Cobalt-Catalyzed Methoxycarbonylation of Substituted Dichlorobenzenes as an Example of a Facile Radical Anion Nucleophilic Substitution in Chloroarenes

Abstract
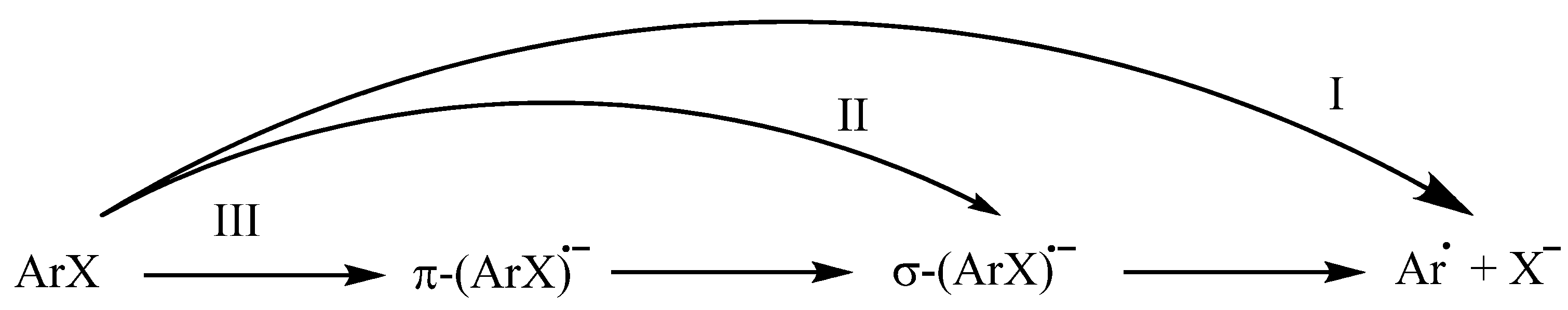
:1. Introduction


2. Results and Discussion
2.1. The Relative Reactivity of Substituted Dichlorobenzenes

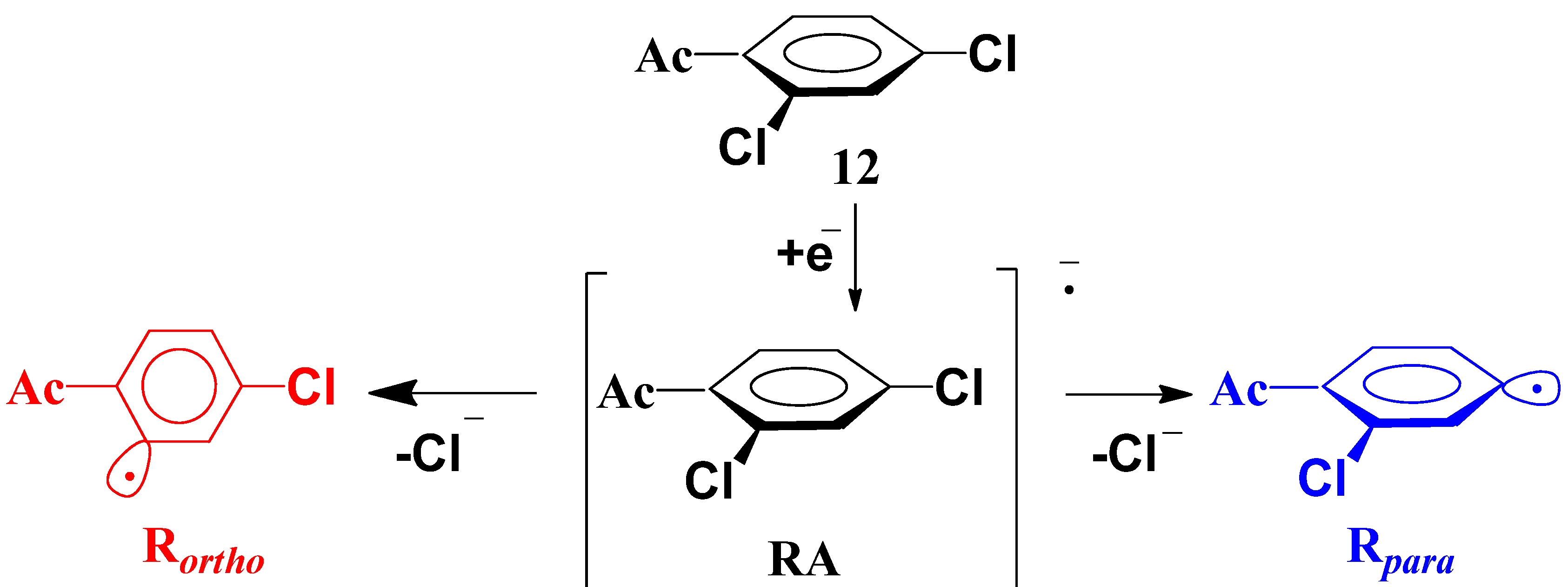
2.2. Regioselectivity in the Methoxycarbonylation of Substituted Dichlorobenzenes



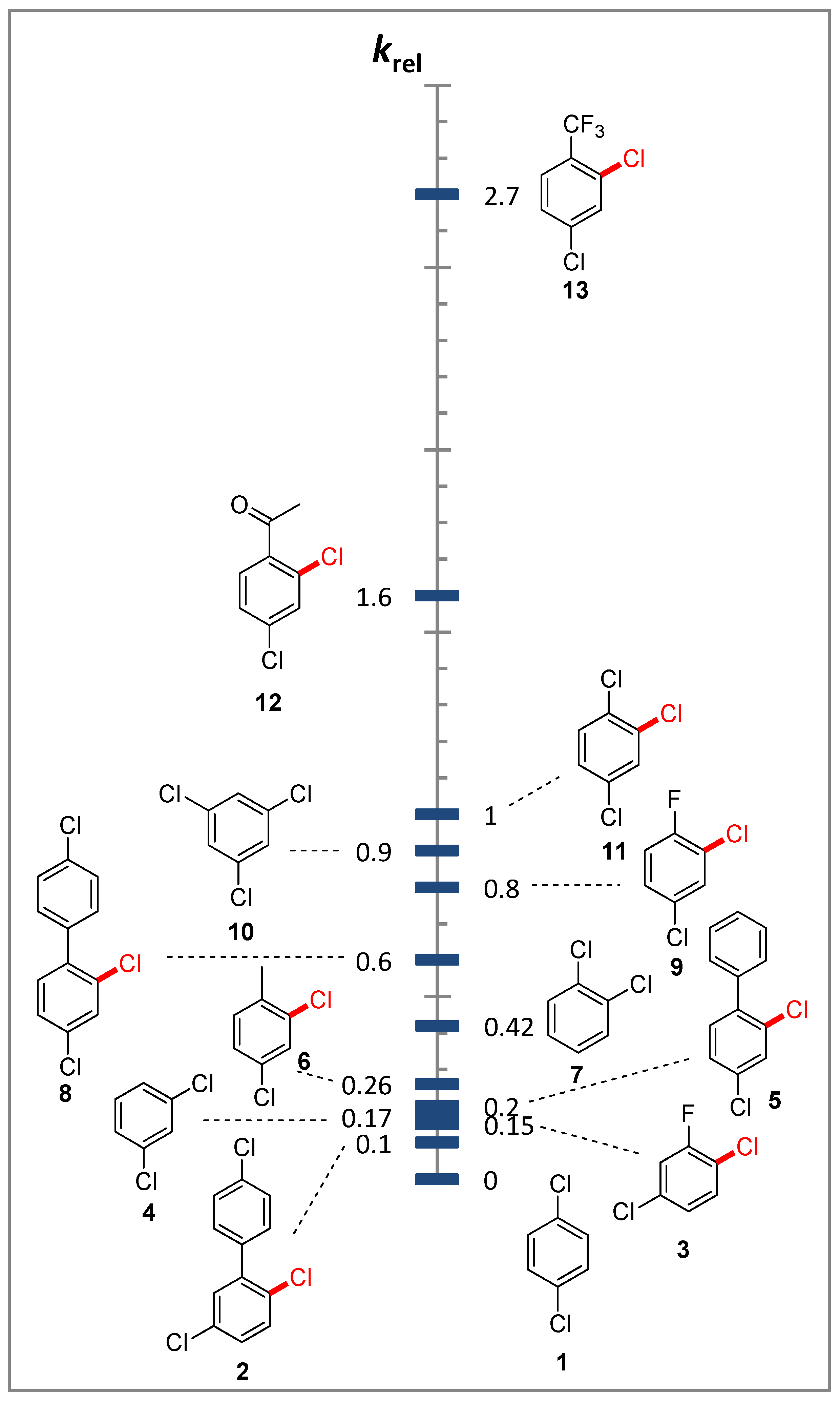{kind=link}
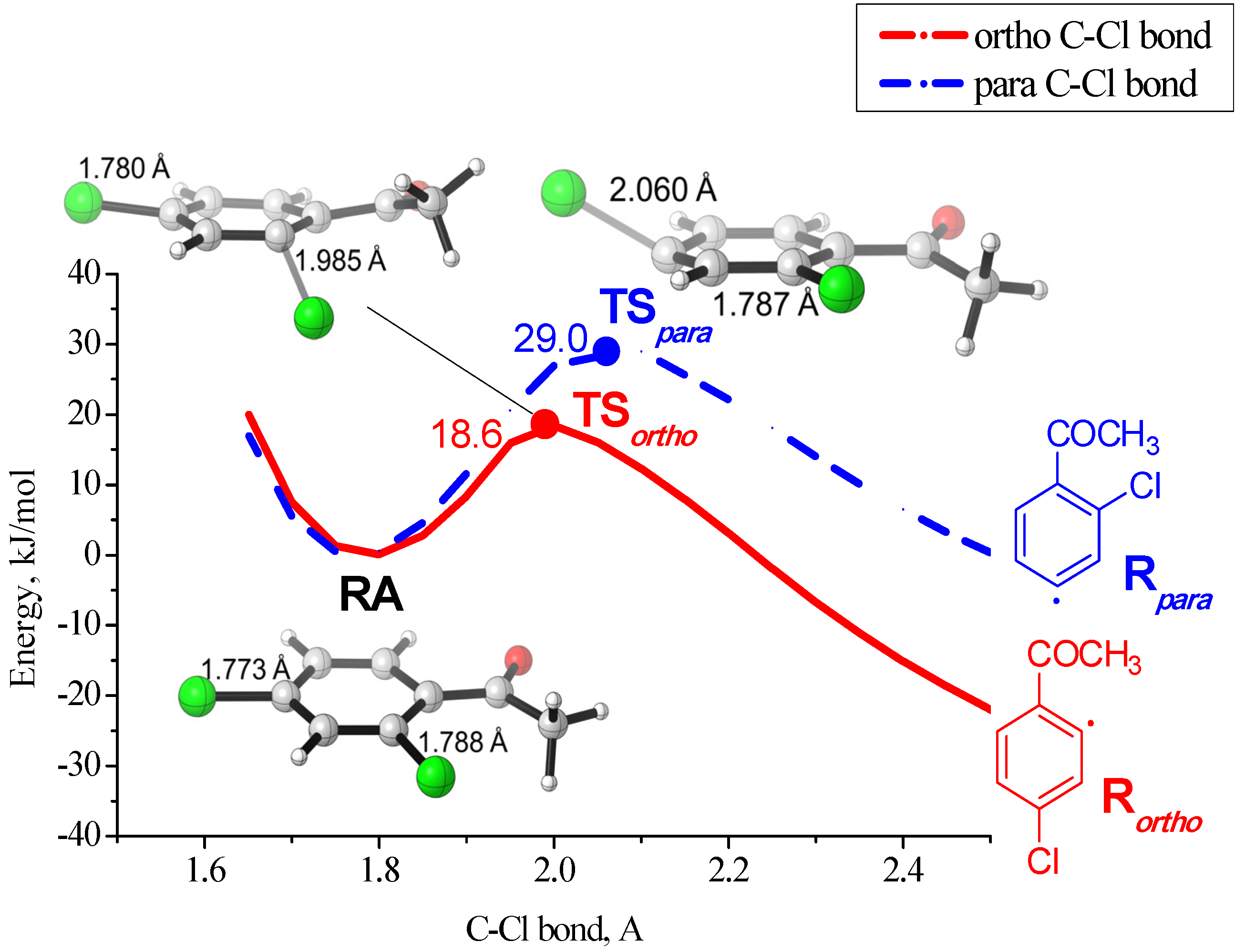{kind=link}
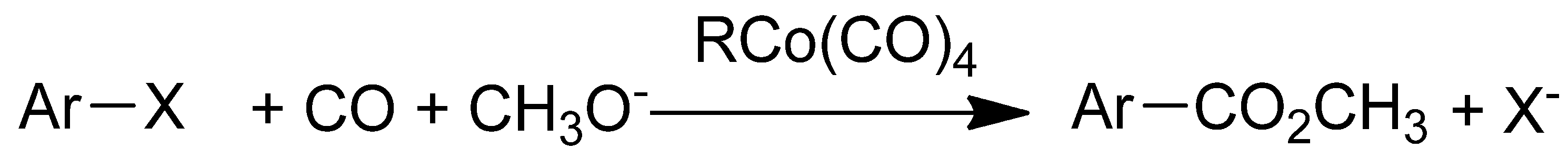{kind=link}
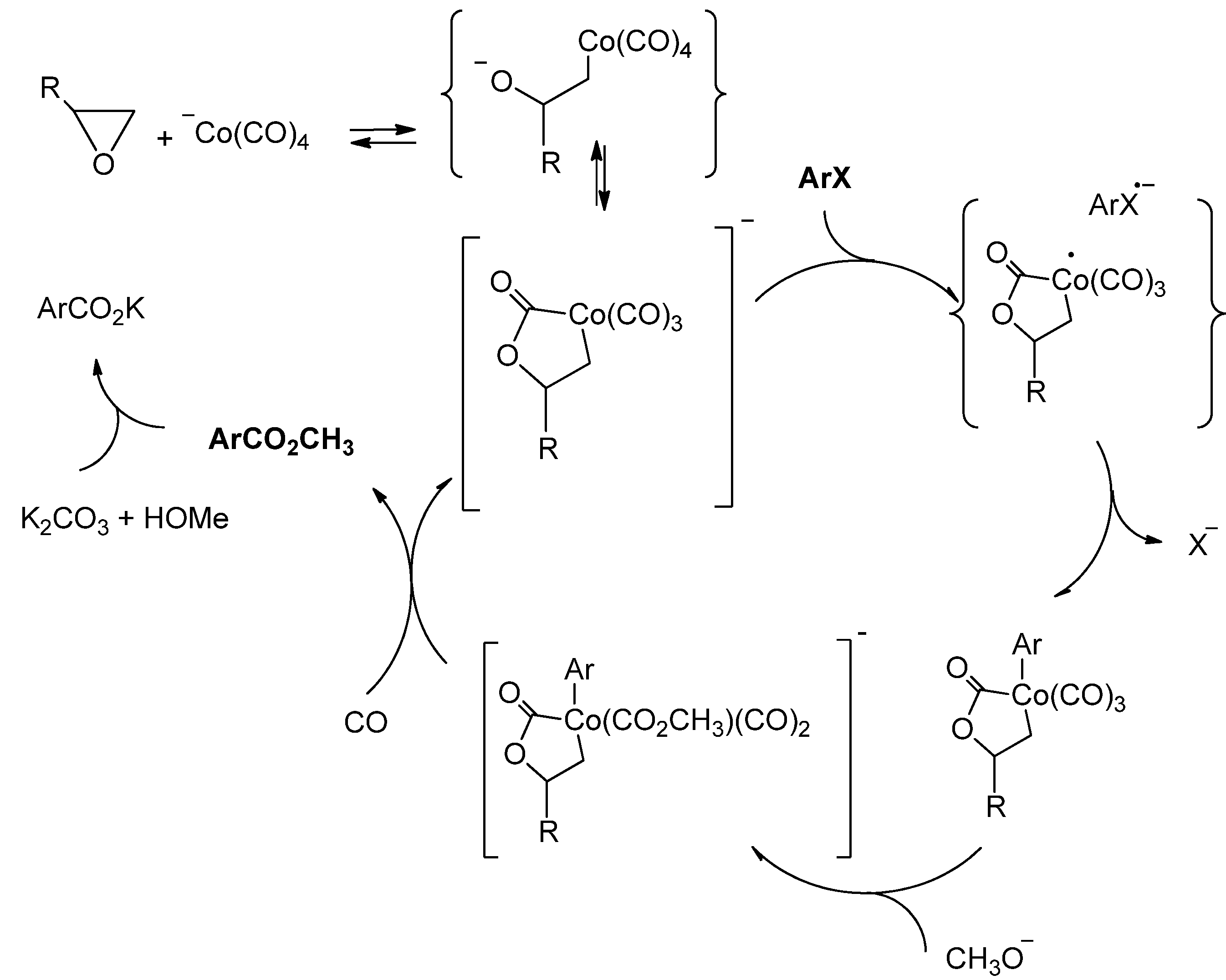{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | Product | Isolated yield, % |
|---|---|---|---|
| 1 |  |  | 82 |
| 2 |  |  | 73 |
| 3 |  |  | 63 |
| 4 |  |  | 80 |
2.3. Possible Driving Forces of the Ortho-Effect



2.4. Synthetic Importance
3. Experimental
3.1. General Information
3.2. Quantum Chemical Calculations
3.3. General Procedure for Blank Experiments
3.4. General Procedure I for Competitive Methoxycarbonylation
3.4.1. Competitive Methoxycarbonylation of 1,2-Dichlorobenzene (7) and 1,3-Dichlorobenzene (4)
3.4.2. Competitive Methoxycarbonylation of 1,2-Dichlorobenzene (7) and 1,4-Dichlorobenzene (1)
3.4.3. Competitive Methoxycarbonylation of 1,2-Dichlorobenzene (7) and 1,2,4-Trichlorobenzene (11)
3.4.4. Competitive Methoxycarbonylation of 1,2,4-Trichlorobenzene (11) and 2,4-Dichloro-1-Fluoro-benzene (9)
3.4.5. Competitive Methoxycarbonylation of 1,2,4-Trichlorobenzene (11) and 1,4-Dichloro-2-Fluoro-benzene (3)
3.4.6. Competitive Methoxycarbonylation of 1,2,4-Trichlorobenzene (11) and 1,3,5-Trichlorobenzene (10)
3.4.7. Competitive Methoxycarbonylation of 1,2,4-Trichlorobenzene (11) and 2,4-Dichlorobiphenyl (5)
3.4.8. Competitive Methoxycarbonylation of 2,4-Dichlorobiphenyl (5) and 2,4,4'-Trichlorobiphenyl (8)
3.4.9. Competitive Methoxycarbonylation of 2,4-Dichlorobiphenyl (5) and 2,4',5-Trichlorobiphenyl (2)
3.4.10. Competitive Methoxycarbonylation of 1,2,4-Trichlorobenzene (11) and 2,4-Dichloroaceto-phenone (12)
3.4.11. Competitive methoxycarbonylation of 2,4-Dichlorobiphenyl (5) and 2,4-Dichloroaceto-phenone (12)
3.4.12. Competitive Methoxycarbonylation of 1,3-Dichlorobenzene (4) and 2,4-Dichlorotoluene (6)
3.4.13. Competitive Methoxycarbonylation of 1,2,4-Trichlorobenzene (11) and 2,4-Dichlorotoluene (6)
3.4.14. Competitive Methoxycarbonylation of 1,2,4-Trichlorobenzene (11) and 2,4-Dichloro-1-(trifluoromethyl)benzene (13)
3.5. General Procedure II for Determination of Methoxycarbonylation Regioselectivity
3.5.1. Methoxycarbonylation of 2,4-Dichlorotoluene (6)
3.5.2. Methoxycarbonylation of 2,4-Dichloroacetophenone (12)
3.5.3. Methoxycarbonylation of 2,4-Dichloro-1-(Trifluoromethyl)Benzene (13)
3.6. Preparation of Substituted Benzoic Acids
3.6.1. Methoxycarbonylation of 2,4-Dichloroacetophenone (12) to Acid 12a
3.6.2. Methoxycarbonylation of 2,4-Dichloroanisole (14)
3.6.3. Methoxycarbonylation of 2,5-Dichloropropiophenone (15)
3.6.4. Methoxycarbonylation of 5-Chloro-4-Methoxy-3-Methylacetophenone (16)
3.6.5. Scaled Procedure for Preparative Reaction
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kim, J.K.; Bunnett, J.F. Evidence for a radical mechanism of aromatic “nucleophilic” substitution. J. Am. Chem. Soc. 1970, 92, 7463–7464. [Google Scholar] [CrossRef]
- Smith, M.B.; March, J. Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007. [Google Scholar]
- Rossi, R.A.; Lukach, A.E. Some synthetic applications of the aromatic radical nucleophilic substitution reactions. In Electron Transfer Reactions in Organic Synthesis; Vanelle, P., Ed.; Research Signpost: Trivandrum, Kerala, India, 2002; pp. 43–61. [Google Scholar]
- Rossi, R.A.; Corsico, E.F. Reactions of Me3Sn- ions with aryl chlorides by the SRN1 mechanism followed by Pd(0)-catalyzed reactions. A novel approach in organic synthesis. Main Group Met. Chem. 2002, 25, 99–106. [Google Scholar]
- Sperotto, E.; van Klink, G.P.M.; van Koten, G.; de Vries, J.G. The mechanism of the modified Ullmann reaction. Dalton Trans. 2010, 39, 10338–10351. [Google Scholar] [CrossRef]
- Rossi, R.A. Recent advances in the synthesis of stannanes and the scope of their posterior chemical transformations. J. Organomet. Chem. 2014, 751, 201–212. [Google Scholar] [CrossRef]
- Tiedje, J.M.; Quesen, J.F.; Chee-Sanford, J. Microbial reductive dechlorination of PCBs. Biodegradation 1993, 4, 231–240. [Google Scholar]
- Dolfing, J. Reductive dechlorination of 3-chlorobenzoate is coupled to ATP production and growth in an anaerobic bacterium, strain DCB-1. Arch. Microbiol. 1990, 153, 264–266. [Google Scholar] [CrossRef]
- Mohn, W.W.; Tiedje, J.M. Evidence for chemiosmotic coupling of reductive dechlorination and ATP synthesis in Desulfomonile tiedjei. Arch. Microbiol. 1991, 157, 1–6. [Google Scholar]
- Costentin, C.; Robert, M.; Savéant, J.-M. Electron transfer and bond breaking: Recent advances. Chem. Phys. 2006, 324, 40–56. [Google Scholar] [CrossRef]
- Pop, S.M.; Gupta, N.; Raza, A.S.; Ragsdale, S.W. Transcriptional Activation of Dehalorespiration: Identification of Redox-Active Cysteines Regulating Dimerization and DNA Binding. J. Biol. Chem. 2006, 281, 26382–26390. [Google Scholar] [CrossRef]
- Bhatt, P.; Kumar, M.S.; Mudliar, S.; Chakrabarti, T. Biodegradation of chlorinated compounds—a review. Crit. Rev. Environ. Sci. Technol. 2007, 37, 165–198. [Google Scholar] [CrossRef]
- Field, J.A.; Sierra-Alvarez, R. Microbial transformation of chlorinated benzoates. Rev. Environ. Sci. Bio/Technol. 2008, 7, 191–210. [Google Scholar]
- Pieper, D.H.; Seeger, M. Bacterial Metabolism of Polychlorinated Biphenyls. J. Mol. Microbiol. Biotechnol. 2008, 15, 121–138. [Google Scholar] [CrossRef]
- Stenuit, B.; Lamblin, G.; Cornelis, P.; Agathos, S.N. Aerobic Denitration of 2,4,6-Trinitrotoluene in the Presence of Phenazine Compounds and Reduced Pyridine Nucleotides. Environ. Sci. Technol. 2012, 46, 10605–10613. [Google Scholar]
- Francalanci, F.; Foa, M.; Gardano, A.; Bencini, E. Cobalt-catalyzed carbonylation of aryl halides. J. Organomet. Chem. 1985, 285, 293–303. [Google Scholar] [CrossRef]
- Francalanci, F.; Foa, M. Recent developments in cobalt-catalyzed carbonylation. J. Mol. Catal. 1987, 41, 89–107. [Google Scholar] [CrossRef]
- Miura, M.; Akase, F.; Nomura, M.; Shinohara, M. Carbonylation of aryl halides and vinyl bromides mediated by tetracarbonylcobalt anion. J. Chem. Soc. Perkin Trans. 1 1987, 1021–1025. [Google Scholar]
- Zhesko, T.E.; Boyarskii, V.P.; Beletskaya, I.P. Carbonylation of aromatic halides in the Co2(CO)8-alkyl halide-base system. Metalloorganicheskaya Khim. 1989, 2, 385–387. [Google Scholar]
- Marchal, J.; Bodiguel, J.; Fort, Y.; Caubere, P. A New indirect application of aggregative activation: Synthesis of esters by cobalt-catalyzed carbonylation of aryl, heterocyclic, and vinyl Halides under atmospheric pressure. J. Org. Chem. 1995, 60, 8336–8340. [Google Scholar] [CrossRef]
- Zhesko, T.E.; Boyarskii, V.P.; Nikitina, A.G. Carbonylation of aryl halides catalyzed by modified cobalt carbonyls. Russ. J. Gen. Chem. 1998, 68, 78–82. [Google Scholar]
- Boyarskii, V.P.; Zhesko, T.E.; Lanina, S.A. Synthesis of Aromatic carboxylic acids by carbonylation of aryl halides in the presence of epoxide-modified cobalt carbonyls as Catalysts. Russ. J. Appl. Chem. 2005, 78, 1844–1848. [Google Scholar] [CrossRef]
- Boyarskii, V.P.; Larionov, E.V.; Polyakova, S.M.; Boyarskaya, I.A.; Zhesko, T.E. Mechanism of the catalytic carbonylation of aryl halides with a modified cobalt carbonyl. Russ. J. Gen. Chem. 2007, 77, 915–922. [Google Scholar] [CrossRef]
- Brunet, J.J.; Sidot, C.; Caubere, P.; Loubinoux, B. Activation of reducing agents. Sodium hydride containing complex reducing agents. 10. NaH-RONa-Co(OAc)2-CO, a new reagent for the carbonylation of aryl halides at atmospheric pressure. J. Org. Chem. 1979, 44, 2199–2202. [Google Scholar] [CrossRef]
- Brunet, J.J.; Sidot, C.; Caubere, P. Activation of reducing agents. Sodium hydride containing complex reducing agents: XIV. NaCoCO4 as an SRN1 nucleophile in the carbonylation of aryl halides by CoCRACO at atmospheric pressure. New preparation of NaCoCO4 by NaH reduction of dicobalt octacarbonyl. J. Organomet. Chem. 1980, 204, 229–241. [Google Scholar] [CrossRef]
- Brunet, J.J.; Sidot, C.; Caubere, P. Cobalt carbonyl catalyzed sRN1 carbonylation of aryl and vinyl halides by phase transfer catalysis. Tetrahedron Lett. 1981, 22, 1013–1016. [Google Scholar] [CrossRef]
- Brunet, J.J.; Sidot, C.; Caubere, P. Sunlamp-irradiated phase-transfer catalysis. 1. Cobalt carbonyl catalyzed SRN1 carbonylations of aryl and vinyl halides. J. Org. Chem. 1983, 48, 1166–1171. [Google Scholar] [CrossRef]
- Vanderesse, R.; Marchal, J.; Caubere, P. An efficient cobalt catalyzed alkoxycarbonylation of aryl and heteroaryl halides. Synth. Commun. 1993, 23, 1361–1370. [Google Scholar] [CrossRef]
- Kashimura, T.; Kudo, K.; Mori, S.; Sugita, N. Cobalt carbonyl catalyzed polycarbonylation of polyhalogenated aromatics under photostimulation. Chem. Lett. 1986, 15, 299–302. [Google Scholar] [CrossRef]
- Kashimura, T.; Kudo, K.; Mori, S.; Sugita, N. Cobalt carbonyl-catalyzed double-carbonylation of o-halogenated benzoic acids under photostimulation. Chem. Lett. 1986, 15, 483–486. [Google Scholar] [CrossRef]
- Kashimura, T.; Kudo, K.; Mori, S.; Sugita, N. Cobalt carbonyl-catalyzed polycarbonylation of aryl halides in sodium methoxide/methanol under photostimulation. Chem. Lett. 1986, 15, 851–854. [Google Scholar] [CrossRef]
- Kashimura, T.; Kudo, K.; Mori, S.; Sugita, N. Cobalt salt-catalyzed carbonylation of aromatic halides under photostimulation. Chem. Lett. 1987, 16, 577–580. [Google Scholar]
- Boyarskii, V.P.; Boyarskaya, I.A.; Duka, G.G. Calculation of the possibility of formation of a cyclic metallolactone anionic complex in the methyloxirane-potassium tetracarbonylcobaltate system. Russ. J. Gen. Chem. 2008, 78, 1380–1381. [Google Scholar] [CrossRef]
- Boyarskii, V.P.; Duka, G.G.; Boyarskaya, I.A. Participation of cyclic cobaltolactone anionic complex in the catalytic cycle of aryl halides carbonylation. Russ. J. Gen. Chem. 2009, 79, 2449–2451. [Google Scholar] [CrossRef]
- Lanina, S.A.; Boyarskii, V.P.; Zhesko, T.E.; Nikiforov, V.A.; Bart, T.Ya. Carbonylation of polychlorinated biphenyls over cobalt carbonyl catalyst modified with propylene oxide. Russ. J. Gen. Chem. 2008, 78, 127–132. [Google Scholar] [CrossRef]
- Boyarskii, V.P.; Zhesko, T.E.; Larionov, E.V.; Polukeev, V.A. Synthesis of heteroaromatic carboxylic acids by carbonylation of heteroaryl halides with catalysts based on cobalt carbonyl modified with epoxides. Russ. J. Appl. Chem. 2007, 80, 571–575. [Google Scholar] [CrossRef]
- Boyarskiy, V.P.; Fonari, M.S.; Khaybulova, T.S.; Gdaniec, M.; Simonov, Y.A. Chemoselectivity of cobalt-catalysed carbonylation—A reliable platform for the synthesis of fluorinated benzoic acids. J. Fluor. Chem. 2010, 131, 81–85. [Google Scholar] [CrossRef]
- Evstifeev, A.V.; Shein, S.M. Nucleophilic aromatic substitution. XXXIV Medium effect on the rate of reactions between ortho- and para-substituted chlorobenzenes and sodium alcoholates. Zh. Org. Khim. 1969, 5, 919–923. [Google Scholar]
- Evstifeev, A.V.; Shein, S.M. Mechanism of electrophilic and basic catalysis in reactions of o- and p-substituted halobenzenes with alkali metal alcoholates in an alcohol medium. Zh. Org. Khim. 1972, 8, 746–751. [Google Scholar]
- Behar, D.; Neta, P. Intramolecular electron transfer and dehalogenation of anion radicals. 4. Haloacetophenones and related compounds. J. Am. Chem. Soc. 1981, 103, 2280–2283. [Google Scholar] [CrossRef]
- Tanner, D.D.; Chen, J.J.; Chen, L.; Luelo, C. Fragmentation of substituted acetophenones and halobenzophenone ketyls. Calibration of a mechanistic probe. J. Am. Chem. Soc. 1991, 113, 8074–8081. [Google Scholar] [CrossRef]
- Neta, P.; Behar, D. Intramolecular electron transfer and dehalogenation of anion radicals. Calibration of a mechanistic probe. J. Am. Chem. Soc. 1981, 103, 103–106. [Google Scholar] [CrossRef]
- Basso, S.M.; Montañez, J.P.; Santiago, A.N. Synthesis and evaluation of phytotoxicity of disugran analogs. Lett. Org. Chem. 2008, 5, 633–639. [Google Scholar]
- Montañez, J.P.; Uranga, J.G.; Santiago, A.N. Regioselectivity of methyl chlorobenzoate analogues with trimethylstannyl anions by radical nucleophilic substitution: Theoretical and experimental study. New J. Chem. 2010, 34, 1170–1175. [Google Scholar] [CrossRef]
- Uranga, J.G.; Montañez, J.P.; Santiago, A.N. Theoretical and experimental study on the reactivity of methyl dichlorobenzoates with sulfur-centered nucleophiles by electron transfer reactions. Tetrahedron 2012, 68, 584–589. [Google Scholar] [CrossRef]
- Zhao, H.; Fu, H.; Qiao, R. Copper-Catalyzed Direct Amination of Ortho-Functionalized Haloarenes with Sodium Azide as the Amino Source. J. Org. Chem. 2010, 75, 3311–3316. [Google Scholar]
- Rossi, R.A.; Pierini, A.B.; Peñéñory, A.B. Nucleophilic Substitution Reactions by Electron Transfer. Chem. Rev. 2003, 103, 71–167. [Google Scholar] [CrossRef]
- Robert, M.; Costentin, C.; Savéant, J.-M. Fragmentation of Aryl Halide π Anion Radicals. Bending of the Cleaving Bond and Activation vs Driving Force Relationships. J. Am. Chem. Soc. 2004, 126, 16051–16057. [Google Scholar] [CrossRef]
- Pierini, A.B.; Duca, J.S. Theoretical study on haloaromatic radical anions and their intramolecular electron transfer reactions. J. Chem. Soc. Perkin Trans. 2 1995, 9, 1821–1828. [Google Scholar] [CrossRef]
- Muthukrishnan, A.; Sangaranarayanan, M.V.; Boyarskiy, V.P.; Boyarskaya, I.A. Regioselective electrochemical reduction of 2,4-dichlorobiphenyl – Distinct standard reduction potentials for carbon–chlorine bonds using convolution potential sweep voltammetry. Chem. Phys. Lett. 2010, 490, 148–153. [Google Scholar] [CrossRef]
- Muthukrishnan, A.; Boyarskiy, V.; Sangaranarayanan, M.V.; Boyarskaya, I. Mechanism and Regioselectivity of the Electrochemical Reduction in Polychlorobiphenyls (PCBs): Kinetic Analysis for the Successive Reduction of Chlorines from Dichlorobiphenyls. J. Phys. Chem. C 2012, 116, 655–664. [Google Scholar] [CrossRef]
- Mullins, M.D.; Pochini, C.M.; McCrindle, S.; Romkes, M.; Safe, S.H.; Safe, L.M. High-resolution PCB analysis: Synthesis and chromatographic properties of all 209 PCB congeners. Environ. Sci. Technol. 1984, 18, 468–476. [Google Scholar] [CrossRef]
- Nikiforov, V.; Wightman, R. Reaction of nitroarenes and arenesulfonylchlorides with hexachlorocyclopentadiene. Chimia 1997, 51, 452. [Google Scholar]
- Frisch, A.M.J.; Trucks, G.W.; Shelelgel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03; Revision C.02; Gaussian, Inc.: Wallingford, CT, USA, 2003. [Google Scholar]
- Hebrard, F.; Kalck, P. Cobalt-Catalyzed Hydroformylation of Alkenes: Generation and Recycling of the Carbonyl Species, and Catalytic Cycle. Chem. Rev. 2009, 109, 4272–4282. [Google Scholar] [CrossRef]
- Peltier, D.; Pichevin, A. The carboxyl group. Infrared absorption and ionization. Bull. Soc. Chim. France 1960, 1141–1147. [Google Scholar]
- Stempel, G.H., Jr.; Greene, C.; Rongone, R.; Sobel, B.; Odioso, R. Some Disubstituted α-Methylstyrenes and their Polymerization Characteristics and a Comparative Study of the Refractive Indexes of Substituted Styrenes and α-Methylstyrenes. J. Am. Chem. Soc. 1951, 73, 455–456. [Google Scholar] [CrossRef]
- Egbertson, M.S.; Vassallo, L.M.; Hartman, G.D.; Halczenko, W.; Whitman, D.B.; Perkins, J.J.; Krause, A.E.; Ihle, N.; Claremon, D.A.; Hoffman, W.; et al. Fibrinogen Receptor Antagonists. U.S. Patent 5648368 A1, 15 July 1997. [Google Scholar]
- Springfield, S.A.; Doubleday, W.W.; Buono, F.; Couturier, M.A.; Lear, Y.; Favreau, D.; Levesque, K.; Manchand, P.S.; Frieser, M.; Cocuzza, A.J.; et al. Process for preparation of 1,7-dichloro-4-methoxy-isoquinoline. PCT Int. Appl. WO 2010027889 A2; 20100311, 2010. [Google Scholar]
- Lukaćs, G.; Porcs-Makkay, M.; Simig, G. Lithiation of 2-(chloroaryl)-2-methyl-1,3-dioxolanes and application in synthesis of new ortho-functionalized acetophenone derivatives. Tetrahedron Lett. 2003, 44, 3211–3214. [Google Scholar] [CrossRef]
- Bhatt, R.; Gong, B.; Hong, F.; Jenkins, S.A.; Klein, J.P.; Kumar, A.M.; Tulinsky, J. Preparation of 6-phenyl-N-phenyl-[1,3,5]triazine-2,4-diamine derivatives and related compounds with lysophosphatidic acid acyltransferase β (LPAAT-β) inhibitory activity for use in the treatment of cancer. PCT Int. Appl WO 2003037346 A1; 20030508, 2003. [Google Scholar]
- Alessi, T.R.; Ellingboe, J.W. Preparation of novel substituted 3H-1,2,3,5-oxathiadiazole 2-oxides as hypoglycemics. U.S. Patent US 4895860 A, 23 January 1990. [Google Scholar]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Khaibulova, T.S.; Boyarskaya, I.A.; Larionov, E.; Boyarskiy, V.P. Cobalt-Catalyzed Methoxycarbonylation of Substituted Dichlorobenzenes as an Example of a Facile Radical Anion Nucleophilic Substitution in Chloroarenes. Molecules 2014, 19, 5876-5897. https://doi.org/10.3390/molecules19055876
Khaibulova TS, Boyarskaya IA, Larionov E, Boyarskiy VP. Cobalt-Catalyzed Methoxycarbonylation of Substituted Dichlorobenzenes as an Example of a Facile Radical Anion Nucleophilic Substitution in Chloroarenes. Molecules. 2014; 19(5):5876-5897. https://doi.org/10.3390/molecules19055876
Chicago/Turabian StyleKhaibulova, Tatyana S., Irina A. Boyarskaya, Evgeny Larionov, and Vadim P. Boyarskiy. 2014. "Cobalt-Catalyzed Methoxycarbonylation of Substituted Dichlorobenzenes as an Example of a Facile Radical Anion Nucleophilic Substitution in Chloroarenes" Molecules 19, no. 5: 5876-5897. https://doi.org/10.3390/molecules19055876
APA StyleKhaibulova, T. S., Boyarskaya, I. A., Larionov, E., & Boyarskiy, V. P. (2014). Cobalt-Catalyzed Methoxycarbonylation of Substituted Dichlorobenzenes as an Example of a Facile Radical Anion Nucleophilic Substitution in Chloroarenes. Molecules, 19(5), 5876-5897. https://doi.org/10.3390/molecules19055876