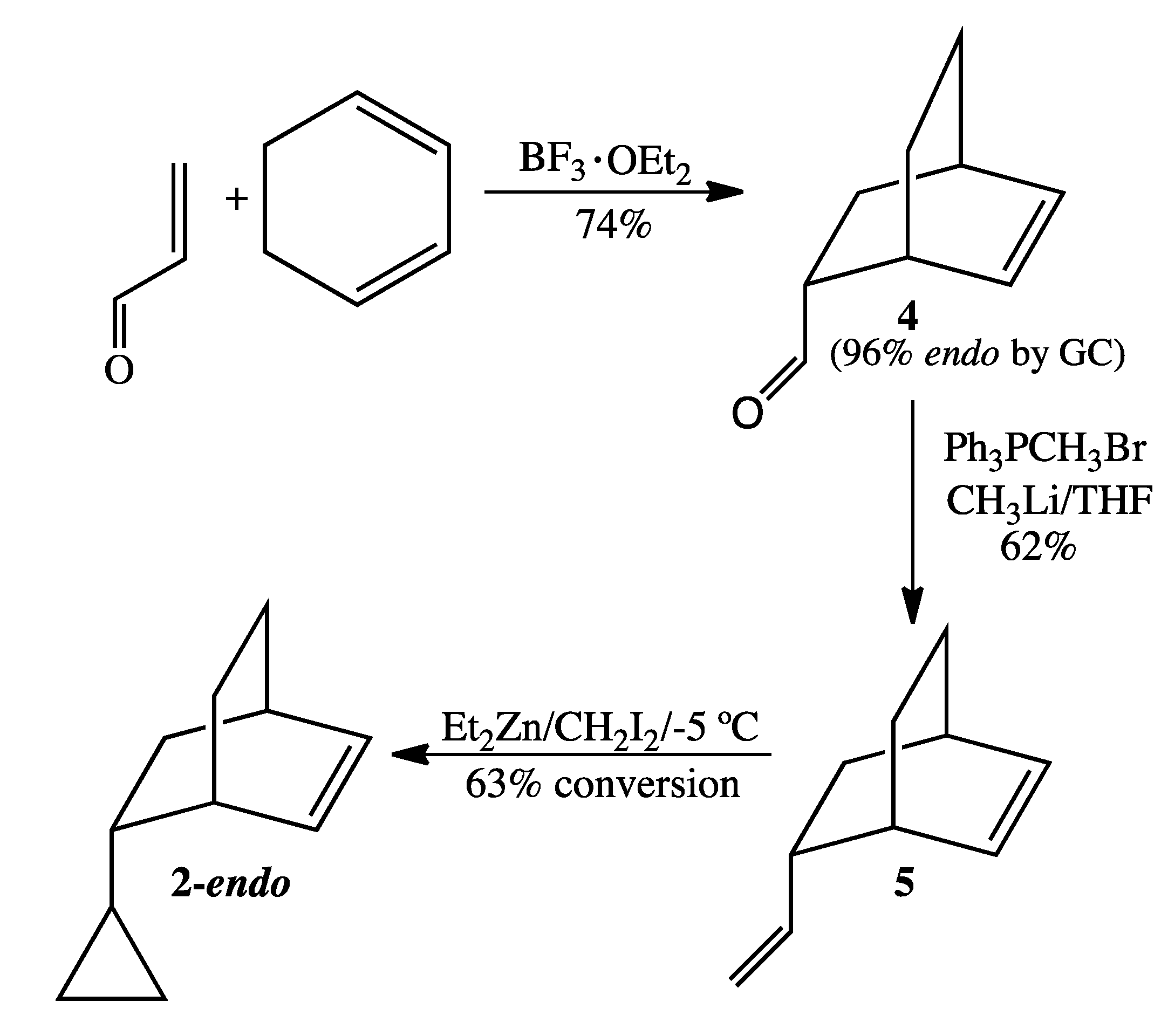
3.4. Preparation of 2-endo (Scheme 5)
Bicyclo[2.2.2]oct-2-en-5-carboxaldehyde (4). The catalyst BF3.OEt2 (2.0 mL, 16 mmol) was added under inert atmosphere to a solution of 1,3-cyclohexadiene (97%, 7.5 mL, 76 mmol) and acrolein (95%, 2.7 mL, 38 mmol) dissolved in anhydrous ether (50 mL). After stirring for 4 h, the reaction was quenched with ice-cold distilled water and then extracted with ether. The combined organic layers were washed sequentially with water, saturated sodium bicarbonate (aq), water, and brine and then dried over MgSO4. Concentration via rotary evaporation resulted in crude product (3.7 g, 71% yield), 96% of the endo epimer by GC analysis. 1H-NMR (500 MHz, CDCl3) δ 9.44 (d, 1H), 6.32 (t, 1H), 6.10 (t, 1H), 2.95 (br m, 1H), 2.63 (br m, 1H), 2.55 (br m, 1H), 1.72 (m, 1H), 1.65 (m, 2H), 1.55 (m, 1H), 1.35 (tt, 1H), 1.27 (m, 1H). 13C-NMR (125 MHz, CDCl3) δ 203.6 (O=CH), 136.0 (=CH), 130.6 (=CH), 50.8 (CH), 30.6 (CH), 29.2 (CH), 26.6 (CH2), 25.1 (CH2), 24.7 (CH2). FTIR (neat) νmax 3044, 2937, 1721, 701 cm−1. LRMS (EI) m/z 136 (M+, C9H12O, 11), 108 (17), 93 (8), 91 (10), 80 (53), 79 (100).
5-Vinylbicyclo[2.2.2]oct-2-ene (5). A solution of recrystallized methyltriphenylphosphonium bromide (5.7 g, 16 mmol) in anhydrous THF (100 mL) was cooled to −78 °C and was treated slowly with 1.6 M CH3Li in ether (10.0 mL, 16 mmol). The resultant yellow suspension was then allowed to stir at 0 °C for 1 h. After again cooling the reaction mixture to −78 °C, bicyclo[2.2.2]oct-2-en-5-carboxaldehyde (2.0 g, 14.7 mmol) dissolved in anhydrous THF (5 mL) was added dropwise via syringe over 1 h. The reaction mixture, which was allowed to warm gradually to rt and to stir overnight, was then quenched with ice-cold water and extracted twice with pentane. The combined organic layers were washed with water and brine, dried over MgSO4, and concentrated by short-path simple distillation to yield crude diolefin product (1.5 g, 75%) as a yellow oil. The endo:exo ratio, as determined by GC analysis, was 96:4. 1H-NMR (500 MHz, CDCl3) δ 6.28 (t, 1H), 6.14 (t, 1H), 5.55 (pent, 1H), 4.86 (d, 1H), 4.79 (d, 1H), 2.46 (m, 2H), 1.75 (m, 1H), 1.55 (m, 1H), 1.46 (m, 1H), 1.24 (m, 3H), 1.06 (m, 1H). 13C-NMR (125 MHz, CDCl3) δ 145.4 (=CH), 134.8 (=CH), 132.0 (=CH), 111.6 (=CH2), 42.4 (CH), 35.7 (CH), 33.6 (CH2), 29.8 (CH), 26.1 (CH2), 24.4 (CH2). FTIR (neat) νmax 3040, 2934, 1638, 996, 906, 693 cm−1. LRMS (EI) m/z 134 (M+, 8), 92 (8), 91 (15), 80 (100), 79 (76); HRMS (EI) calcd for C10H14 (M+) 134.1096, found 134.1093.
5-Cyclopropylbicyclo[2.2.2]oct-2-ene (2). A solution of 1.0 M diethylzinc in hexanes (10.0 mL, 10.0 mmol) was added under argon to a dry round-bottomed flask at −40 °C. Sequential dropwise addition of 5-vinylbicyclo[2.2.2]oct-2-ene (1.0 g, 7.5 mmol) and diiodomethane (0.5 mL, 7.5 mmol) was performed. The reaction mixture was then allowed to warm to −5 °C and the temperature was held between −5 °C and 0 °C for 16 h, during which time additional aliquots of diethylzinc (4.0 mL) and diiodomethane (0.2 mL) were added to facilitate selective cyclopropanation of 5-vinylbicyclo-[2.2.2]0ct-2-ene at the exocyclic vinyl group. After 16 h, the reaction vessel was sealed with parafilm and stored overnight in a freezer at 0 °C. GC analysis the following day showed a 1.0:1.7 ratio of 5-vinylbicyclo[2.2.2]oct-2-ene to 2, corresponding to a 63% conversion. The reaction was quenched with ice-cold water and extracted twice with pentane. The combined organic layers were washed with water and brine, dried over MgSO4, and concentrated by short-path simple distillation. The dominant epimer 2-endo was obtained in >98% purity by prep GC (on a 12' × ¼'' DC710 packed column at 128 °C). 1H-NMR (500 MHz, CDCl3) δ 6.26 (dt, 1H), 6.20 (t, 1H), 2.49 (br m, 1H), 2.45 (br m, 1H), 1.72 (dq, 1H), 1.37 (m, 2H), 1.20 (m, 2H), 1.07 (m, 1H), 0.82 (m, 1H), 0.42 (m, 1H), 0.29 (m, 2H), 0.01 (m, 2H). 13C-NMR (125 MHz, CDCl3) δ 134.5 (=CH), 132.8 (=CH), 44.1 (CH), 35.3 (CH), 34.1 (CH2), 30.2 (CH), 26.4 (CH2), 24.6 (CH2), 18.1 (CH), 3.8 (CH2), 3.3 (CH2). FTIR (neat) νmax 3075, 3043, 3000,1640, 703 cm−1. LRMS (EI) m/z 148 (M+, 1), 92 (4), 91 (7), 80 (100), 79 (35); HRMS (EI) calcd for C11H16 (M+) 148.1252, found 148.1253.
A selective cyclopropanation of 5-vinylbicyclo[2.2.2]oct-2-ene to 2 at 20 °C resulted in 69% conversion. Selectivity for the desired product 2 was achieved regardless of temperature. The ratio of 2 to dicyclopropanated side product was 4.2:1 for the reaction at 20 °C compared to 5.0:1 for the reaction at 0 °C.
The thermal stability of 2-endo was assessed by determining the 2-endo:dodecane (ISTD) ratio over a period of 30 h at 275 °C; sampling at 10 h increments afforded a fundamentally invariant ratio of 5.2 ± 0.2.
A preparative 120-h thermal reaction of 1-exo at 275 °C afforded a ca. 2:1 mixture of 2-endo:2-exo. The LRMS of 2-exo was virtually indistinguishable from that of 2-endo. The 13C-NMR signals due to 2-exo were identified from the NMR spectrum of this epimeric mixture: δ 136.2 (=CH), 133.7 (=CH), 42.5 (CH), 34.8 (CH), 32.6 (CH2), 30.2 (CH), 26.3 (CH2), 20.1 (CH2), 15.6 (CH), 4.0 (CH2).
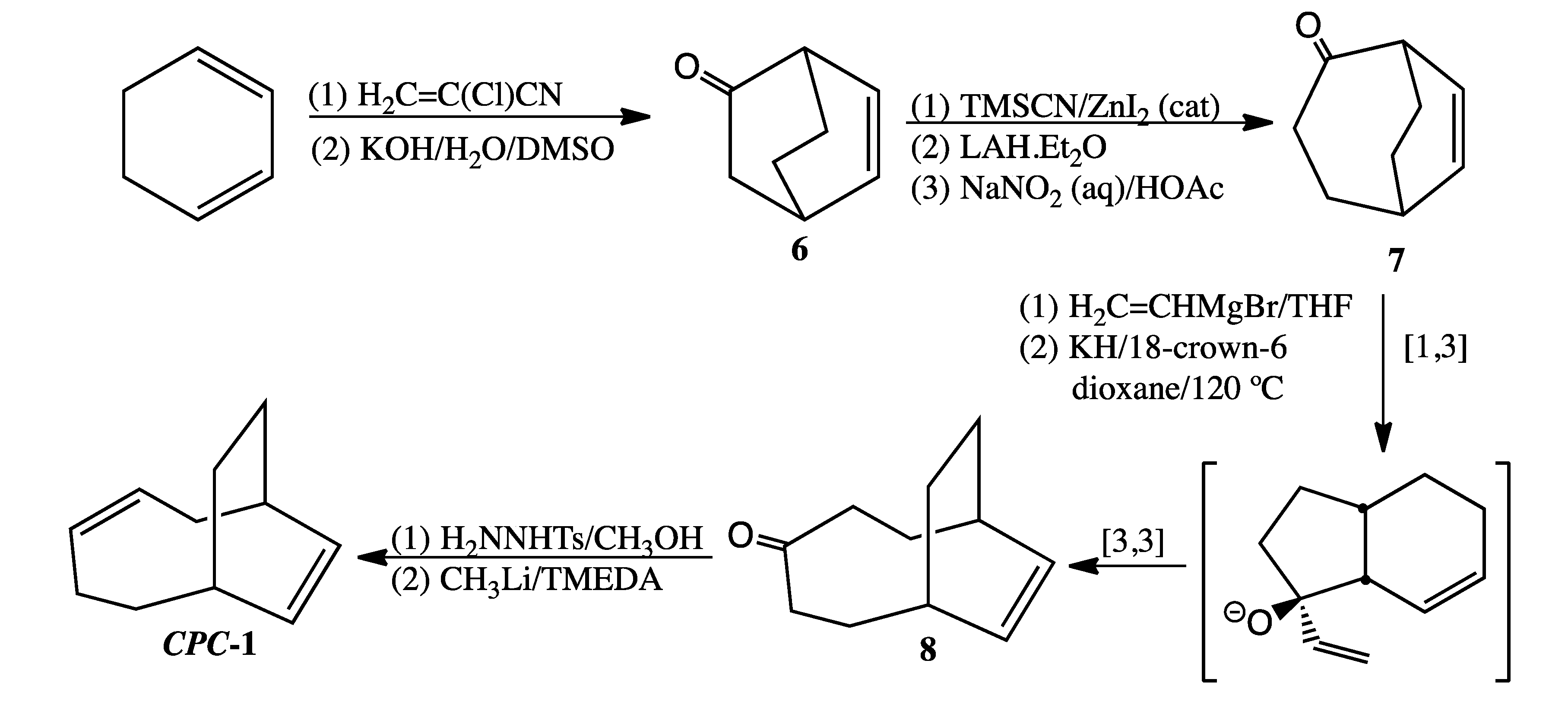
3.5. Preparation of CPC-1 (Scheme 6)
Bicyclo[2.2.2]oct-5-en-2-one (
6). Compound
6, a low melting solid prone to sublimation, was prepared as previously reported [
4].
1H-NMR (500 MHz, CDCl
3) δ 6.47 (t, 1H), 6.11 (t, 1H), 3.12 (br m, 1H), 2.97 (br m, 1H), 2.04 (s, 2H), 1.84 (m, 1H), 1.69 (m, 1H), 1.57 (m, 1H), 1.50 (m, 1H).
13C-NMR (125 MHz, CDCl
3) δ 213.1 (C=O), 137.1 (=CH), 128.5 (=CH), 48.6 (CH), 40.5 (CH
2), 32.4 (CH), 24.3 (CH
2), 22.5 (CH
2). FTIR (neat) ν
max 3051, 2945, 1717, 700 cm
−1. LRMS (EI)
m/z 122 (M
+, 22), 80 (100), 79 (81); HRMS (EI) calcd for C
8H
10O (M
+) 122.0732, found 122.0729.
Bicyclo[3.2.2]non-6-en-2-one (
7). Subjecting 1.12 g (9.18 mmol) of
6 to Tiffaneau-Demjanov ring expansion [
14] afforded 0.36 g (29% yield) of
7 (contaminated with 7% bicyclo[3.2.2]non-6-en-3-one) after purification by column chromatography.
1H-NMR (500 MHz, CDCl
3) δ 6.35 (t, 1H), 6.08 (t, 1H), 3.05 (br t, 1H), 2.69 (br m, 1H), 2.56 (t, 2H), 1.97 (m, 1H), 1.89 (m, 1H), 1.82 (m, 4H).
13C-NMR (125 MHz, CDCl
3) δ 209.9 (C=O), 136.7 (=CH), 127.9 (=CH), 49.3 (CH), 39.4 (CH
2), 31.2 (CH), 30.7 (CH
2), 24.9 (CH
2), 24.1 (CH
2). FTIR (neat) ν
max 3039, 2940, 1693, 705 cm
−1. LRMS (EI)
m/z 136 (M
+, 20), 118 (50), 117 (38), 92 (85), 80 (57), 79 (100).
Bicyclo[5.2.2]undec-8-en-4-one (
8). A sample of 0.34 g (2.5 mmol)
7 was allowed to react gradually with 4.0 mL (4.0 mmol) of 1.0 M vinylmagnesium bromide in THF under argon at −65 °C. Workup by treatment with 1 M NH
4Cl (aq) followed by successive extraction with ether and subsequent drying over MgSO
4 afforded 0.32 g (2.0 mmol, 78%) of crude vinyl alcohol as a pair of diastereomers. FTIR (neat) ν
max 3424, 3032, 2975, 1631, 998, 916, 720 cm
−1. LRMS (EI)
m/z 164 (M
+, 2), 146 (14), 131 (17), 104 (64), 92 (81), 91 (71), 83 (100), 79 (100). The vinyl alcohols were treated with 0.29 g (2.1 mmol) of KH (30 wt% in mineral oil) and 18-crown-6 (1.09 g, 4.1 mmol) dissolved in 20 mL anhydrous dioxane under reflux at 120 °C for 1.5 h. The major product in the crude mixture (0.16 g, 47%) corresponded to
8, as confirmed by
13C-NMR spectral comparison with reported literature values [
16].
13C-NMR (125 MHz, CDCl
3) δ 219.0 (C=O), 133.1 (=CH), 41.0 (CH
2), 31.7 (CH
2), 30.5 (CH), 25.4 (CH
2). FTIR (neat) ν
max 3019, 2918, 1697, 702 cm
−1. LRMS (EI)
m/z 164 (M
+, 23), 136 (75), 118 (82), 117 (50), 92 (100), 91 (69), 79 (100).
Bicyclo[5.2.2]undeca-3,8-diene (CPC-1). To a solution of 150 mg (0.81 mmol) of p-toluenesulfonyl hydrazide in 1 mL of methanol was added crude 8 (150 mg, 0.92 mmol). The resultant crystals were filtered and washed with cold 1:1 pentane-ether (1 mL). The tosylhydrazone derivative of 8 (0.02 g, 0.09 mmol) was then suspended in TMEDA (anhydrous, 0.2 mL). After cooling the mixture in a dry ice/acetone bath, 1.6 M CH3Li in diethyl ether (4.4 equiv., 0.25 mL, 0.4 mmol) was added dropwise via syringe. The resultant solution turned dark orange in color. After stirring overnight, the reaction was quenched with 1:1 pentane-water, washed successively with 2 M HCl (aq) and 2M NaOH (aq), and then dried over MgSO4. Removal of solvent by short-path distillation gave a trace amount of liquid. Major component LRMS (EI) m/z 148 (M+, 11), 133 (13), 120 (29), 106 (29), 93 (30), 92 (36), 91 (50), 79 (100). This mass spectrum is virtually identical to that of the thermal product eluting at 12.3 min (see SI).
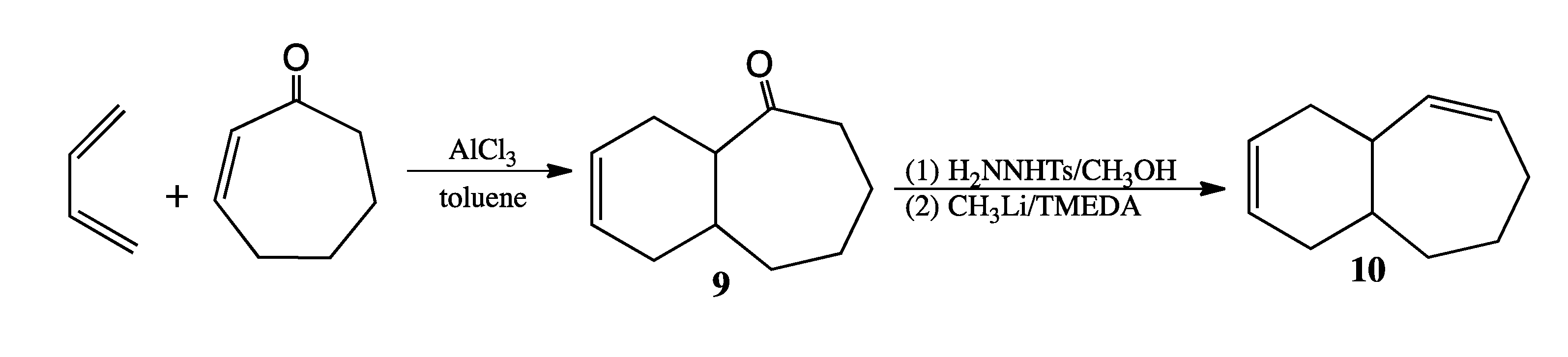
3.6. Preparation of Bicyclo[5.4.0]undeca-2,9-diene (Scheme 7)
Bicyclo[5.4.0]undec-9-en-2-one (9). A sample of 1.63 g (12.2 mmol) of AlCl3 was weighed into a thick-walled flask under argon in a glovebag. A solution of 2-cycloheptenone (1.5 mL, 0.988 g/mL, 13.6 mmol) in anhydrous toluene (7.6 mL) was transferred to the flask via syringe. After complexation between the ketone and Lewis acid had proceeded for 40 min at rt, a solution of 1,3-butadiene in toluene (27.4 mL, 20 wt%, 0.806 g/mL, 81.7 mmol) was added via syringe through a septum, which was then replaced with Teflon screw cap. After stirring the reaction at rt for 22 h, the reaction mixture was extracted with ether, and the organic layer was washed twice with water and once with brine. Elution of the ketone from a silica column with 98:2 pentane:ether afforded 1.04 g (6.4 mmol, 47%) of pure ketone 9. 1H-NMR (500 MHz, CDCl3) δ 5.68 (m, 1H), 5.62 (m, 1H), 2.76 (dt, 1H), 2.65 (m, 1H), 2.45 (dd, 1H), 2.29 (br m, 3H), 2.17 (m, 1H), 1.89 (m, 1H), 1.80 (m, 2H), 1.66 (m, 4H). 13C-NMR (125 MHz, CDCl3) δ 215.2 (C=O), 125.4 (=CH), 125.3 (=CH), 49.7 (CH), 43.7 (CH2), 34.0 (CH), 33.3 (CH2), 31.7 (CH2), 27.5 (CH2), 26.2 (CH2), 24.1 (CH2). FTIR (neat) νmax 3023, 2921, 1691, 1654, 653 cm−1. LRMS (EI) m/z 164 (M+, 45), 146 (29), 135 (27), 117 (46), 104 (55), 91 (49), 79 (100), 77 (47).
Bicyclo[5.4.0]undeca-2,9-diene (10). Ketone 9 (0.52 g, 3.2 mmol) was added to a solution of p-toluenesulfonyl hydrazide (0.45 g, 2.4 mmol) in methanol (4 mL). After sitting overnight, a crop of crystals (0.75 g, 68%) was obtained. The tosylhydrazone of ketone 9 was dried in a vacuum oven: mp 137–139 °C. Dropwise addition of a solution of 1.6 M CH3Li in pentane (4.0 mL, 6.4 mmol) at −55 °C over 15 min to a suspension of the tosylhydrazone (0.60 g, 1.7 mmol) suspended in 4 mL anhydrous TMEDA produced a dark red solution. After stirring overnight at rt, the reaction was quenched with cold water and extracted with pentane. Removal of the solvent by short-path distillation yielded product 10 (0.13 g, 0.88 mmol, 52%), which was purified via prep GC (on a 12' × ¼'' DC710 packed column at 130 °C) for purposes of spectral analysis. 1H-NMR (500 MHz, CDCl3) δ 5.79 (m, 1H), 5.66 (m, 2H), 5.44 (dd, 1H), 2.68 (br s, 1H), 2.24 (br d, 1H), 2.11 (m, 3H), 1.99 (br d, 1H), 1.88 (m, 2H), 1.85 (m, 1H), 1.72 (m, 1H), 1.54 (m, 1H), 1.48 (m, 1H). 13C-NMR (125 MHz, CDCl3) δ 135.8 (=CH), 132.0 (=CH), 127.2 (=CH), 125.5 (=CH), 37.5 (CH), 35.3 (CH2), 34.6 (CH), 32.5 (CH2), 29.1 (CH2), 28.9 (CH2), 23.4 (CH2). LRMS (EI) m/z 148 (M+, 41), 133 (13), 119 (14), 105(21), 94 (84), 92 (31), 91 (48), 79 (100), 77 (25).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




