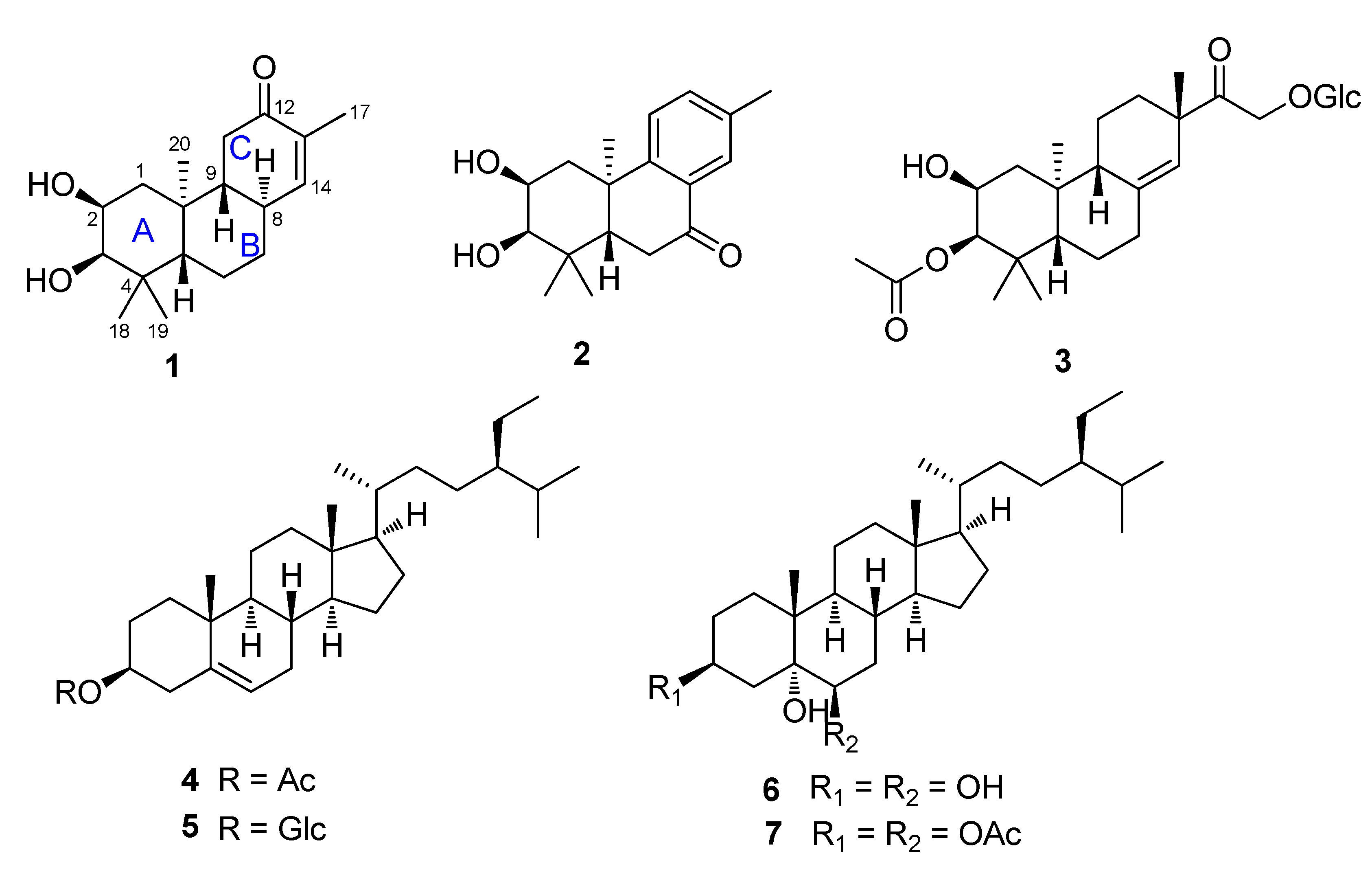
2. Results and Discussion
The air-dried powder of the leaves of F. fimbriata was extracted with 95% EtOH at room temperature to give a crude extract, which was then suspended in H2O and successively partitioned with petroleum ether, EtOAc, and n-BuOH, respectively. Various column chromatographic separations of the petroleum ether, EtOAc extract afforded compounds 1–7.
Compound
1 was obtained as a colorless oil with the molecular formula C
18H
28O
3 as established by an
m/
z of 315.1943 [M + Na]
+ (calcd for C
18H
28O
3Na, 315.1936) from HRESIMS. The
1H-NMR spectrum of
1 showed four tertiary methyls [at δ
H 2.27, 1.00, 0.94, and 0.87 (each 3H, s)], two oxygenated methines [at δ
H 4.05 (ddd,
J = 2.8, 4.4, and 11.8 Hz, H-2) and 3.45 (d,
J = 2.8 Hz, H-3)], and an olefinic proton [at δ
H 6.65 (t,
J = 1.6 Hz, H-14)]. The
13C-NMR spectrum in combination with DEPT experiments showed 18 carbon resonances, including four quaternary carbons (one conjugated carbonyl and one olefinic), six methines (two oxygenated and one olefinic), four methylenes, and four quaternary methyls. The double bond and the carboxyl group accounted for two out of the five degrees of unsaturation, and the leftover double-bond equivalents required a tricyclic nature of
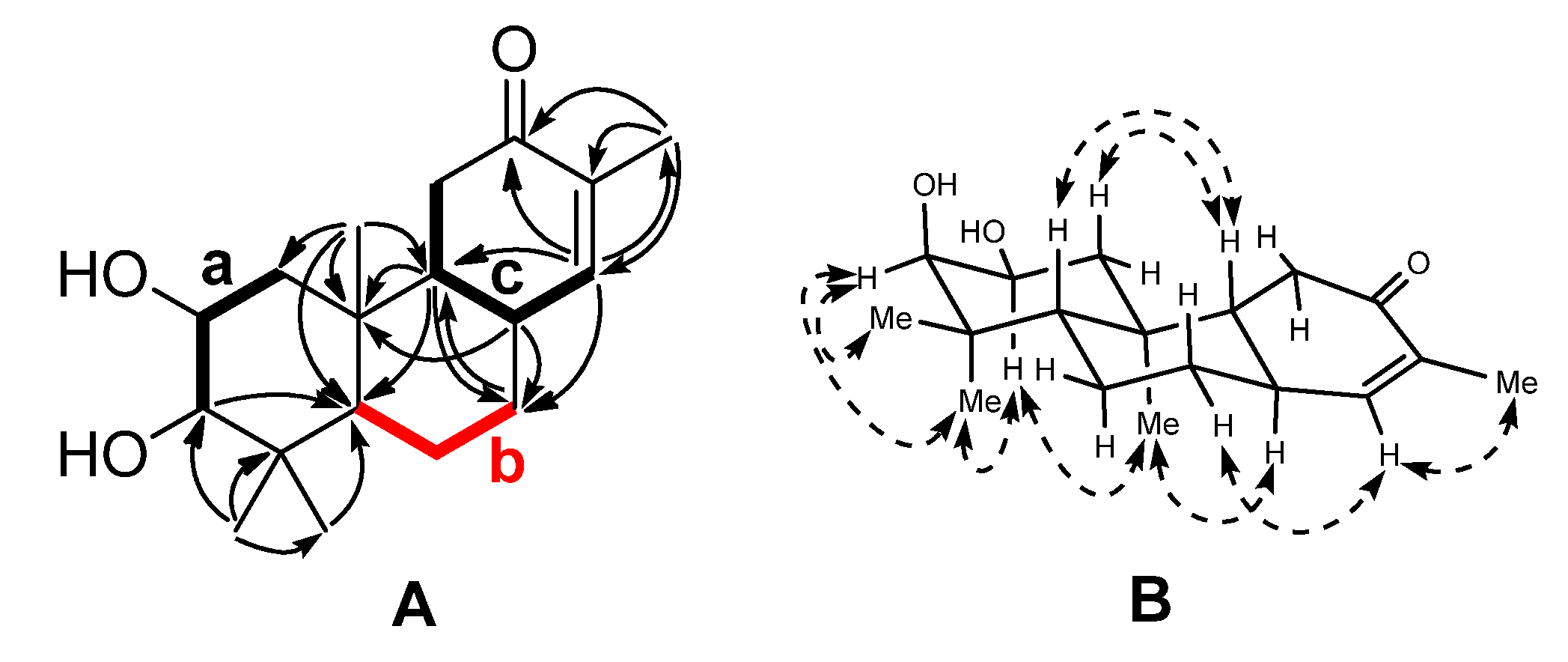
1. The gross structure was constructed by two-dimensional (2D) NMR analysis. Three fragments (
Figure 2),
a (C-1 to C-3),
b (C-5 to C-7), and
c (C-14, C-8, C-9, and C-11) were established by the correlations observed in the
1H-
1H COSY spectrum of
1. The connectivity of these fragments, quaternary carbons and other substitutes was accomplished mainly by analysis of the HMBC spectrum (
Figure 2). Fragment
a, C-10 bearing the angular methyl group (C-20), the C-5 methine, and the
gem-dimethyl group could construct ring A by the HMBC correlations from H
3-20 to C-1, C-5, and C-10 and from H
3-18/19 to C-3, C-4, and C-5. The HMBC correlations of H-9 to C-5, C-7, and C-10, H-7 to C-9, and H-8 to C-7, C-9 and C-10 supported that ring B was constructed by C-8–C-10 and fragment
b, and fused with ring A via C-5 and C-10. Fragment
c and the α,β-unsaturated ketone group provide ring C by the HMBC correlations from H-14 to C-8, C-9, C-12, and C-13. The planar structure of
1 thus emerged from the above spectra analysis.
Figure 2.
(
A) Selected
1H-
1H COSY (▬) and HMBC (→) correlations of
1; (
B) Selected NOESY correlations of
1 (
![Molecules 19 05863 i001]()
).
Figure 2.
(
A) Selected
1H-
1H COSY (▬) and HMBC (→) correlations of
1; (
B) Selected NOESY correlations of
1 (
![Molecules 19 05863 i001]()
).
The relative configuration of
1 was determined by analysis of the NOESY correlations (
Figure 2) and coupling constants. The NOESY spectrum showed cross peaks between the proton pairs H-2/H
3-19, H-2/H
3-20, indicating that these protons were cofacial and axial oriented, which were arbitrarily assigned as α orientation. Accordingly, a chair conformation for ring A was assigned. Based on the small coupling constant (
J = 2.6 Hz) between H-2 and H-3, the OH-3 was assigned as β orientation. The β assignment of coplanar protons H-5 and H-9 were deduced from the NOE correlations of H-1β/H-5, H-5/H-7β, and H-5/H-9.
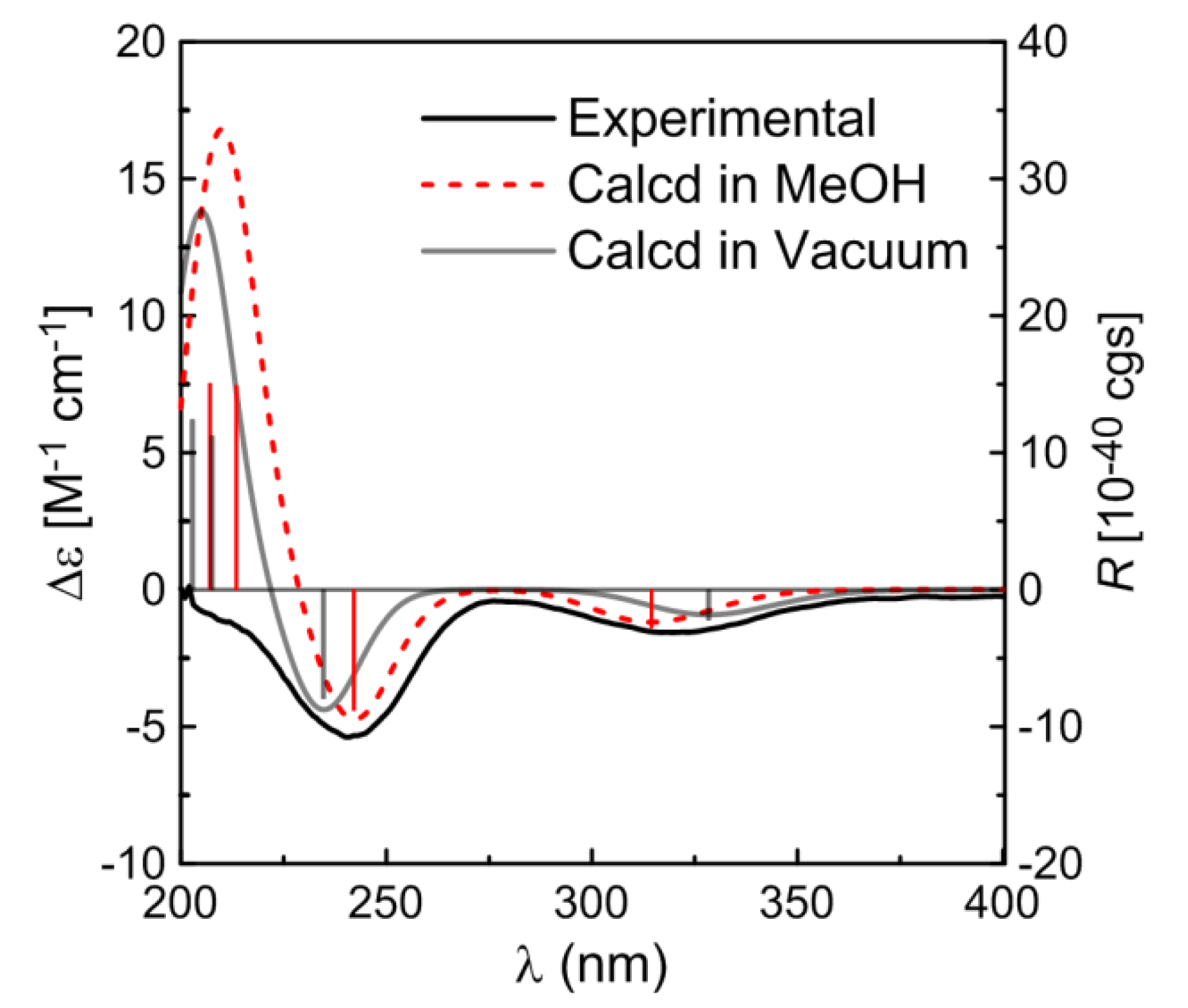
The absolute configuration of
1 was established by comparing the experimental ECD spectrum with the calculated data. The TDDFT calculations were performed using the B3LYP functional and TZVP basis set. Then conformer of
1 were subjected to TDDFT calculations for solution CD in MeOH and also calculated for the gas phase CD. The simulated CD spectra (
Figure 3) exhibited negative Cotton effects (CEs) around 320 nm and 240 nm and a positive CE around 210 nm, all generally consistent with the experimental spectrum, and the weighted spectra in both the gas phase and MeOH solution (
Figure 3) provided the excellent fit with the experimental data, giving a firm support to the determined absolute configuration of
1.
Figure 3.
Calculated CD spectra of compound 1 in MeOH solution (red) and in vacuum (gray) and comparison between the calculated and experimental CD (black) spectra. Vertical bars represent rotational strengths R. σ = 0.20 eV.
Figure 3.
Calculated CD spectra of compound 1 in MeOH solution (red) and in vacuum (gray) and comparison between the calculated and experimental CD (black) spectra. Vertical bars represent rotational strengths R. σ = 0.20 eV.
Compound
2 was obtained as a white amorphous powder, and its molecular formula was determined to be C
18H
24O
3 (seven degrees of unsaturation) from the quasi-molecular ion peak at
m/
z 311.1987 [M + Na]
+ (calcd for C
18H
24O
3Na, 311.1989) in the HRESIMS spectrum. The IR absorption bands at 3440, 1708, 1600, 1500, and 1476 cm
−1 indicated the presence of hydroxyl, carbonyl, and aromatic groups. The
1H-NMR spectrum of
2 (
Table 1) showed the signals of four tertiary methyl groups [at δ
H 2.34, 1.25, 1.19, and 1.03 (each 3H, s)], two oxygenated methines [at δ
H 4.26 (ddd,
J = 2.6, 4.5, and 11.5 Hz, H-2) and 3.61 (d,
J = 2.6 Hz, H-3)], and three aromatic protons [at δ
H 7.80 (d,
J = 1.3 Hz, H-14), 7.34 (dd,
J = 1.3, 8.0 Hz, H-12), and 7.27 (d,
J = 8.0 Hz, H-11)]. The
13C-NMR spectrum in combination with DEPT experiments showed 18 carbon resonances, including six quaternary carbons (one conjugated carbonyl and three olefinic carbons), six methines (two oxygenated and three olefinic carbons), two methylenes, and four methyls. The double bonds and ketone group accounted for four out of the seven degrees of unsaturation, the remaining three double-bond equivalents required
2 to be tricyclic. The aforementioned data resembled those of compound
1. The major structural differences were due to the location of the conjugated ketone group and C-ring aromatized in
2. The structure of
2 was further demonstrated by HMBC and NOESY spectra (See
supplementary materials Figure S27). Compound
2 represents the first C-ring aromatized dinor-
ent-pimarane reported hitherto.
The absolute configuration of compound
2 was postulated on the basis of comparison between the experimental ECD spectrum and the calculated data. As can be seen in
Figure 4, the Boltzmann weighted CD spectra in both the gas phase and MeCN solution, in particular the solution spectrum, are in good agreement with the experimental spectrum.
Compound
3, a colorless oil, had a molecular formula of C
28H
44O
10 as determined by HRESIMS
m/
z 585.2929 [M + HCOO]
− (calcd for C
29H
45O
12, 585.2923). Closely inspection of the
1H and
13C-NMR spectra indicated compound
3 was a diterpenoid glycoside. Comparison of the NMR data (
Table 1) and molecular formula of
3 with those of ephemeranthoside [
4] demonstrated that
3 had an additional acetyl group. [δ
H2.10 s (3H); δ
C 21.3 and 173.0], which was located at C-3 from analysis of the HMBC spectrum. The relative configuration of 3 was established by analysis of the NOESY spectrum.
Table 1.
1H-NMR (400 MHz) and 13C-NMR (100 MHz) data for compounds 1–3 (δ in ppm).
Table 1.
1H-NMR (400 MHz) and 13C-NMR (100 MHz) data for compounds 1–3 (δ in ppm).
| No. | 1 a | 2 b | 3 a |
|---|
| δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type |
|---|
| 1α | 1.54, m | 40.4, CH2 | 2.31, m | 40.3, CH2 | 1.56, m | 41.3, CH2 |
| 1β | 1.44, t (11.8) | | 1.99, m | | 1.53, m | |
| 2 | 4.05, ddd (2.8, 4.4, 11.8) | 66.3, CH | 4.26, ddd (2.6, 4.5, 11.5) | 66.2, CH | 4.00, ddd (2.6, 4.7, 11.7) | 65.9, CH |
| 3 | 3.45, d (2.8) | 79.2, CH | 3.61, d (2.6) | 78.8, CH | 4.91, d (2.6) | 81.5, CH |
| 4 | | 38.2, C | | 38.8, C | | 39.9, C |
| 5 | 1.37, m | 47.6, CH | 2.29, m | 42.8, CH | 1.44, m | 49.9, CH |
| 6α | 1.58, m | 21.7, CH2 | 2.64, d (1.4) | 36.0, CH2 | 1.42, m | 22.6, CH2 |
| 6β | 1.37, m | | 2.62, m | | 1.57, m | |
| 7α | 2.13, m | 30.2, CH2 | | 199.4, C | 2.12, m | 36.5, CH2 |
| 7β | 2.07, m | | | | 2.42, d (14.2) | |
| 8 | 2.49, t (12.0) | 45.8, CH | | 131.1, C | | 142.9, C |
| 9 | 1.59, m | 61.3, CH | | 153.7, C | 1.92, t (9.1) | 52.0, CH |
| 10 | | 37.1, C | | 39.2, C | | 40.5, C |
| 11α | 2.37, dd (3.8, 11.6) | 29.2, CH2 | 7.27, d (8.0) | 124.2, C | 1.65, m | 21.3, CH2 |
| 11β | 2.15 br, s | | | | 1.24, m | |
| 12α | | 197.4, C | 7.34, dd (1.3, 8.0) | 135.0, C | 1.10, m | 33.5, CH2 |
| 12β | | | | | 2.31, d | |
| 13 | | 145.3, C | | 135.8, C | | 48.7, C |
| 14α | 6.65, t (1.6) | 148.0, CH | 7.80, d (1.3) | 127.7, C | 5.54, s | 125.5, CH |
| 14β | | | | | | |
| 15 | | | | | | 213.5, C |
| 16a | | | | | 4.49, d (18.4) | 72.4, CH2 |
| 16b | | | | | 4.88, d (18.4) | |
| 17 | 2.27, s | 26.1, CH3 | 2.34, s | 20.6, CH3 | 1.14, s | 27.5, CH3 |
| 18 | 0.87, s | 21.3, CH3 | 1.03, s | 21.5, CH3 | 0.89, s | 28.5, CH3 |
| 19 | 1.00, s | 28.7, CH3 | 1.19, s | 28.4, CH3 | 0.98, s | 22.4, CH3 |
| 20 | 0.94, s | 16.0, CH3 | 1.25, s | 24.3, CH3 | 0.77, s | 16.0, CH3 |
| CH3CO | | | | | 2.10, s | 21.3, CH3 |
| CH3CO | | | | | | 173.0, C |
| 1’ | | | | | 4.24, d (7.6) | 104.2, CH |
| 2’ | | | | | 3.16, dd (7.6, 9.2) | 75.0, CH |
| 3’ | | | | | 3.27, dd (2.7, 5.0) | 78.2, CH |
| 4’ | | | | | 3.28, d (8.8) | 71.5, CH |
| 5’ | | | | | 3.24, dd (2.7, 5.0) | 77.6, CH |
| 6’a | | | | | 3.87, dd (3.6, 12.0) | 72.4, CH2 |
| 6’b | | | | | 3.64, dd (3.6, 12.0) | |
Figure 4.
Calculated CD spectra of compound 2 in MeCN solution (red) and in vacuum (gray) and comparison between the calculated and experimental CD (black) spectra. Vertical bars represent rotational strengths R. σ = 0.24 eV.
Figure 4.
Calculated CD spectra of compound 2 in MeCN solution (red) and in vacuum (gray) and comparison between the calculated and experimental CD (black) spectra. Vertical bars represent rotational strengths R. σ = 0.24 eV.
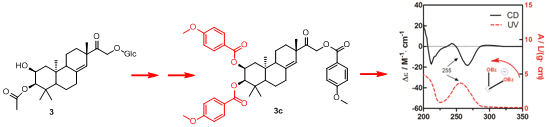
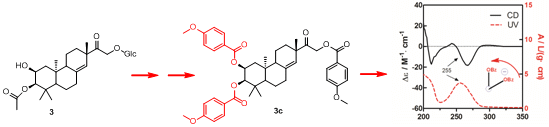
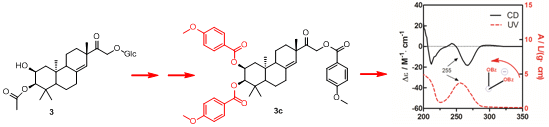
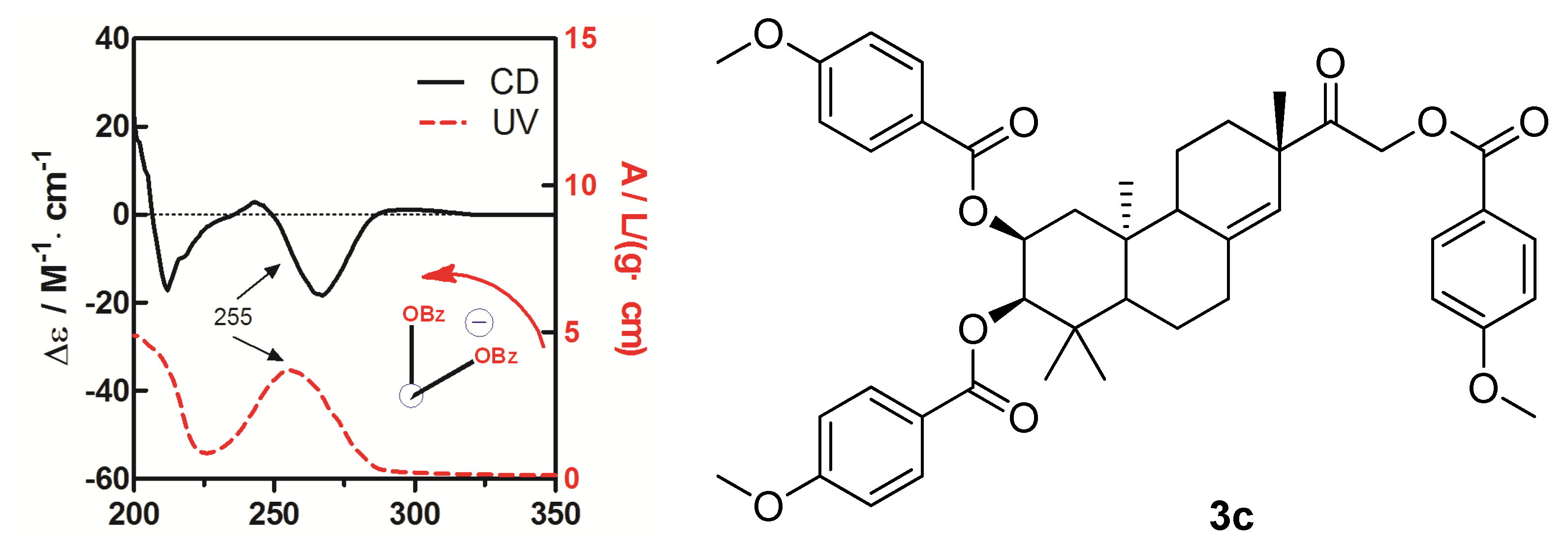
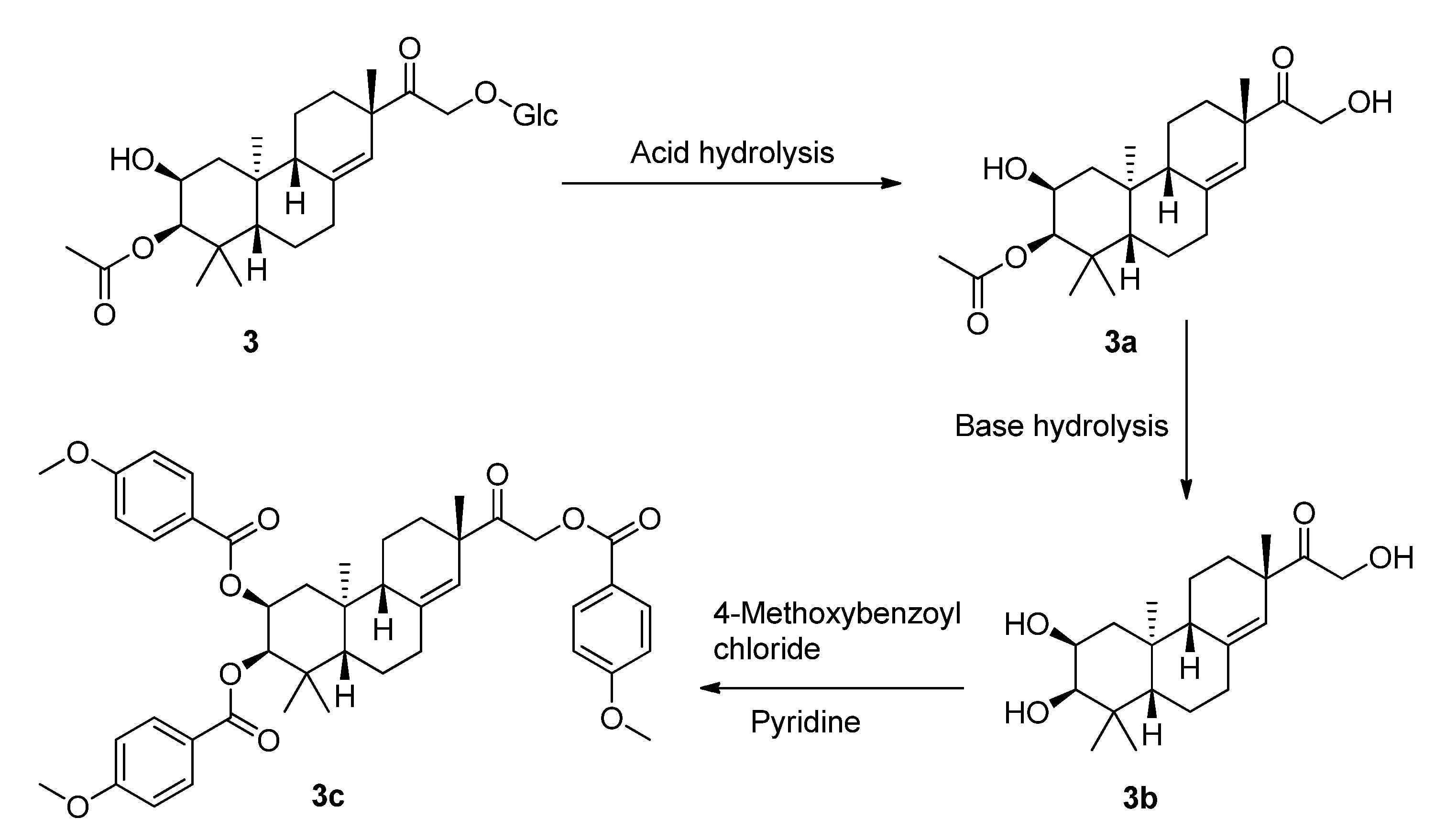
To determine the absolute configuration of compound
3, the chemical transformation from
3 to
3c was performed (as shown in
Scheme 1), then the exciton chirality method was applied on 2,3,16-tri-
p-methoxybenzoate derivative (
3c). The negative chirality resulting from the exciton coupling between the two chromophores of
p-methoxybenzoate at 267 nm (∆ε −18.41, π-π* transition) and 243 nm (∆ε +2.85, π-π* transition) indicated that the transition dipole moments of the two chromophores were oriented in a counterclockwise manner (
Figure 5) [
11]. Thus, the absolute configurations of C-2 and C-3 of
3c were determined to be 2
S and 3
R, respectively. The absolute configuration of the β-glucose was identified to be
d-configuration by HPLC analysis. Therefore, compound
3 was established as 2,16-dihydroxyl-15-keto-2-acetoxy-
ent-pimar-8(14)-ene-16-
O-β-
d-glucopyranoside.
Scheme 1.
The chemical transformation from 3 to 3c.
Scheme 1.
The chemical transformation from 3 to 3c.
The known compounds sitost-5-en-3β-ol acetate (
4) [
12], β-sitosterol-3-
O-β-
d-glucopyranoside (
5) [
13], 3,5,6-trihydroxysitostane (
6) [
14], stigmastane-3β,5α,6β-triol-3,6-diacetate (
7) [
15] were identified by comparison of their spectroscopic data with the literature values.
The anti-inflammatory activity of all isolates were evaluated. The results (
Table 2) showed that compounds
1–
3 exhibited potent inhibitory activity against NO and TNF-α production in RAW264.7 cells and the steroids were inactive with the IC
50 values more than 20 µM.
Figure 5.
CD and UV spectra of compound 3c in MeOH. The arrow denotes the electric transition dipole of the chromophores.
Figure 5.
CD and UV spectra of compound 3c in MeOH. The arrow denotes the electric transition dipole of the chromophores.
Table 2.
IC50 values of the active compounds 1–3 against NO and TNF-α production in RAW264.7 cells.
Table 2.
IC50 values of the active compounds 1–3 against NO and TNF-α production in RAW264.7 cells.
| Compound | IC50 (μM) |
|---|
| NO | TNF-α |
|---|
| 1 | 19.2 | 6.2 |
| 2 | 6.7 | 5.6 |
| 3 | 13.8 | 8.9 |
| Celastrol a | 1.1 | 0.9 |
3. Experimental
3.1. General
Optical rotations were measured on a Rudolph Autopol I automatic polarimeter. IR spectra were determined on a Bruker Tensor 37 infrared spectrophotometer. NMR spectra were measured on a Bruker AM-400 spectrometer at 25 °C. ESIMS was measured on a Finnigan LC QDECA instrument, and HRESIMS was performed on a Waters-Micromass Q-TOF. A Shimadzu LC-20 AT equipped with a SPD-M20A PDA detector was used for HPLC. A YMC-pack ODS-A column (250 × 10 mm, S-5 µM, 12 nm) was used for semipreparative HPLC separation. Silica gel (300–400 mesh, Qingdao Haiyang Chemical Co., Ltd., Qingdao city, China), C18 reversed-phase silica gel (12 nm, S-50 µM, YMC Co., Ltd., Taiwan Chu-Pei city, China), Sephadex LH-20 gel (Amersham Biosciences, Piscataway, NJ, USA) and MCI gel (CHP20P, 75–150 µM, Mitsubishi Chemical Industries Ltd., Tokyo, Japan.) were used for column chromatography. All solvents used were of analytical grade (Guangzhou Chemical Reagents Company, Ltd., Guangzhou, China). All cell lines were obtained from the China Center for Type Culture Collection of the Chinese Academy of Sciences.
3.2. Plant Material
Plant of F. fimbriata were collected in July 2010 from Yunnan Province, China, and were identified by one of the authors (Prof. D. P. Yang). A voucher specimen (accession number: LSJSH201010) has been deposited at the School of Pharmaceutical Sciences, Sun Yat-sen University.
3.3. Extraction and Isolation
The air-dried powder of the leaves of F. fimbriata (1 kg) was extracted with 95% EtOH (3 × 10 L) at room temperature (rt) to give 80 g of crude extract. The extract was suspended in H2O (1 L) and successively partitioned with petroleum ether (PE, 3 × 1 L), EtOAc (3 × 1 L) and n-BuOH (3 × 1 L), respectively. The PE extract (15 g) was subjected to silica gel column chromatography (CC) and eluted with PE/dichloromethane (0:10→10:0) successively to afford three fractions (I–III). Fraction I (2.5 g) was chromatographed over an silica gel column eluting with PE/CH2Cl2 (0:10→10:0) to afford five fractions (Fr. Ia–Ie), Fr. Ia was then subjected to silica gel column eluting with PE/CH2Cl2 (0:10→10:0) and obtained compound 4 (113 mg). Fraction III (3.5 g) was chromatographed over an silica gel column eluting with PE/Acetone (0:10→10:0) to afford five fractions (Fr. IIIa–IIIe). Fr. IIIa (1.0 g) was separated by silica gel CC (PE/CH2Cl2, 50:1→10:1) to afford five fractions (Fr. IIIaa–IIIae). Fr. IIIac was subjected to Rp-C18 CC using a gradient of MeOH/H2O (v/v from 7:3 to 10:0) to yield 6 (22 mg) and another fraction, which after chromatography on a Sephadex LH-20 column using CHCl3/MeOH (1:1) as eluent to obtain 7 (23 mg). The EtOAc extract (42 g) was subjected to MCI gel CC eluted with a MeOH/H2O gradient (3:7→10:0) to afford three fractions (XI–XIII). Fraction XI (6.5 g) was subjected to silica gel CC (PE/EtOAc, 2:1→0:1) to give three fractions (XIa–XIc). Fr. XIa (2.1 g) was separated by silica gel CC (PE/EtOAc, 2:1), followed by semi-preparative HPLC (CH3OH/H2O, 8:2, 3 mL/min) to give 1 (6 mg). Fr. XIc (1.3 g) was separated by Rp-C18 silica gel CC (MeOH/H2O, 5:5→0:0) to yield 2 (8 mg). Fraction XII (5.5 g) was subjected to silica gel CC (CHCl3/MeOH, 30:1→0:1) to give three fractions (XIIa–XIIc). Fr. XIIb (2.5 g) was subjected to silica gel CC (PE/CHCl3, 1:1→0:1) to give three fractions (XIIb1–XIIb4). Fr. XIIb1 (0.9 g) was subjected to Rp-C18CC (MeOH/H2O, 2:8→10:0) and followed by silica gel CC (EtOAc/acetone, 3:1→0:1) to afford 5 (38 mg). Fr. XIIb4 (1.2 g) was subjected to Rp-C18 CC (MeOH/H2O, 0:10→10:0) and Sephadex LH-20 (EtOH) to yield 3 (25 mg).
3.4. Spectral Data
Flickinflimilin A (
1). Colorless oil;
![Molecules 19 05863 i002]()
−46.2° (
c 0.24, CHCl
3); UV (MeOH) λ
max (log ε) 242 (2.18) nm, 203 (2.46) nm; CD (
c 3.4 × 10
−3 M, CH
3CN),
λmax (∆ε) 242 (−0.26), 278 (−0.01), and 323 (−0.07); IR (KBr) ν
max 3437, 1718, 1591, 1461, 1378, 1129, 1038, 949, and 764 cm
−1;
1H and
13C-NMR data, see
Table 1; positive ESIMS
m/
z 315.3 [M + Na]
+, 607.6 [2M + Na]
+; HREIMS
m/
z 315.1943 [M + Na]
+ (calcd for C
18H
8O
3Na, 315.1936).
Flickinflimilin B (
2). White amorphous powder;
![Molecules 19 05863 i002]()
+16.0° (
c 0.50, MeOH); UV (MeOH) λ
max (log ε) 207 (2.94), 251 (2.45) nm; CD (
c 1.7 × 10
−3 M, CH
3CN), λ
max (∆ε) 207 (−6.64), 251 (+2.29), 270 (−0.34), 300 (+2.19), and 337 (−3.08); IR (KBr) ν
max 3440, 1708, 1600, 1500, 1476, 1129, 767, and 750 cm
−1;
1H and
13C-NMR data, see
Table 1; positive ESIMS
m/
z 289.1 [M + H]
+; negative ESIMS
m/
z 333.2 [M + HCOO]
−; HRESIMS
m/
z 311.1987 [M + Na]
+ (calcd for C
18H
24O
3Na, 311.1989).
Flickinflimilin C (
3). Colorless oil;
![Molecules 19 05863 i002]()
−55.0° (
c 0.22, CHCl
3); IR (KBr) ν
max 3445, 1714, 1646, 1461, 1379, 1257, 1126, 1085, and 950 cm
−1;
1H and
13C-NMR data, see
Table 1; positive ESIMS
m/
z 563.3 [M + Na]
+, 1103.5 [2M + Na]+; negative ESIMS
m/
z 585.3 [M + HCOO]
−, 1125.8 [2M + HCOO]−; HRESIMS
m/z 585.2929 [M + HCOO]
− (calcd for C
29H
45O
12, 585.2923).
3.5. Computational Methods for Electronic Circular Dichroism
Molecular mechanics calculations were carried out with Spartan’14 software package (Wavefunction Inc., Irvine, CA, USA, 2013) and quantum chemical computations were run with Gaussian 09 program package (Gaussian, Inc., Pittsburgh PA, USA, 2011) using default grids and convergence criteria. MMFF conformational search generated conformers within a 10 kcal/mol energy window were optimized using DFT method at B3LYP/6-31G (d) level. Frequency calculations were run at the same level to verify that each optimized conformer was a true minimum and to estimate their relative thermal free energies (∆G) at 298.15K. The TDDFT calculations were performed using the hybrid B3LYP functional, and Ahlrichs’ basis sets SVP (split valence plus polarization) and TZVP (triple zeta valence plus polarization). The number of excited states per each molecule was 20–30. Solvent effects were taken into account by using polarizable continuum model (PCM). CD spectra were generated by the program SpecDis (University of Würzburg, Würzburg, Germany, 2012) using a Gaussian band shape with 0.28 eV exponential half-width from dipole-length dipolar and rotational strengths; the difference with dipole-velocity values was negligible (<10%) for most transitions.
3.6. Determination of Sugar Configuration [16]
Compound 3 (6 mg) was refluxed with 2 M HCl (2 mL, dioxane/H2O, 1:1) at 100 °C for 4 h. After removing the dioxane under vacuum, the solution was then diluted with H2O and then extracted with EtOAc (3 × 1 mL). The EtOAc layer was evaporated under vacuum, then subjected to CC over silica gel eluteing with CHCl3:MeOH (30:1) to afford 3a. The aqueous layer was evaporated under vacuum, diluted repeatedly with H2O, evaporated under vacuum to obtain neutral residue, and then analyzed by TLC over silica gel (Me2CO/n-BuOH/H2O, 6:3:1) together with authentic sugar sample (glucose, Rf = 0.49). The remaining residue was dissolved in pyridine (200 µL), to which 2 mg of l-cysteine methyl ester hydrochloride was added. The mixture was stirred at 60 °C for 1 h; then 50 µL of o-tolyl isothiocyanate was added, and the mixture was stirred at 60 °C for another 1 h. The reaction mixture was directly analyzed by standard C18 HPLC [a YMC-pack ODS-A column (250 × 10 mm, S-5 µM, 12 nm), CH3CN/H2O, 25:75, 3 mL/min]. The peak (tR = 19.0 min) coincided with a derivative of d-glucose, as compared with authentic d-glucose with tR at 19.1 min.
3.7. Chemical Transformation of 3a to 3c [17]
To a stirred solution of 3a (10 mg) in MeOH (2 mL), NaOH (1 mg) was added. The mixture was stirred at room temperature for 0.5 h to obtain 3b, and then 3b (7.2 mg) was transferred into a clean NMR tube and was dried completely under the vacuum of an oil pump. Deuterated pyridine (0.55 mL) and 4-methoxybenzoyl chloride (12 µL) were added into the NMR tube immediately under dry conditions, and then the NMR tube was shaken carefully to mix the sample and 4-methoxybenzoyl chloride evenly. The reaction NMR tube was permitted to stand at 45 °C and monitored by 1H-NMR. The reaction was found to be completed after 2 h. 1H-NMR data of the 4-methoxybenzoate derivative (3c) of 3b was obtained from the reaction NMR tube directly. The reaction mixtures were transferred from the NMR tube and subjected to Sephadex LH-20 CC eluting with CHCl3:MeOH (1:1). CD (CH3OH, ∆ε) 212 (−17.20), 243 (+2.85), 267 (−18.41), 297 (+1.12) nm; 1H-NMR (CDCl3, 400 MHz) δH 5.43, (1H, s, H-14), 5.40, [1H, (ddd, J = 2.1, 4.3, 12.4 Hz, H-2)], 4.15, [1H, (dd, J = 7.6, 14.2 Hz, H-16a)], 3.67, [1H, (d, J = 2.1 Hz, H-3)], 3.15, [1H, (dd, J = 7.6, 14.2 Hz, H-16b)], 2.29–2.42, (4H, m, H-6, H-7), 1.96–2.14, (4H, m, H-1, H-12), 1.83, (1H, m, H-9), 1.44, (1H, m, H-5), 1.07, (3H, s, H-17), 1.01, (3H, s, H-18), 0.92, (3H, s, H-19), 0.88, (3H, s, H-20), [7.97–8.05, (6H, m), 6.91–6.96, (6H, d, J = 8.5 Hz ), 3.86–3.87, (9H, s, CH3O-p), p-methoxybenzoyl]; 13C-NMR (CDCl3, 100 MHz) δC 207.2 (C-15), 142.1 (C-8), 124.1 (C-14), 76.8 (C-3), 71.3 (C-2), 66.7 (C-16), 50.8 (C-9), 47.7 (C-5), 47.3 (C-13), 39.5 (C-10), 38.7 (C-4), 36.2 (C-7), 35.6 (C-12), 32.6 (C-1), 28.6 (C-18), 27.2 (C-17), 22.6 (C-19), 21.6 (C-11), 20.2 (C-6), 15.4 (C-20), (165.7, 165.5, 163.6, 163.5, 132.0, 131.6, 113.7, 113.6, 55.4) (p-methoxybenzoyl); negative ESIMS m/z 737.3 [M − H]−.
3.8. Biological Assays
3.8.1. Cytotoxic Assay [18]
The RAW264.7 cell line was obtained from ATCC (Manassas, VA, USA), and was cultured in DMEM medium (Hyclone, Logan, UT, USA), supplemented with 10% fetal bovine serum (FBS, Hyclone) at 37 °C in a humidified atmosphere with 5% CO
2. Cell viability was assessed by MTT (Sigma, St. Louis, MO, USA). Briefly, 100 µL of adherent cells with an initial density of 1 × 10
5 cells/mL were seeded into a 96-well plate and allowed to adhere for 24 h. Cells were exposed to the test compounds at various concentrations for 48 h. After the incubation, MTT (5 mg/mL) was added to each well, and the incubation continued for 4 h at 37 °C. The cells were lysed with 100 µL of 20% SDS–50% DMF after removal of the medium. The optical density of the lysate was measured at 595 nm in a 96-well microtiter plate reader (Bio-Rad 680, Hercules, CA, USA). The IC
50 value of each compound was calculated by Reed and Muench’s method [
19].
3.8.2. Nitric Oxide Inhibitory Assay [20]
Inhibition of NO production was determined in a LPS-stimulated RAW264.7 macrophage cell line. Cells were seeded in 96-well plates (1 × 105 cells/well) and allowed to adhere for 24 h at 37 °C in a humidified atmosphere containing 5% CO2. The medium was then replaced with fresh medium containing LPS (2 µg/mL) and test compounds at 10 µM, and the cells were incubated for 24 h. NO production was determined by measuring the accumulation of nitrite in the culture supernatant with Griess reagent (0.5% sulfanilamide and 0.05% naphthylene-diamide dihydrochloride in 2.5% H3PO4) and then allowed to stand for 5 min at rt. The absorbance at 540 nm was measured using a HTS 7000 microplate reader. The nitrite concentration in the medium was determined from the calibration curve (r = 0.9998) obtained by using different concentrations of sodium nitrite (NaNO2) in the culture medium as the standard. Blank correction was performed by subtracting the absorbance due to medium only from the absorbance reading of each well.
3.8.3. Assay for the Production of Pro-inflammatory Cytokines (TNF-α) [21]
Compounds were dissolved in DMSO, and suspensions of RAW264.7 cells were cultured in complete RPMI 1640 medium (Hyclone) containing 10% FBS. The cultured cells were incubated with the tested compounds (10 µM) for 24 h, followed by LPS stimulation (2 µg/mL). Supernatants were collected to analyze cytokine levels. The mouse TNF-α ELISA Kit was used to determine the cytokine concentration in the culture supernatants.


 ).
).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



 −46.2° (c 0.24, CHCl3); UV (MeOH) λmax (log ε) 242 (2.18) nm, 203 (2.46) nm; CD (c 3.4 × 10−3 M, CH3CN), λmax (∆ε) 242 (−0.26), 278 (−0.01), and 323 (−0.07); IR (KBr) νmax 3437, 1718, 1591, 1461, 1378, 1129, 1038, 949, and 764 cm−1; 1H and 13C-NMR data, see Table 1; positive ESIMS m/z 315.3 [M + Na]+, 607.6 [2M + Na]+; HREIMS m/z 315.1943 [M + Na]+ (calcd for C18H8O3Na, 315.1936).
−46.2° (c 0.24, CHCl3); UV (MeOH) λmax (log ε) 242 (2.18) nm, 203 (2.46) nm; CD (c 3.4 × 10−3 M, CH3CN), λmax (∆ε) 242 (−0.26), 278 (−0.01), and 323 (−0.07); IR (KBr) νmax 3437, 1718, 1591, 1461, 1378, 1129, 1038, 949, and 764 cm−1; 1H and 13C-NMR data, see Table 1; positive ESIMS m/z 315.3 [M + Na]+, 607.6 [2M + Na]+; HREIMS m/z 315.1943 [M + Na]+ (calcd for C18H8O3Na, 315.1936).