Molecular Disorder in (‒)-Encecanescin

Abstract
:
1. Introduction

2. Results and Discussion
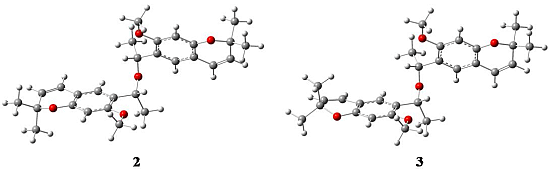
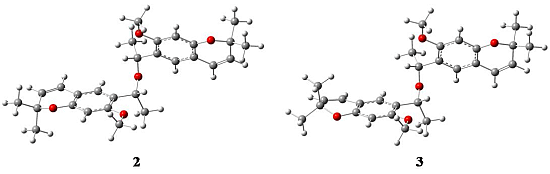
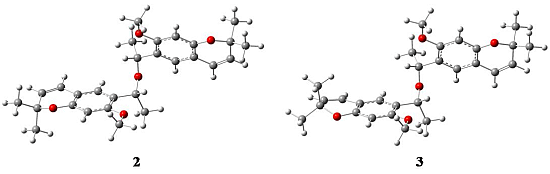
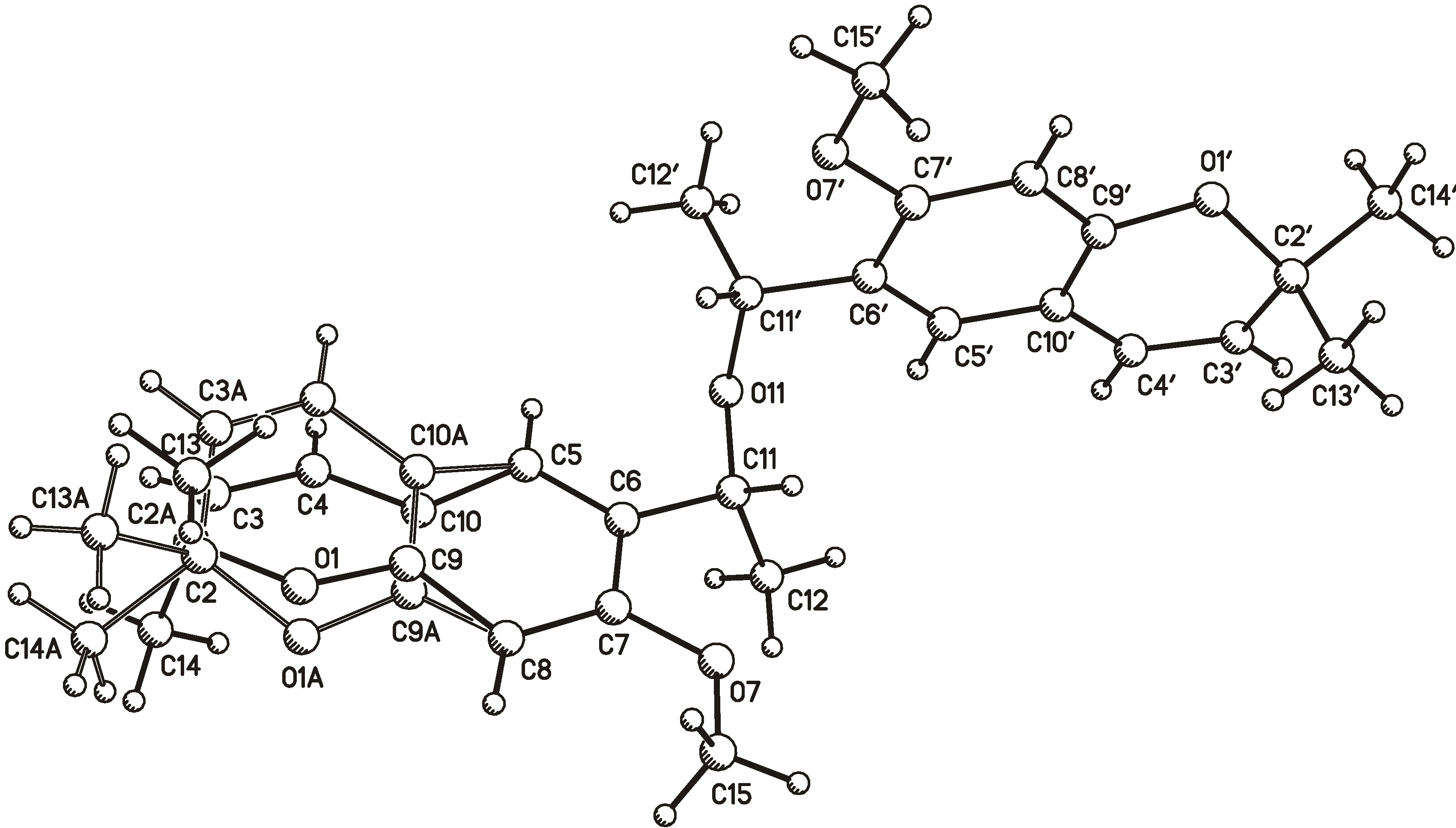
2.1. X-ray Crystallography

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical formula | C28H34O5 |
|---|---|
| Formula weight | 450.2402 |
| Temperature | 293(2) K |
| Wavelength | 0.71073 Å |
| Crystal system, space group | monoclinic, P21/c |
| Unit cell dimensions | a = 11.0930(2) Å alpha = 90° |
| b = 8.4352(2) Å beta = 94.622(11)° | |
| c = 27.5559(11) Å gamma = 90° | |
| Volume | 2570.07(14) Å3 |
| ZCalculated density | 40.851 Mg/m3 |
| Absorption coefficient | 0.061 mm−1 |
| F(000) | 724 |
| Crystal size | 0.24 × 0.20 × 0.18 mm |
| Theta range for data collection | 2.46° to 27.49° |
| Limiting indices | −14 ≤ h ≤ 13, −10 ≤ k ≤ 10, −35 ≤ l ≤ 28 |
| Reflections collected/ unique | 14836/5814 [R(int) = 0.0892] |
| Completeness to theta = 27.49 | 98.7% |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 5814/216/379 |
| Goodness-of-fit on F2 | 0.918 |
| Final R indices | [I > 2sigma(I)] R1 = 0.0671, wR2 = 0.1293 |
| R indices (all data) | R1 = 0.2389, wR2 = 0.1772 |
| Largest diff. peak and hole | 0.160 and −0.166 e.A−3 |
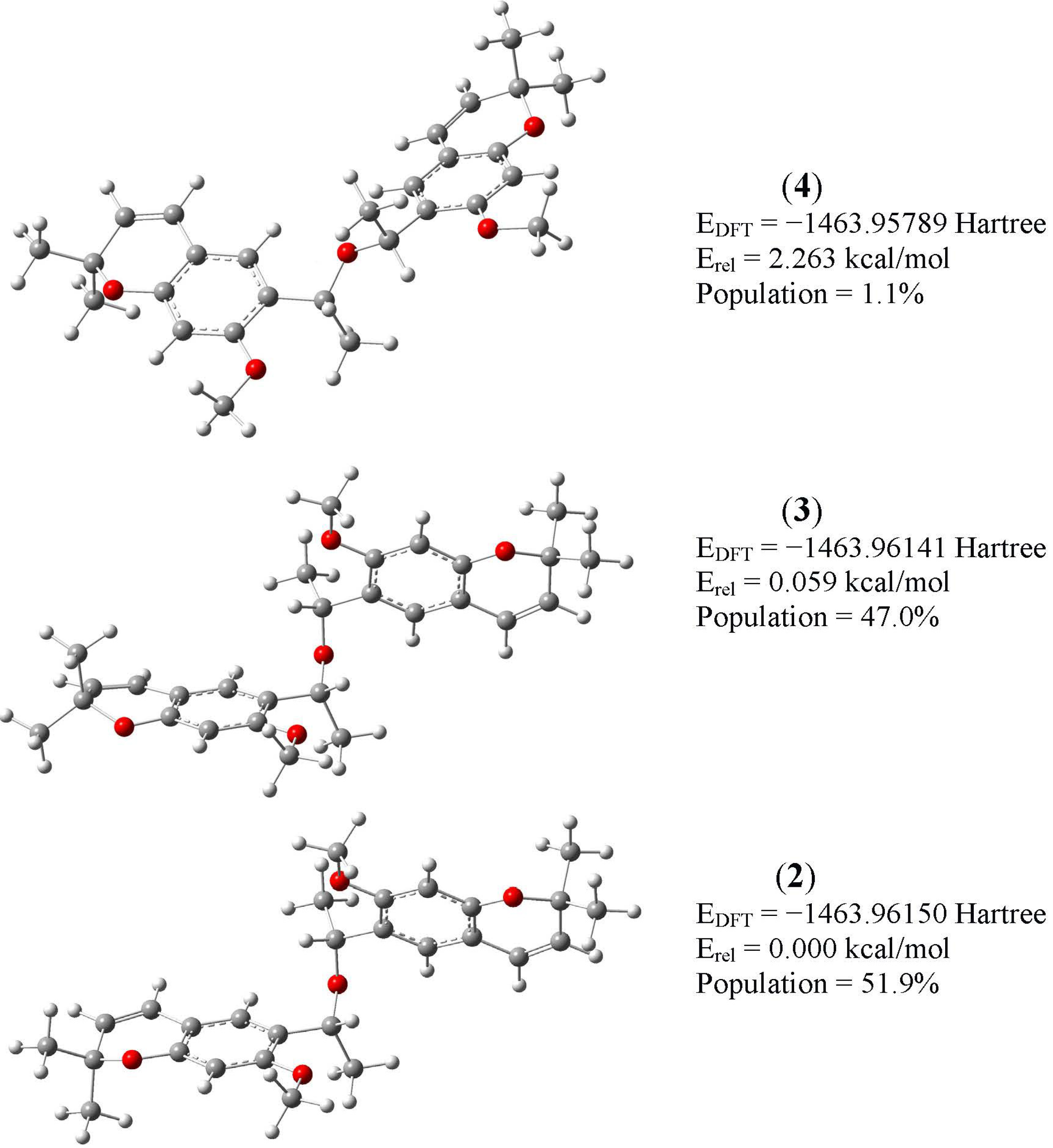
2.2. B3LYP Calculations

| Parameter | 1 | 2 | 3 |
|---|---|---|---|
| Bond length | |||
| C(10)-C(5) | 1.401(15) | 1.40026 | 1.4005 |
| C(10)-C(4) | 1.436(15) | 1.45559 | 1.4568 |
| C(10)-C(9) | 1.482(16) | 1.40225 | 1.4032 |
| C(4)-C(3) | 1.310(10) | 1.33793 | 1.3386 |
| C(3)-C(2) | 1.38(2) | 1.51363 | 1.5129 |
| C(2)-C(13) | 1.446(15) | 1.52796 | 1.53535 |
| C(2)-C(14) | 1.451(16) | 1.53726 | 1.52713 |
| C(2)-O(1) | 1.486(13) | 1.46334 | 1.46937 |
| O(1)-C(9) | 1.36(2) | 1.36414 | 1.36453 |
| C(9)-C(8) | 1.44(2) | 1.39292 | 1.39302 |
| C(5)-C(6) | 1.377(4) | 1.38931 | 1.39071 |
| C(6)-C(7) | 1.396(4) | 1.40947 | 1.41067 |
| C(3')-C(2') | 1.494(4) | 1.51363 | 1.51290 |
| C(2')-C(13') | 1.521(4) | 1.52796 | 1.53535 |
| C(2')-C(14') | 1.521(4) | 1.53726 | 1.52713 |
| Bond angle | |||
| C(5)-C(10)-C(4) | 126.7(9) | 124.31413 | 124.28231 |
| C(5)-C(10)-C(9) | 113.3(13) | 118.07731 | 118.01906 |
| C(4)-C(10)-C(9) | 112.7(14) | 117.54869 | 117.66232 |
| C(3)-C(4)-C(10) | 122.6(8) | 120.34825 | 120.33249 |
| C(4)-C(3)-C(2) | 125.4(10) | 121.02151 | 121.32649 |
| C(3)-C(2)-C(13) | 123.9(16) | 111.64217 | 110.71254 |
| C(3)-C(2)-C(14) | 76.0(9) | 110.62884 | 111.60611 |
| C(13)-C(2)-C(14) | 157.0(19) | 111.16920 | 111.19836 |
| C(3)-C(2)-O(1) | 115.7(14) | 110.60774 | 110.50808 |
| C(13)-C(2)-O(1) | 75.0(8) | 104.5417 | 108.01913 |
| C(14)-C(2)-O(1) | 108.6(10) | 108.03583 | 104.57564 |
| C(9)-O(1)-C(2) | 118.6(13) | 118.74609 | 119.00702 |
| O(1)-C(9)-C(8) | 119.7(11) | 117.56225 | 117.58616 |
| O(1)-C(9)-C(10) | 123.7(17) | 121.35132 | 121.26106 |
| C(3')-C(2')-C(13') | 111.4(3) | 110.60783 | 110.71254 |
| C(3')-C(2')-C(14') | 111.2(3) | 111.56751 | 111.60611 |
| C(13')-C(2')-C(14') | 111.0(3) | 111.18802 | 111.19836 |
| C(13)-C(2)-O(1) | 106.7(3) | 107.99372 | 108.01913 |
| Dihedral angle | |||
| C(4)-C(3)-C(2)-C(13) | 80.0(17) | 140.83879 | 93.83749 |
| C(4)-C(3)-C(2)-C(14) | −112.9(12) | −94.753 | −141.72175 |
| C(13)-C(2)-O(1)-C(9) | −115.3(12) | −156.22747 | −84.38296 |
| C(14)-C(2)-O(1)-C(9) | 88.5(17) | 85.28967 | 157.09634 |
| C(13')-C(2')-C(3')-C(4') | 97.5(3) | 94.55224 | 93.83749 |
| C(14')-C(2')-C(3')-C(4') | −138.1(3) | −141.02837 | −141.72175 |
| C(13')-C(2')-O(1')-C(9') | −88.3(3) | −85.26823 | −84.38296 |
| C(14')-C(2')-O(1')-C(9') | 153.8(2) | 156.2661 | 157.09634 |
2.3. FTIR and Raman Spectra
| Raman | IRexp | IRcalc | INT | IRcalc | INT | Proposed assignment |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | ||||
| 3043.66 | 3041 w | 3177 | 15.7 | 3176 | 10.4 | υC-H Ar |
| 2983.39 | 2974 m | 3131 | 17.8 | 3131 | 35.9 | υas CH3 methoxy |
| 2938.11 | 2932 w | 3106 | 24.7 | 3106 | 23.8 | υas CH3 gem |
| 1645.95 | 1644 vw | 1651 | 298.0 | 1651 | 3.2 | υs HC=CH ring |
| 1621.54 | 1615 m | 1599 | 31.4 | 1600 | 26 | υs HC=CH ring |
| 1578.38 | 1576 s | 1523 | 155.4 | 1524 | 181.6 | βC-H Ar+CH3 |
| 1499.45 | 1492 w | 1493 | 18.7 | 1493 | 23.3 | γCH3 methoxy |
| 1462 vw | 1479 | 1.8 | 1479 | 2.4 | ωCH3 methoxy | |
| 1432.21 | 1443 vw | 1457 | 2.4 | 1456 | 1.1 | βring, Ar-O-R |
| 1380 m | 1382 | 52.6 | 1383 | 49.3 | ρHC=CH ring/ωAr-C-H-OCH3 | |
| 1362.52 | 1360 m | 1372 | 12.6 | 1374 | 26.1 | γC-H |
| 1307.86 | 1303 s | 1303 | 154.2 | 1303 | 159.1 | υas HC=CH Ar |
| 1279 m | 1284 | 35.4 | 1285 | 27.8 | βAr, ring | |
| 1239.18 | 1230 m | 1231 | 33.3 | 1232 | 40.8 | βHC=CH ring/υas CH3 gem |
| 1196 s | 1214 | 158.7 | 1214 | 167.4 | ρCH3 methoxy, CH3 gem | |
| 1173.11 | 1163 m | 1178 | 58.3 | 1180 | 89.3 | βHC=CH ring, C-H Ar |
| 1114.62 | 1123 vs | 1145 | 625.3 | 1145 | 615.6 | υasC-O-C |
| 1093 m | 1104 | 100.6 | 1099 | 37.7 | τCH3-C-O-C-CH3 | |
| 1074 vs | 1087 | 218.5 | 1087 | 224.4 | ρCH3/υas C-O-C | |
| 1029 m | 1048 | 39.8 | 1048 | 44.3 | υCH3-CH,CH3-O | |
| 1013 s | 1033 | 88.9 | 1033 | 77.6 | υasAr-O-CH3/τHC-CH3 | |
| 950.96 | 959 m | 967 | 28.5 | 967 | 43.7 | υsHC-C(CH3)2-O, CH3-CH-O |
| 884.95 | 894 m | 902 | 55.6 | 902 | 55.5 | τCH3 gem |
| 804.36 | 801 vw | 809 | 3.1 | 809 | 1.1 | τAr-O-R, ring |
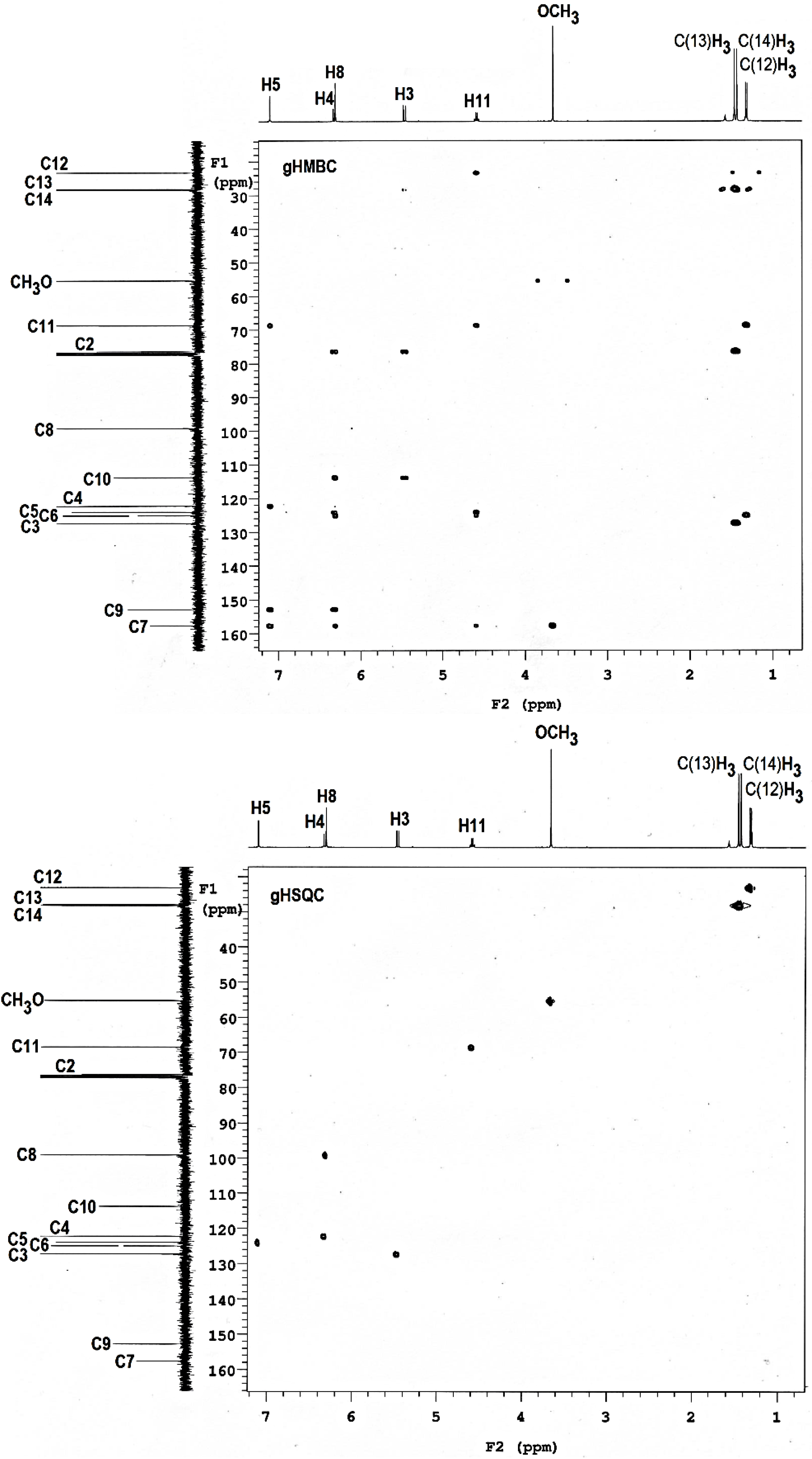
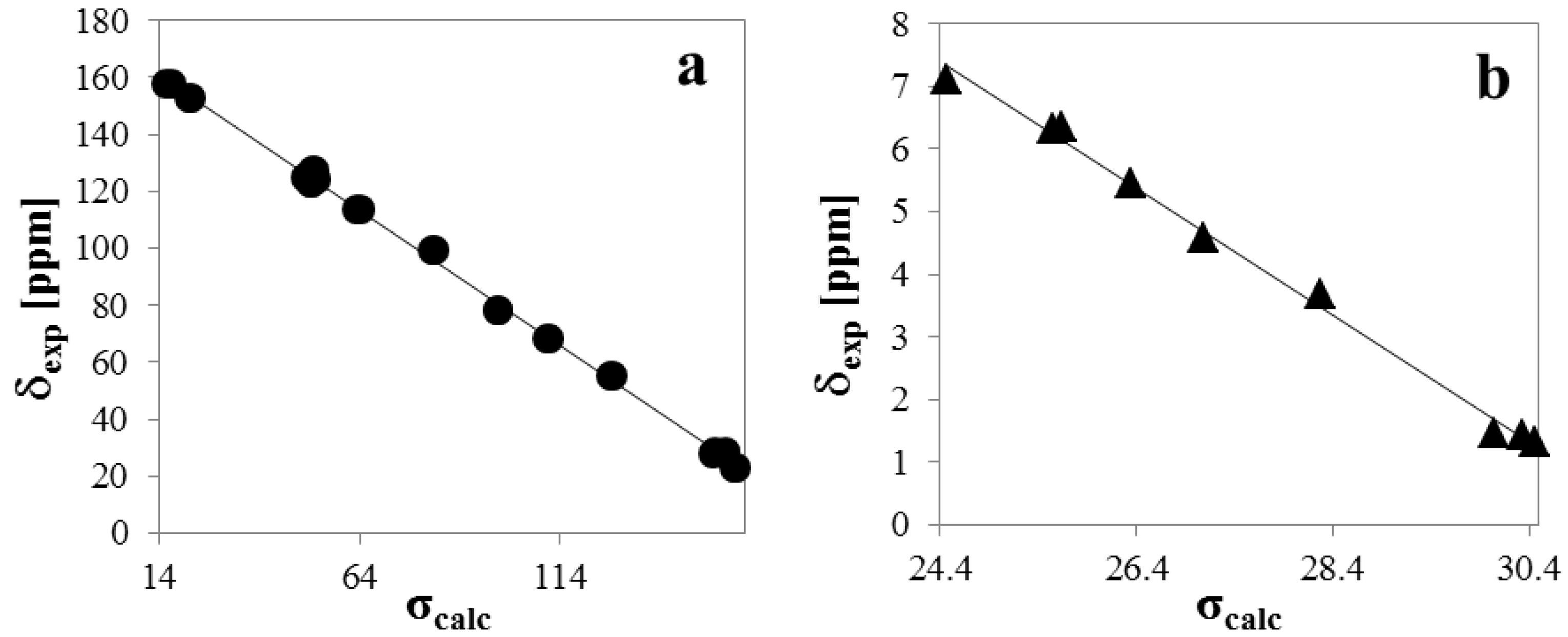
2.4. 1H- and 13C-NMR Spectra

| Atom | δexp | δpred (2) | δpred (3) | σcalc (2) | σcalc (3) |
|---|---|---|---|---|---|
| C(2) | 78.3 | 80.7 | 80.2 | 98.47 | 98.84 |
| C(3) | 127.3 | 124.1 | 124.5 | 52.53 | 52.00 |
| C(4) | 122.7 | 124.9 | 124.3 | 51.68 | 52.19 |
| C(5) | 124.0 | 124.4 | 123.9 | 52.22 | 52.58 |
| C(6) | 125.1 | 126.3 | 125.7 | 50.27 | 50.70 |
| C(7) | 157.7 | 158.8 | 157.6 | 15.85 | 17.03 |
| C(8) | 99.2 | 96.2 | 95.7 | 82.07 | 82.46 |
| C(9) | 152.8 | 153.6 | 154.5 | 21.32 | 20.25 |
| C(10) | 113.8 | 113.4 | 114.0 | 63.91 | 63.12 |
| C(11) | 68.6 | 68.5 | 69.3 | 111.29 | 110.30 |
| C(12) | 23.2 | 24.7 | 24.5 | 157.67 | 157.66 |
| C(13) | 28.3 | 29.5 | 27.0 | 152.57 | 155.04 |
| C(14) | 28.1 | 26.8 | 29.5 | 155.41 | 152.43 |
| C(15) | 55.3 | 53.8 | 53.7 | 126.87 | 126.85 |
| C(2') | 78.3 | 80.7 | 80.2 | 98.47 | 98.84 |
| C(3') | 127.3 | 124.5 | 124.5 | 52.16 | 52.00 |
| C(4') | 122.7 | 125.0 | 124.3 | 51.64 | 52.19 |
| C(5') | 124.0 | 123.7 | 123.9 | 53.01 | 52.58 |
| C(6') | 125.1 | 125.0 | 125.7 | 51.61 | 50.70 |
| C(7') | 157.7 | 157.8 | 157.6 | 16.96 | 17.03 |
| C(8') | 99.2 | 95.7 | 95.7 | 82.58 | 82.46 |
| C(9') | 152.8 | 153.3 | 154.5 | 21.67 | 20.25 |
| C(10') | 113.8 | 113.9 | 114.0 | 63.28 | 63.12 |
| C(11') | 68.6 | 68.8 | 69.3 | 110.98 | 110.30 |
| C(12') | 23.2 | 24.4 | 24.5 | 157.92 | 157.66 |
| C(13') | 28.3 | 27.0 | 27.0 | 155.15 | 155.04 |
| C(14') | 28.1 | 29.6 | 29.5 | 152.41 | 152.43 |
| C(15') | 55.3 | 53.7 | 53.7 | 126.98 | 126.85 |
| A | 173.81 | 173.68 | |||
| B | −0.946 | −0.946 | |||
| r2 | 0.9986 | 0.9986 |
| Atom | δexp | δpred (2) | δpred (3) | σcalc (2) | σcalc (3) |
|---|---|---|---|---|---|
| H(3) | 5.46 | 5.44 | 5.44 | 26.34 | 26.34 |
| H(4) | 6.34 | 6.25 | 6.39 | 25.54 | 25.33 |
| H(5) | 7.10 | 7.34 | 7.21 | 24.46 | 24.46 |
| H(8) | 6.32 | 6.15 | 6.05 | 25.64 | 25.69 |
| H(11) | 4.59 | 4.68 | 4.69 | 27.08 | 27.15 |
| H(12) | 1.31 | 1.26 | 1.29 | 30.46 | 30.78 |
| H(13) | 1.46 | 1.69 | 1.29 | 30.04 | 30.77 |
| H(14) | 1.43 | 1.41 | 1.64 | 30.32 | 30.39 |
| H(15) | 3.67 | 3.48 | 3.68 | 28.28 | 28.22 |
| A | 32.076 | 30.141 | |||
| B | −1.0114 | −0.9376 | |||
| r2 | 0.9956 | 0.9956 |

3. Experimental
3.1. General
3.2. Materials
3.3. Methods
3.4. Computational Calculations
4. Conclusions
Supplementary Materials
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Bowers, W.S.; Ohta, T.; Cleere, J.S.; Marsella, P.A. Discovery of insect anti-juvenile hormones in plants. Plants yield a potential fourth-generation insecticide. Science 1976, 193, 542–547. [Google Scholar]
- Salamon, E.; Mannhold, R.; Weber, H.; Lemoine, H.; Frank, W. 6-Sulfonylchromenes as highly potent KATP-channel openers. J. Med. Chem. 2002, 45, 1086–1097. [Google Scholar] [CrossRef]
- Fang, N.; Yu, S.; Mabry, T.J. Chromenes from ageratina arsenii and revised structures of two epimeric chromene dimers. Phytochemistry 1988, 27, 1902–1905. [Google Scholar] [CrossRef]
- Bohlmann, F.; Tsankova, E.; Jakupovic, J.; King, R.M.; Robinson, H. Dimeric chromenes and mixed dimers of a chromene with euparin from encelia canescens. Phytochemistry 1983, 22, 557–560. [Google Scholar] [CrossRef]
- Abboud, K.A.; Simonsen, S.A.; Fang, N.; Yu, S.; Mabry, T.J. Structure of (−/+)-encecanescin. Acta Crystallogr. 1990, C46, 1563–1566. [Google Scholar]
- Sánchez-Mendoza, M.E.; Reyes-Trejo, B.; Sánchez-Gómez, P.G.; Cervantes-Cuevas, H.; Castillo-Henkel, C.; Arrieta, J. Bioassay-guided isolation of an anti-ulcer benzochromene from Eupatorium aschembornianum: Role of nitric oxide, prostaglandins and sulfhydryls. Fitoterapia 2010, 81, 66–71. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar]
- Alcolea Palafox, M. Scaling factors for the prediction of vibrational spectra. I. Benzene molecule. Int. J. Quantum Chem. 2000, 77, 661–684. [Google Scholar] [CrossRef]
- Alcolea Palafox, M.; Rastogi, V.K. Quantum chemical predictions of the vibrational spectra of polyatomic molecules. The uracil molecule and two derivatives. Spectrochim. Acta A 2002, 58, 411–440. [Google Scholar] [CrossRef]
- Simpson, J.H. Organic Structure Determination Using 2-D NMR Spectroscopy; Elsevier Academic Press: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Osmiałowski, B.; Kolehmainen, E.; Gawinecki, R. GIAO/DFT calculated chemical shifts of tautomeric species. 2-Phenacylpyridines and (Z)-2-(2-hydroxy-2-phenylvinyl)pyridines. Magn. Reson. Chem. 2001, 39, 334–340. [Google Scholar] [CrossRef]
- Chang, G.; Guida, W.C.; Still, W.C. An internal-coordinate Monte Carlo method for searching conformational space. J. Am. Chem. Soc. 1989, 111, 4379–4386. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. V. Systematic optimization of exchange correlation functionals. J. Chem. Phys. 1997, 107, 8554–8560. [Google Scholar] [CrossRef]
- Hehre, W.J.; Random, L.; Schleyer, P.V.R.; Pople, J.A. Ab Initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 13, 16378–16396. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Wolinski, K.; Hilton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Sample Availability: A sample of (−)-encecanescin (1) is available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Reyes-Trejo, B.; Guerra-Ramírez, D.; Zuleta-Prada, H.; Santillán, R.; Sánchez-Mendoza, M.E.; Arrieta, J.; Reyes, L. Molecular Disorder in (‒)-Encecanescin. Molecules 2014, 19, 4695-4707. https://doi.org/10.3390/molecules19044695
Reyes-Trejo B, Guerra-Ramírez D, Zuleta-Prada H, Santillán R, Sánchez-Mendoza ME, Arrieta J, Reyes L. Molecular Disorder in (‒)-Encecanescin. Molecules. 2014; 19(4):4695-4707. https://doi.org/10.3390/molecules19044695
Chicago/Turabian StyleReyes-Trejo, Benito, Diana Guerra-Ramírez, Holber Zuleta-Prada, Rosa Santillán, María Elena Sánchez-Mendoza, Jesús Arrieta, and Lino Reyes. 2014. "Molecular Disorder in (‒)-Encecanescin" Molecules 19, no. 4: 4695-4707. https://doi.org/10.3390/molecules19044695
APA StyleReyes-Trejo, B., Guerra-Ramírez, D., Zuleta-Prada, H., Santillán, R., Sánchez-Mendoza, M. E., Arrieta, J., & Reyes, L. (2014). Molecular Disorder in (‒)-Encecanescin. Molecules, 19(4), 4695-4707. https://doi.org/10.3390/molecules19044695