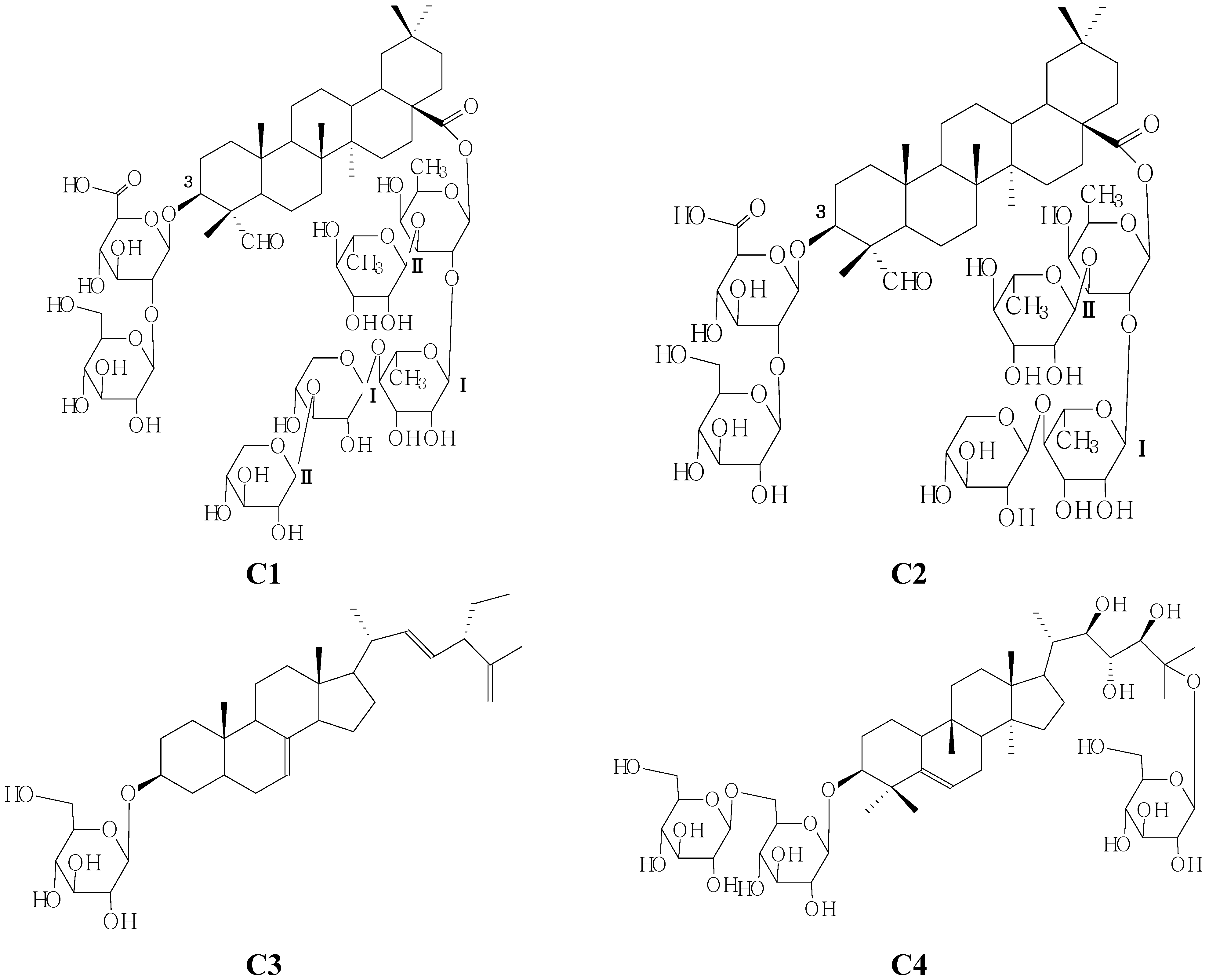
2. Results and Discussion
The n-BuOH-soluable portions of the 95% EtOH extract of seeds of M. charantia L. was subjected to silica gel, RP-18, and Sephadex LH-20 column chromatographies and semipreparative HPLC to yield two new oleanane-type triterpenoids C1 and C2 and two known compounds identified as 3-O-β-d-glucopyranosyl-24β-ethyl-5α-cholesta-7-trans-22E,5(7)trien-3β-ol (C3) and momordicoside S (C4).
Compound
C1 was an amorphous white powder, which gave a positive result in the Liebermann- Burchard test. Acid hydrolysis of compound
C1 with 2 mol/L HCl–1,4-dioxane (1:1,
v/v) furnished
d-xylose,
l-rhamnose,
d-glucose, and
d-fucose in the ratio of 2:2:1:1 by HPLC analysis of the corresponding thiazolidine derivatives following conversion to the 1-[(
S)-N-acetyl-(
R)-methylbenzylamino]-1-deoxyalditol acetate derivatives [
7]. In the (‒)- and (+)-ESI-MS of
C1, quasimolecular ion peaks were observed at
m/z 1,509 [M–H]
− and
m/z 1,533 [M+Na]
+ respectively, HR-ESI-MS (
m/z 1,533.7190 [M+Na]
+) analysis revealed the molecular formula of
C1 to be C
69H
112O
33Na (calcd. 1,533.7168). The five fragment ions at
m/z 1,377 [M–132–H], 1,329 [M–162–H], 1,171 [M–162–176–H], 807 [M–146 × 3–132 × 2–H] and 469 [M–146 × 3–132 × 2–162–176–H], indicated the sequential losses of seven sugar moieties (five hexoses and two pentoses).
The
1H (pyridine-
d5) spectra of
C1 revealed the presence of nine methyl proton signals at
δH 0.76 (Me-25), 0.89 (Me-29), 0.92 (Me-26), 0.96 (Me-30), 1.18 (Me-27), 1.36 (Me-24), 9.89 (H-29) and included signals due to a gyosogenin skeleton [
8], 1.54, 3H, d,
J = 5.5 Hz (Me-Fuc), 1.61, 3H, d,
J = 5.5 Hz (Me-Rha
II), 1.67, 3H, d,
J = 5.5 Hz,(Me-Rha
I), as well as an olefinic proton at δ
H 5.37, 1H, s (H-12); The signals at δ
C 122.4, 143.8 in the
13C-NMR spectrum were assigned to a 12(13)-ene grouping by comparison with literature data [
5].
The
1H and
13C-NMR (
Table 1) spectra of
C1 exhibited seven sugar anomeric protons assignable to a
β-
d-glucopyranosiduronic acid moiety [
δH 4.76, 1H, d, 6.5 Hz], a
β-
d-glucopyranosyl moiety[
δH 5.38, 1H, s], a
β-
d-fucopyranosyl moiety [
δH 6.30, 1H, d, 5.5 Hz ], two
α-
l-rhamnopyranosyl moieties [
δH 5.74, 1H, s (Rhm
I-1-H);
δH 5.67, 1H, s (Rhm
II-1-H)], two
β-
d-xylopyranosyl moieties [
δH 5.08, 1H, d, 7.0 Hz (Xyl
I-1-H);
δH 5.21, 1H, d, 7.0 Hz (Xyl
II-1-H)], and a gyosogenin moiety [
δH 3.04, 1H, dd-like, (18-H),
δH 4.04, 1H, m (3-H),
δH 5.44, 1H, s, (12-H);
δH 9.89, 1H, s (23-H)]. The identities of the monosaccharides and the oligosaccharide sequence were determined by a combination of DEPT and two-dimensional NMR experiments (such as HMQC, HMBC, TOCSY NMR). The sequence of the glycan part was deduced from the following HMBC correlations: the anomeric proton signals at
δH 4.76 and
δC 83.5 (H-1 of the
β-
d-glucopyranosiduronic acid attached to the C-3 of the aglycone),
δH 5.38 and
δC 82.6 (H-1 of the
β-
d-glucopyranosyl moiety attached to the C-2 of the
β-
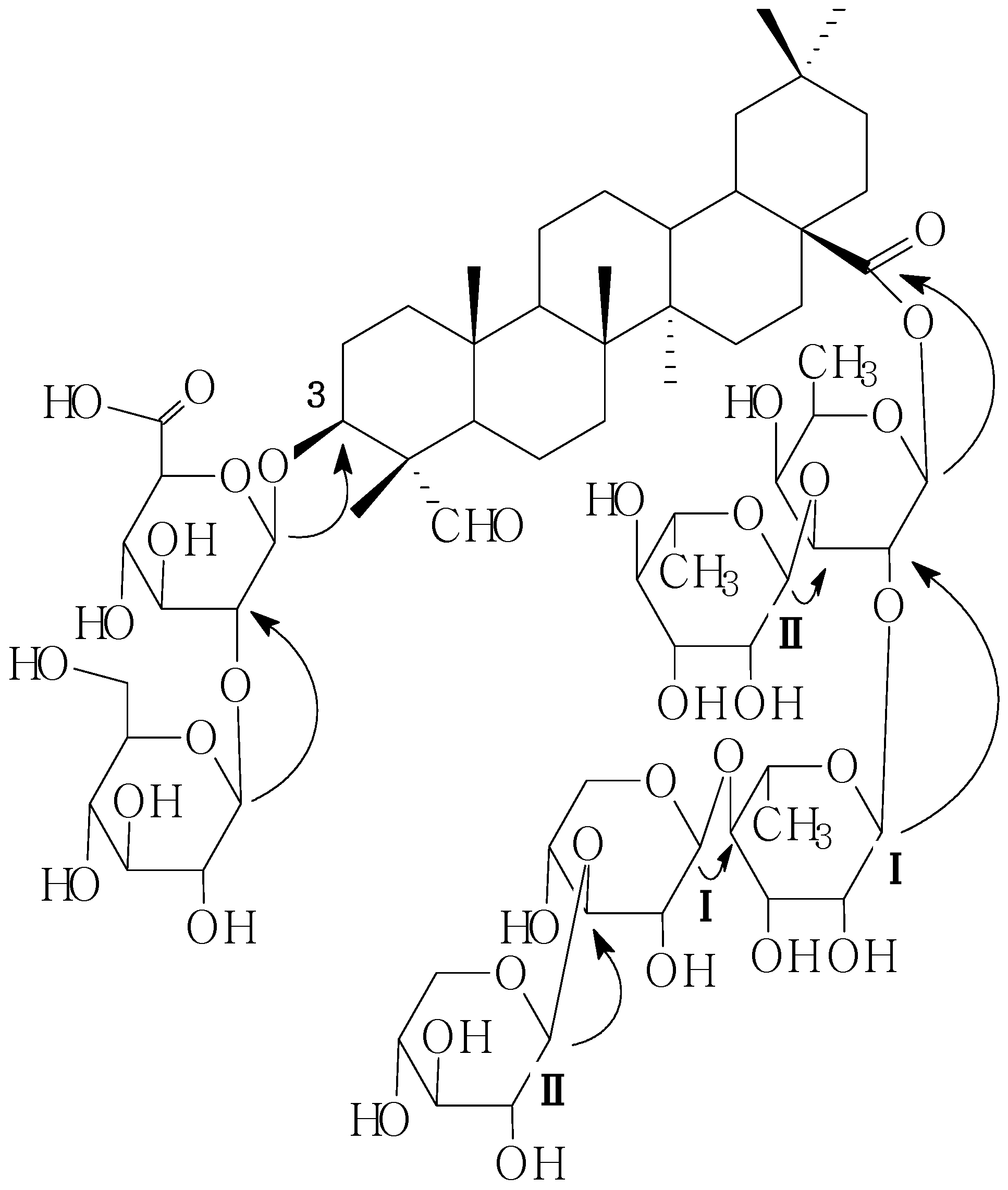
d-gluco-pyranosiduronic acid), and on the other hand, the pentasaccharide part at C-28 was established by the following HMBC information between the following protons and carbons: Fuc-1-H and 28-C, RhmI-1-H and Fuc-2-C, RhmII-1-H and Fuc-3-C, XylI-1-H and RhmI-4-C, XylII-1-H and XylI-3-C (
Figure 2). On the basis of the foregoing evidence, the structure of
C1 was determined as 28-O-β-
d-xylopyranosyl(1→3)-β-
d-xylopyranosyl(1→4)-α-
l-rhamnopyranosyl(1→2)-[α-
l-rhamnopyranosyl-(1→3)]-β-
d-fucopyranosylgypsogenin 3-O-β-
d-glucopyranosyl (1→2)-β-
d-glucopyranosiduronic acid.
Table 1.
1H-NMR (500 MHz) and 13C-NMR (125 MHz) data for C1 in pyridine-d5 (δ, ppm).
Table 1.
1H-NMR (500 MHz) and 13C-NMR (125 MHz) data for C1 in pyridine-d5 (δ, ppm).
| Position | C1-Aglycone | Position | C1-Sugar Chain |
|---|
| 13C | 1H | 13C | 1H |
|---|
| 1 | 39.8 | | GlcA-1 | 102.7 | 4.76, 1H, d, 6.5 Hz |
| 2 | 25.8 | | 2 | 82.6 | 4.32, 1H, m |
| 3 | 83.5 | 4.04, 1H, m | 3 | 77.8 | 4.65, 1H, m |
| 4 | 54.7 | ------ | 4 | 72.4 | 4.20, 1H, m |
| 5 | 48.1 | | 5 | 76.9 | 4.50, 1H, m |
| 6 | 18.7 | | 6 | 175.9 | |
| 7 | 32.9 | | Glc-1 | 105.9 | 5.38, 1H, s |
| 8 | 39.8 | | 2 | 75.1 | 4.15, 1H, m |
| 9 | 47.5 | | 3 | 77.8 | 4.25, 1H, m |
| 10 | 35.9 | | 4 | 71.4 | 4.02, 1H, m |
| 11 | 23.5 | | 5 | 77.9 | 4.20, 1H, m |
| 12 | 122.4 | 5.44, 1H, s | 6 | 61.8 | 3.65, 2H, m |
| 13 | 143.8 | ------- | Fuc-1 | 93.43 | 6.30, 1H, d, 5.5 Hz |
| 14 | 41.7 | ------ | 2 | 76.7 | 4.25, 1H, m |
| 15 | 27.9 | | 3 | 83.7 | 3.95, 1H, m |
| 16 | 23.5 | | 4 | 72.4 | 3.86, 1H, m |
| 17 | 46.7 | ------- | 5 | 74.0 | 3.80, 1H, m |
| 18 | 41.8 | 3.04, 1H, dd-like | 6 | 18.3 | 1.54, 3H, d, 5.5 Hz |
| 19 | 46.7 | | RhmI-1 | 100.9 | 5.74, 1H, s |
| 20 | 30.6 | ------ | 2 | 71.4 | 4.33, 1H, m |
| 21 | 32.1 | | 3 | 72.3 | 4.28, 1H, m |
| 22 | 32.9 | | 4 | 82.9 | 4.50, 1H, m |
| 23 | 209.7 | 9.89, 1H, s | 5 | 68.7 | 4.01, H, m |
| 24 | 10.9 | 1.36, 3H, s | 6 | 18.3 | 1.67, 3H, d, 5.5 Hz |
| 25 | 15.4 | 0.76, 3H, s | RhmII-1 | 101.2 | 5.67, 1H, s |
| 26 | 17.3 | 0.92, 3H, s | 2 | 71.4 | 3.26, 1H, m |
| 27 | 26.1 | 1.18, 3H, s | 3 | 72.1 | 4.32, 1H, m |
| 28 | 175.9 | ------ | 4 | 70.6 | 4.71, 1H, m |
| 29 | 32.1 | 0.89, 3H, s | 5 | 68.7 | 4.51, 1H, m |
| 30 | 23.5 | 0.96, 3H, s | 6 | 18.3 | 1.61, 3H, d, 5.5 Hz |
| | | | XylI-1 | 106.2 | 5.08, 1H, d, 7.0 Hz |
| | | | 2 | 74.8 | 4.01, 1H, m |
| | | | 3 | 86.7 | 4.18, 1H, m |
| | | | 4 | 70.2 | 4.13, 1H, m |
| | | | 5 | 66.6 | 4.28/3.45, 2H, m |
| | | | XylII-1 | 105.6 | 5.21, 1H, d, 7.0 Hz |
| | | | 2 | 76.3 | 4.04, 1H, m |
| | | | 3 | 77.9 | 4.16, 1H, m |
| | | | 4 | 70.6 | 4.13, 1H, m |
| | | | 5 | 67.0 | 4.28/3. 61, 2H, m |
Figure 2.
Key HMBC correlations of compound C1.
Figure 2.
Key HMBC correlations of compound C1.
Compound
C2 was obtained as a white, amorphous powder, In the (−)- and (+)-ESI-MS of
C2, quasimolecular ion peaks were observed at
m/z 1,377 [M-H]
− and,
m/z 1,401 [M+Na]
+, respectively, and HR-ESI-MS analysis revealed the molecular formula of
C2 to be C
65H
102O
31 (calcd. 1401.6297, observed
m/z 1,401.6345 [M+Na]
+). Eight fragment ions at 1,247 [M–132+H], 1,217 [M–162+H], 1,085 [M–162–132+H], 939 [M–132–162–146+H], 909 [M–162–132–176+H], 793 [M–162–132–146–146+H], 647 [M–132–162–146–146–146+H] and 436 [M–132–162–146–146–146–176+H], indicated the sequential losses of one pentose and five hexoses. Acid hydrolysis of compound
C2 with 2 mol/L HCl–1,4-dioxane (1:1,
v/v) furnished
d-xylose,
l-rhamnose,
d-glucose,
d-fucose in the ratio of 1:2:1:1 which were identified by HPLC analysis of the thiazolidine derivatives following conversion to the corresponding 1-[(
S)-
N-acetyl-(
R)-methylbenzylamino]-1-deoxyalditol acetate derivatives [
8].
The
1H-NMR spectrum of
C2 revealed the presence of nine methyl proton signals at
δH 0.75 (Me-25), 0.88 (Me-29), 0.93 (Me-26), 0.95 (Me-30), 1.18 (Me-27), 1.37 (Me-24), 9.90 (H-29) and it included signals due to a gyosogenin skeleton [
8], 1.53, 3H, d,
J = 6.0 Hz (Me-Fuc), 1.62, 3H, d
, J = 6.5Hz (Me-Rha
II), 1.72, 3H, d,
J = 6.0 Hz (Me-Rha
I), as well as an olefinic proton at
δH 5.37, 1H, s (H-12); The signals at
δC 122.5, 143.9 in the
13C-NMR spectrum were assigned to the 12(13)-ene bond by comparison with literature data [
5].
Comparison of the
1H-NMR and
13C-NMR spectral data of compound
C2 (
Table 2) with that of
C1 indicated the compound
C2 lacked the xylpyranosyl moiety of
C1. The identities of the monosaccharides and the oligosaccharide sequence were determined by a combination of DEPT and two-dimensional NMR experiments (such as HMQC, HMBC, TOCSY NMR). The sequence of the glycan part was deduced from the following HMBC correlations: the anomeric proton signals at
δH 4.77 and
δC 83.9 (H-1 of the
β-
d-glucopyranosiduronic acid attached to the C-3 of the aglycone),
δH 5.37 and
δC 82.2 (H-1 of the
β-
d-glucopyranosyl moiety attached to the C-2 of the
β-
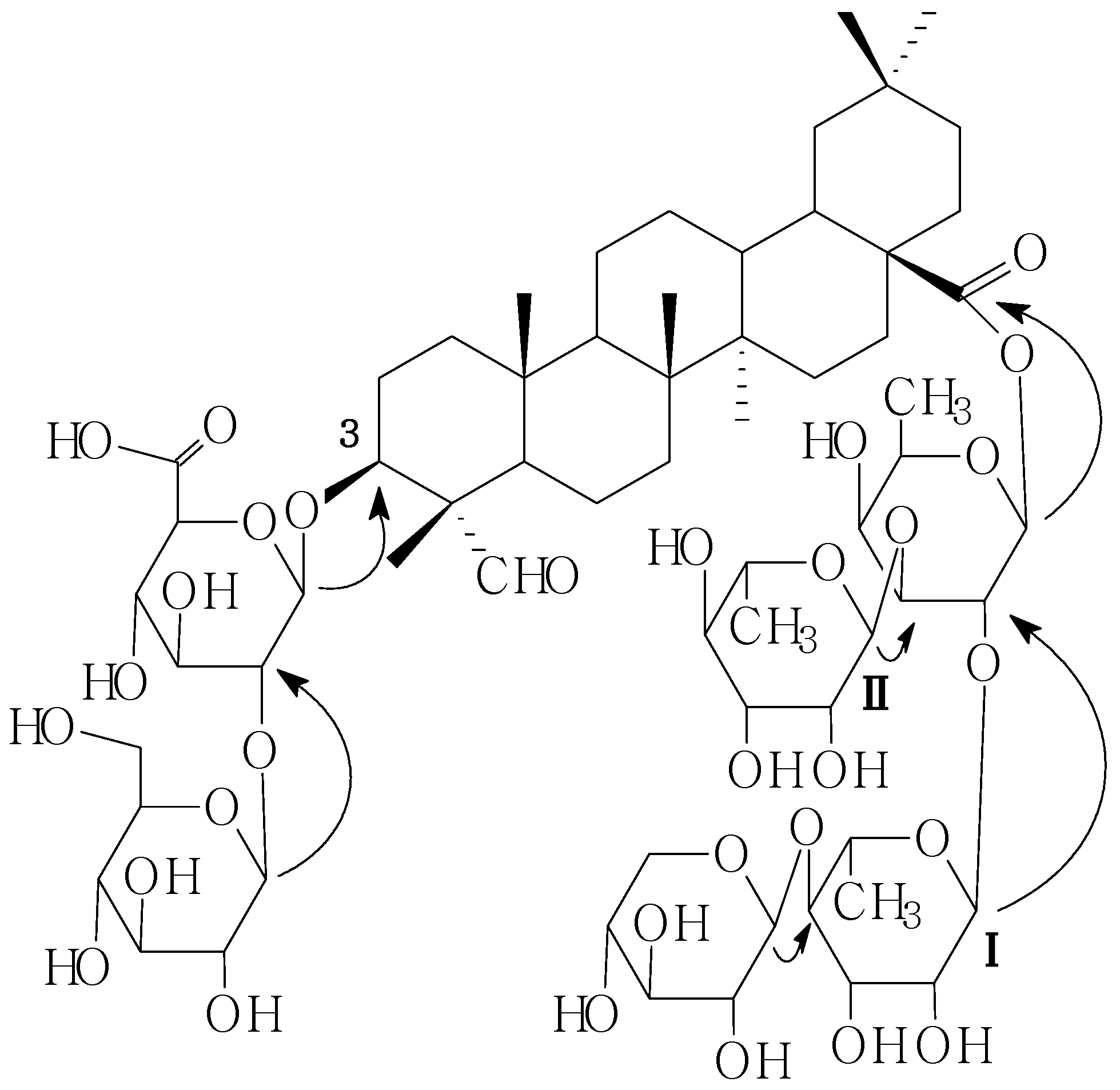
d-gluco-pyranosiduronic acid), and on the other hand, the oligosaccharide part at C-28 was established by the HMBC correlationns between the following protons and carbons: Fuc-1-H and 28-C, Rhm
I-1-H and Fuc-2-C, Rhm
II-1-H and Fuc-3-C, Xyl-1-H and Rhm
I-4-C (
Figure 3). On the basis of the foregoing evidence, the structure of
C2 was established as 28-
O-
β-
d-xylopyranosyl(1→4)-
α-
l-rhamno-pyranosyl(1→2)-[
α-
l-rhamnopyranosyl(1→3)]-
β-
d-fucopyranosyl gypsogenin 3-
O-
β-
d-gluco-pyranosyl(1→2)-
β-
d-glucopyranosiduronic acid.
Table 2.
1H-NMR (500 MHz) and 13C-NMR (125 MHz) data for C2 in pyridine-d5 (δ, ppm).
Table 2.
1H-NMR (500 MHz) and 13C-NMR (125 MHz) data for C2 in pyridine-d5 (δ, ppm).
| Position | C2-Aglycone | Position | C2-Sugar Chain |
|---|
| 13C | 1H | 13C | 1H |
|---|
| 1 | 38.0 | | GlcA-1 | 102.9 | 4.77, 1H, brs |
| 2 | 25.9 | | 2 | 82.2 | 4.36, 1H, m |
| 3 | 83.9 | 4.04, 1H, m | 3 | 77.8 | 4.65, 1H, m |
| 4 | 54.9 | ------ | 4 | 72.4 | 4.20, 1H, m |
| 5 | 48.2 | | 5 | 76.9 | 4.54, 1H, m |
| 6 | 18.6 | | 6 | 175.6 | |
| 7 | 32.4 | | Glc-1 | 106.2 | 5.37, 1H, s |
| 8 | 40.0 | | 2 | 75.4 | 4.51, 1H, m |
| 9 | 47.6 | | 3 | 78.4 | 4.52, 1H, m |
| 10 | 35.6 | | 4 | 71.6 | 4.06, 1H, m |
| 11 | 23.6 | | 5 | 77.9 | 4.36, 1H, m |
| 12 | 122.5 | 5.37, 1H, s | 6 | 62.0 | 4.57, 2H, m |
| 13 | 143.9 | ------- | Fuc-1 | 93.6 | 6.29, 1H, d, 5.5 Hz |
| 14 | 41.8 | ------ | 2 | 76.5 | 3.99, 1H, m |
| 15 | 28.0 | | 3 | 83.3 | 4.31, 1H, m |
| 16 | 23.7 | | 4 | 72.3 | 4.05, 1H, m |
| 17 | 46.9 | ------- | 5 | 74.2 | 4.07, 1H, m |
| 18 | 42.0 | 3.10, 1H, dd-like | 6 | 18.4 | 1.53,3H, d, 6.0 Hz |
| 19 | 46.1 | | RhmI-1 | 100.8 | 5.77, 1H, s |
| 20 | 30.7 | ------ | 2 | 70.8 | 4.33, 1H, m |
| 21 | 32.2 | | 3 | 72.4 | 4.28, 1H, m |
| 22 | 33.0 | | 4 | 83.0 | 4.52, 1H, m |
| 23 | 209.8 | 9.90, 1H, s | 5 | 68.7 | 3.13, 1H, m |
| 24 | 10.9 | 1.37, 3H, s | 6 | 18.8 | 1.72, 3H, d, 6.0 Hz |
| 25 | 15.6 | 0.76, 3H, s | RhmII-1 | 101.5 | 5.68, 1H, s |
| 26 | 17.4 | 0.93, 3H, s | 2 | 71.6 | 3.85, 1H, m |
| 27 | 25.9 | 1.18, 3H, s | 3 | 72.4 | 4.32, 1H, m |
| 28 | 176.2 | ------ | 4 | 70.4 | 4.75, 1H, m |
| 29 | 32.0 | 0.88, 3H, s | 5 | 68.7 | 4.53, 1H, m |
| 30 | 23.1 | 0.95, 3H, s | 6 | 18.6 | 1.62, 3H, d, 6.5 Hz |
| | | | Xyl-1 | 106.9 | 5.15, 1H, d,7.0 Hz |
| | | | 2 | 74.8 | 4.04, 1H, m |
| | | | 3 | 86.9 | 4.21, 1H, m |
| | | | 4 | 70.0 | 4.13, 1H, m |
| | | | 5 | 67.3 | 4.27/3.48, 2H, m |
Figure 3.
Key HMBC correlations of compound C2.
Figure 3.
Key HMBC correlations of compound C2.
3. Experimental Section
3.1. General
Melting points were determined of an XT4-100x micromelting apparatus (Beijing Keyi Electric Light Instrument Factory, Beijing, China) and are uncorrected. Optical rotations were measured with a Perkin-Elmer 241 MC polarimeter (PerkinElmer Inc., Waltham, MA, USA). IR spectra were obtained of Nicolet 5700 IR spectrometer (Thermo Fisher Scientific, Inc., Waltham, MA, USA). NMR spectra were recorded on an Inova 500 (1H, 500 MHz; 13C, 125 MHz) spectrometer (Agilent Technologies, Inc., Santa Clara, CA, USA). ESI-MS was performed with Agilent 1100 LC/MSD (Agilent Technologies, Inc., Santa Clara, CA, USA). For column chromatography, silica gel (200-300 mesh, Qingdao Marine Chemical Inc., Qingdao, China), ODS (40–60 μm, Alltech), and Sephadex LH-20 (Pharmacia Biotech AB, Uppsala, Sweden) were used. The analytical HPLC was performed on an Agilent 1200 LC equipped with a DAD and the preparative HPLC was performed on a Shimadzu LC-20A (Shimadzu LC-20A, Kyoto, Japan) equipped with a YMC-Pack ODS column (20 × 250 mm, 10 μm, YMC Co. Ltd. Kyoto, Japan).
3.2. Plant Material
The seeds of Momordica charantia L. were purchased from Anguo of Hebei Province in 2011, and identified by Lin Ma at Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College. A voucher specimen has been deposited at our lab at the Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College.
3.3. Extraction and Isolation
The seeds of M. charantia L. (15.0 kg) were defatted three times by petroleun ether (90 L each time), and then extracted three times by heating to reflux with 95% ethanol (90 L each time), and the combined solution was concentrated under reduced pressure to yield an extract (1.6 kg). The alcohol extract was partitioned successively with CHCl3, EtOAc and n-BuOH. The n-BuOH-soluble portions (175 g) was subjected to normal phase silica gel column chromatography with gradient elution [CHCl3–MeOH 20:1 (2 L), CHCl3–MeOH 9:1 (2 L), CHCl3–MeOH 4:1 (2 L), CHCl3–MeOH–H2O 7:3:0.5 (2 L), CHCl3–MeOH–H2O 6:4:0.5 (2 L), MeOH (2 L)] to give eleven fractions (Fr1-11). Compound C1 (8.0 mg) was purified from Fr9 (7.6 g) by normal-phase silica gel and Sephadex LH-20 chromatography. Compound C2 (5.0 mg) was purified from Fr10 (20.6 g) by repeated column chromatography as well as compound C1. Compound C3 (210.0 mg) was purified from Fr10 (20.6 g) by repeated column chromatography over silica gel with eluent as CHCl3–MeOH–H2O 4:1:0.1. Compound C4 (102.0 mg) was purified from Fr11 (12.5 g) by repeated column chromatography over silica gel with eluent as CHCl3–MeOH–H2O 6:4:0.5, and further purified by Sephadex LH-20 with eluent as MeOH and preparative HPLC (MeOH–H2O, 60:40).
3.4. Acid Hydrolysis of Compounds C1 and C2
d-Glucose, d-galactose, l-rhamnose, d-fucose, d-xylose and l-arabinose aqueous solution (each 2 mg/mL, 80 μL) PMP CH3OH solution (0.5 mol/L, 80 μL) and aqueous NaOH solution (0.3 mol/L, 80 μL) were heated at 70 °C for 30 min, cooled to room temperature for 10 min, HCl aqueous solution added (0.3 mol/L, 80 μL) and extracted with CHCl3 (0.5 mL, three times). The aqueous fractions were identified by HPLC analysis (Phenomenex C18, 250 mm × 4.6 mm, 5 μm column); flow phase A: CH3CN-20 mmol/L NH4OAc aqueous solution (15:85), B: CH3CN-20 mmol/L NH4OAc aqueous solution (40:60), flow rate: 1.2 mL/min; gradient elution, 0→20 min, volume fraction of B from 0 to 60%; detection wavelengths: 245 nm; sample volume: 20 μL).
Compounds C1 (2 mg) and C2 (2 mg) were heated in an ampule with aqueous 2 mol/L HCl–1,4-dioxane (1:1, 2 mL) at 80 °C for 6 h. The aglycon was extracted with chloroform, and the aqueous layer was evaporated under reduced pressure and subjected to the normal preparation of sugar derivatives. Thus, compound C1 furnished
d-xylose,
l-rhamnose,
d-glucose, and
d-fucose in a ratio of 2:2:1:1, and compound C2 furnished
d-xylose,
l-rhamnose,
d-glucose and
d-fucose in the ratio of 1:2:1:1, which were identified by HPLC analysis of the thiazolidine derivatives following conversion to the 1-[(
S)-
N-acetyl-(
R)-methylbenzylamino]-1-deoxyalditol acetate derivatives [
7].




{kind=link}
{kind=link}
{kind=link}

