In Silico Docking, Molecular Dynamics and Binding Energy Insights into the Bolinaquinone-Clathrin Terminal Domain Binding Site

Abstract
:
1. Introduction

2. Results and Discussion
2.1. Global Docking of Bolinaquinone into Clathrin TD
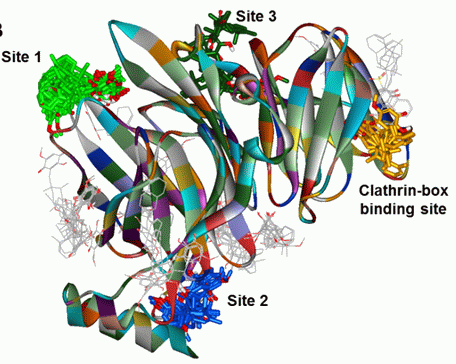
 (clathrin-box binding site),
(clathrin-box binding site),  (site 1),
(site 1),  (site 2) and
(site 2) and  (site 3). (B) Same representation as in A rotated 90° inward, indicating potential 1 binding sites. (C) Bar graph representation of the number of 1 poses in each binding site cluster shown in A and B. The bar colors correspond to the binding sites and 1-clusters identified in A and B.
(clathrin-box binding site), (site 1), (site 2) and (site 3). (B) Same representation as in A rotated 90° inward, indicating potential 1 binding sites. (C) Bar graph representation of the number of 1 poses in each binding site cluster shown in A and B. The bar colors correspond to the binding sites and 1-clusters identified in A and B.
(site 3). (B) Same representation as in A rotated 90° inward, indicating potential 1 binding sites. (C) Bar graph representation of the number of 1 poses in each binding site cluster shown in A and B. The bar colors correspond to the binding sites and 1-clusters identified in A and B.
(clathrin-box binding site), (site 1), (site 2) and (site 3). (B) Same representation as in A rotated 90° inward, indicating potential 1 binding sites. (C) Bar graph representation of the number of 1 poses in each binding site cluster shown in A and B. The bar colors correspond to the binding sites and 1-clusters identified in A and B.
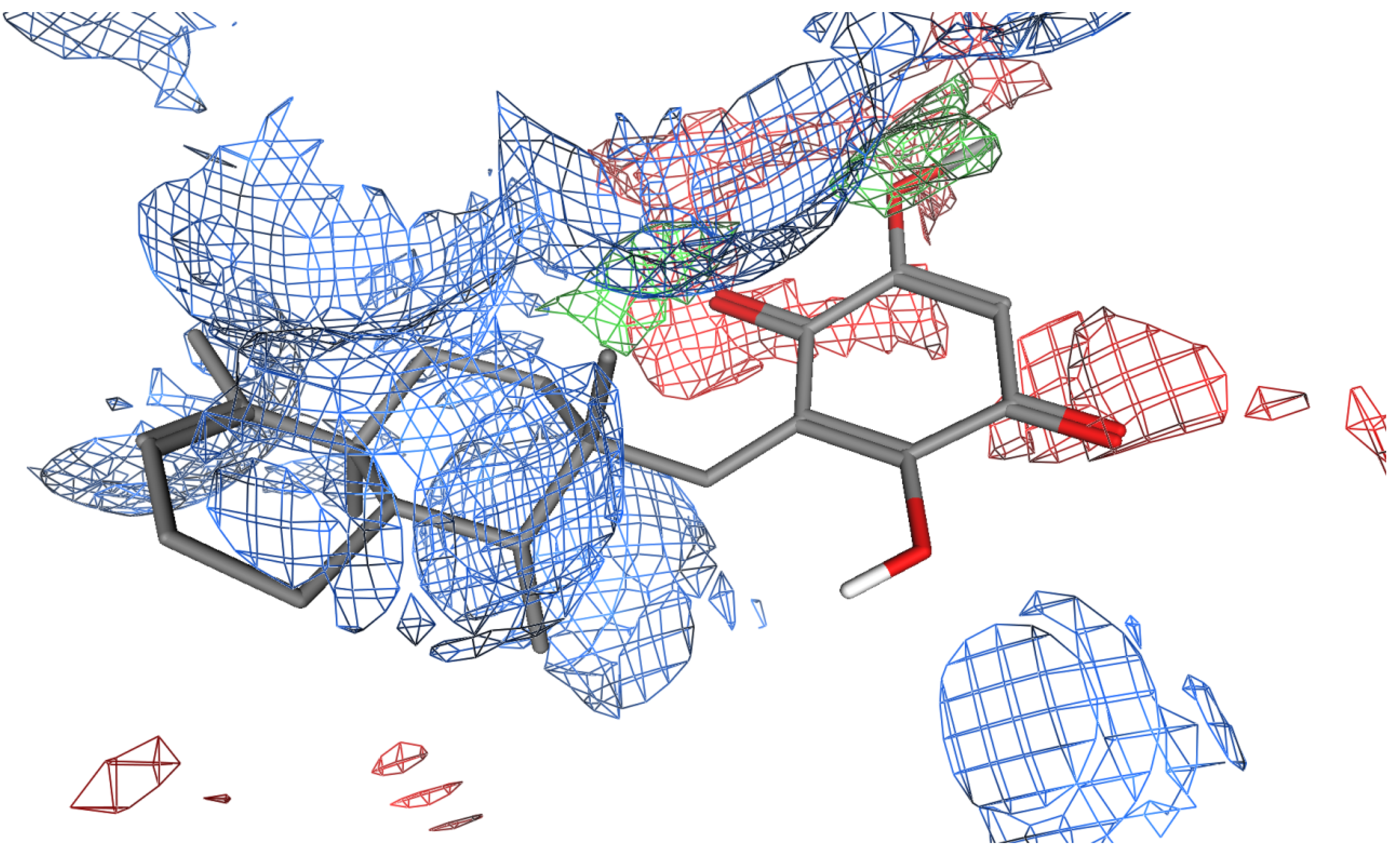
2.2. Flexible Docking of Bolinaquinone into Potential Binding Sites at Clathrin TD

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Potential complex | Flexible residues a | CDocker energy (kcal·mol−1) |
|---|---|---|
| Site 1 | Asn175-Gly179, Arg221-Gln23, Phe252-Phe260 | −20.6 |
| Site 2 | Val253-Leu357, Gln203-Glu207, Gln268-Asp271 | −17.8 |
| Site 3 | Met141-Ser146, Gln182-Tyr184, Lys189-Ser191 | −13.7 |
| Clathrin-box site | Ile52, Ile62-Ser67, Ile93-Ser97 | −18.2 |
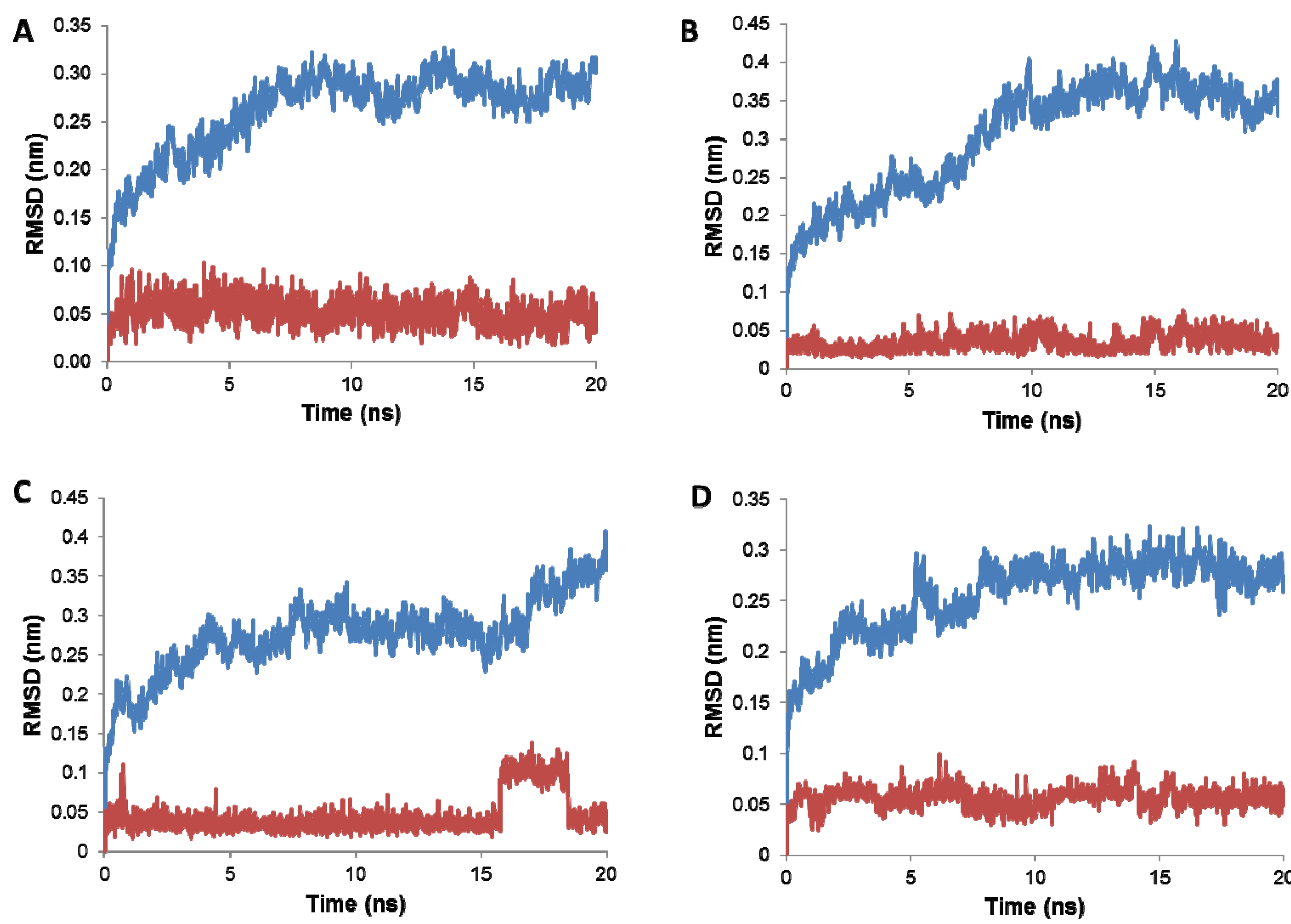
2.3. Stability of the Docked 1 Poses
2.4. Linear Interaction Energy Calculations

| Binding site | van der Waals contribution | Electrostatic contribution | ΔGpred (kcal·mol−1) |
|---|---|---|---|
| Site 1 | −21.5 ± 3.8 | −145.9 ± 8.3 | −5.3 ± 0.6 |
| Site 2 | −16.9 ± 3.2 | −72.5 ± 4.7 | −2.0 ± 0.7 |
| Site 3 | NC a | NC | NC |
| Clathrin-box site | −18.6 ± 2.9 | −103.5 ± 4.5 | −3.6 ± 0.4 |
| Potential binding site | Hydrogen bond average distances (% existence) a | Average number of H-Bonds b | ||
|---|---|---|---|---|
| Carbonyl groups | Hydroxyl group | Methoxy group | ||
| Site 1 | 2.4 (96%) | 1.8 (65%) | 2.1 (27%) | 2.43 |
| Site 2 | 2.9 (68%) | 2.3 (81%) | 3.1 (15%) | 1.72 |
| Clathrin-box site | 2.1 (91%) | 2.6 (38%) | 1.9 (67%) | 2.24 |
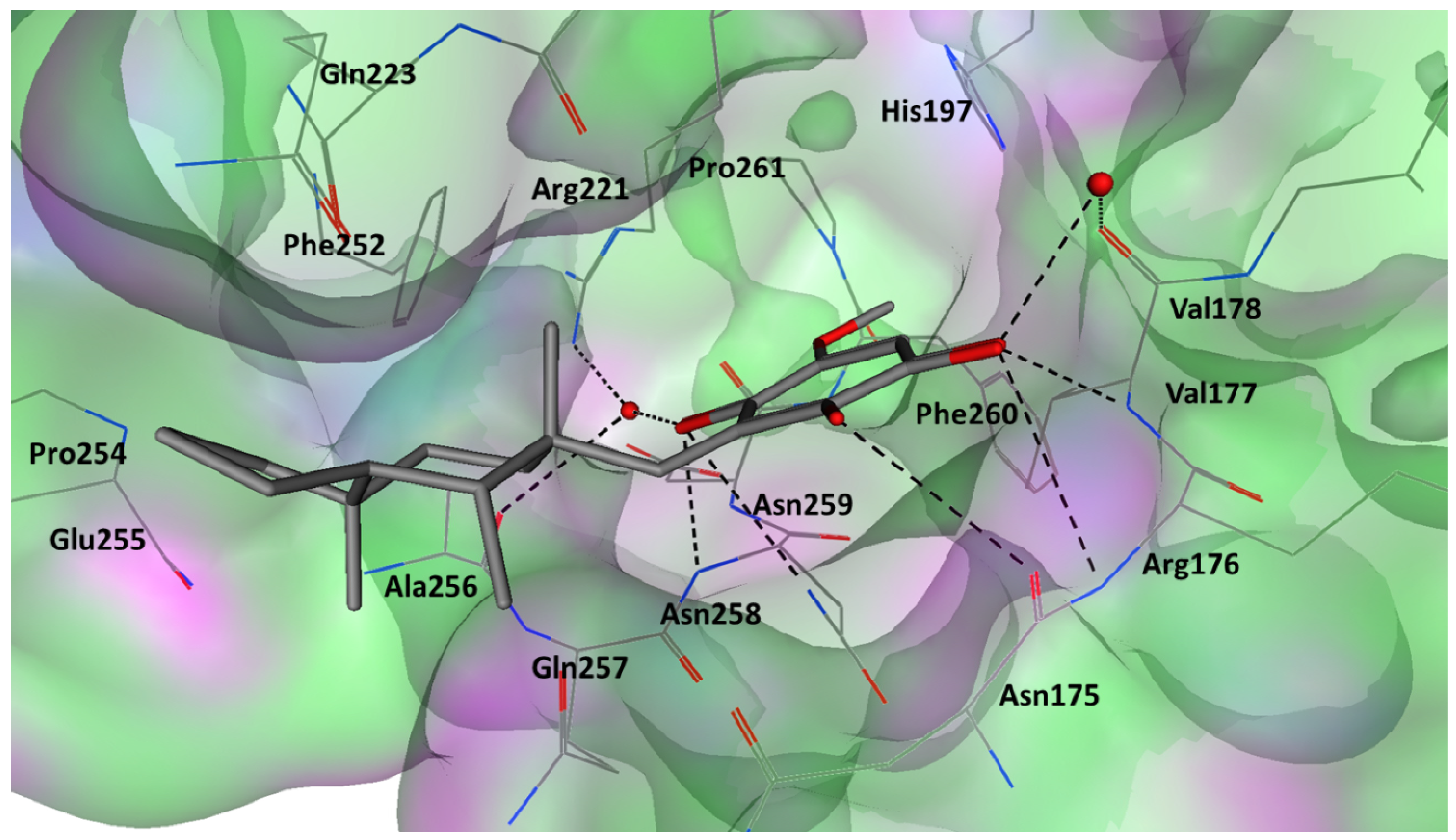
2.5. Key Interactions and Insights for Drug Design

3. Experimental
3.1. Crystal Structure Selection and Preparation

3.2. Initial Global Docking
3.3. Flexible Docking
3.4. Molecular Dynamics Details
3.5. Linear Interaction Energy Calculations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Traub, L.M. Tickets to ride: Selecting cargo for clathrin-regulated internalization. Nat. Rev. Mol. Cell Biol. 2009, 10, 583–596. [Google Scholar] [CrossRef]
- Von Kleist, L.; Stahlschmidt, W.; Bulut, H.; Gromova, K.; Puchkov, D.; Robertson, M.J.; MacGregor, K.A.; Tomilin, N.; Pechstein, A.; Chau, N.; et al. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell 2011, 146, 471–484. [Google Scholar] [CrossRef]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 2009, 137, 433–444. [Google Scholar] [CrossRef]
- Boettner, D.R.; Chi, R.J.; Lemmon, S.K. Lessons from yeast for clathrin-mediated endocytosis. Nat. Cell Biol. 2011, 14, 2–10. [Google Scholar] [CrossRef]
- Saffarian, S.; Cocucci, E.; Kirchhausen, T. Distinct dynamics of endocytic clathrin-coated pits and coated plaques. PLoS Biol. 2009, 7, e1000191. [Google Scholar] [CrossRef]
- Ohmori, K.; Endo, Y.; Yoshida, Y.; Ohata, H.; Taya, Y.; Enari, M. Monomeric but not trimeric clathrin heavy chain regulates p53-mediated transcription. Oncogene 2007, 27, 2215–2227. [Google Scholar]
- Ohata, H.; Ota, N.; Shirouzu, M.; Yokoyama, S.; Yokota, J.; Taya, Y.; Enari, M. Identification of a Function-Specific Mutation of Clathrin Heavy Chain (CHC) Required for p53 transactivation. J. Mol. Biol. 2009, 394, 460–471. [Google Scholar] [CrossRef]
- Endo, Y.; Sugiyama, A.; Li, S.-A.; Ohmori, K.; Ohata, H.; Yoshida, Y.; Shibuya, M.; Takei, K.; Enari, M.; Taya, Y. Regulation of clathrin-mediated endocytosis by p53. Gene Cell. 2008, 13, 375–386. [Google Scholar] [CrossRef]
- Nicot, A.S.; Toussaint, A.; Tosch, V.; Kretz, C.; Wallgren-Pettersson, C.; Iwarsson, E.; Kingston, H.; Garnier, J.M.; Biancalana, V.; Oldfors, A.; et al. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat. Genet. 2007, 39, 1134–1139. [Google Scholar] [CrossRef]
- MacGregor, K.A.; Robertson, M.J.; Young, K.A.; von Kleist, L.; Stahlschmidt, W.; Whiting, A.; Chau, N.; Robinson, P.J.; Haucke, V.; McCluskey, A. Development of 1,8-naphthalimides as clathrin inhibitors. J. Med. Chem. 2013, 57, 131–143. [Google Scholar]
- Margarucci, L.; Monti, M.C.; Fontanella, B.; Riccio, R.; Casapullo, A. Chemical proteomics reveals bolinaquinone as a clathrin-mediated endocytosis inhibitor. Mol. BioSys. 2011, 7, 480–485. [Google Scholar] [CrossRef]
- De Guzman, F.S.; Copp, B.R.; Mayne, C.L.; Concepcion, G.P.; Mangalindan, G.C.; Barrows, L.R.; Ireland, C.M. Bolinaquinone: A novel cytotoxic sesquiterpene hydroxyquinone from a Philippine Dysidea sponge. J. Org. Chem. 1998, 63, 8042–8044. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comp. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Koska, J.R.; Spassov, V.Z.; Maynard, A.J.; Yan, L.; Austin, N.; Flook, P.K.; Venkatachalam, C.M. Fully automated molecular mechanics based induced fit protein−ligand docking method. J. Chem. Inf. Mod. 2008, 48, 1965–1973. [Google Scholar] [CrossRef]
- Spassov, V.Z.; Yan, L.; Flook, P.K. The dominant role of side-chain backbone interactions in structural realization of amino acid code. ChiRotor: A side-chain prediction algorithm based on side-chain backbone interactions. Prot. Sci. 2007, 16, 494–506. [Google Scholar] [CrossRef]
- Diller, D.J.; Merz, K.M. High throughput docking for library design and library prioritization. Proteins 2001, 43, 113–124. [Google Scholar] [CrossRef]
- Wu, G.; Robertson, D.H.; Brooks, C.L.; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comp. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Schuttelkopf, A.W.; van Aalten, D.M.F. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Cryst. Sect. D 2004, 60, 1355–1363. [Google Scholar] [CrossRef]
- Lemkul, J.A.; Allen, W.J.; Bevan, D.R. Practical considerations for building GROMOS-compatible small-molecule topologies. J. Chem. Inf. Model. 2010, 50, 2221–2235. [Google Scholar] [CrossRef]
- Carlsson, J.; Boukharta, L.; Åqvist, J. Combining docking, molecular dynamics and the linear interaction energy method to predict binding modes and affinities for non-nucleoside inhibitors to HIV-1 reverse transcriptase. J. Med. Chem. 2008, 51, 2648–2656. [Google Scholar] [CrossRef]
- Åqvist, J.; Medina, C.; Samuelsson, J.-E. A new method for predicting binding affinity in computer-aided drug design. Prot. Eng. 1994, 7, 385–391. [Google Scholar] [CrossRef]
- Gutiérrez-de-Terán, H.; Åqvist, J. Linear interaction energy: Method and applications in drug design. In Computational Drug Discovery and Design; Baron, R., Ed.; Springer: New York, NY, USA, 2012; volume 819, pp. 305–323. [Google Scholar]
- Nicola, G.; Smith, C.A.; Abagyan, R. New method for the assessment of all drug-like pockets across a structural genome. J. Comput. Biol. 2008, 15, 231–240. [Google Scholar] [CrossRef]
- Stehn, J.R.; Haass, N.K.; Bonello, T.; Desouza, M.; Kottyan, G.; Treutlein, H.; Zeng, J.; Nascimento, P.R.B.B.; Sequeira, V.B.; Butler, T.L.; et al. A novel class of anti cancer compounds which target the actin cytoskeleton of tumor cells. Can. Res. 2013, 73, 5169–5182. [Google Scholar] [CrossRef]
- Odell, L.R.; Howan, D.; Gordon, C.P.; Robertson, M.J.; Chau, N.; Mariana, A.; Whiting, A.E.; Abagyan, R.; Daniel, J.A.; Gorgani, N.N.; et al. The pthaladyns: GTP competitive inhibitors of dynamin I and II GTPase derived from virtual screening. J. Med. Chem. 2010, 53, 5267–5280. [Google Scholar] [CrossRef]
- Cheng, T.; Li, Q.; Zhou, Z.; Wang, Y.; Bryant, S.H. Structure-based virtual screening for drug discovery: A problem-centric review. AAPS J. 2012, 14, 133–141. [Google Scholar] [CrossRef]
- Cavasotto, C.N.; Orry, A.J. Ligand docking and structure-based virtual screening in drug discovery. Curr. Top. Med. Chem. 2007, 7, 1006–1014. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from Abcam Biochemicals (Bristol, UK) or from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Abdel-Hamid, M.K.; McCluskey, A. In Silico Docking, Molecular Dynamics and Binding Energy Insights into the Bolinaquinone-Clathrin Terminal Domain Binding Site. Molecules 2014, 19, 6609-6622. https://doi.org/10.3390/molecules19056609
Abdel-Hamid MK, McCluskey A. In Silico Docking, Molecular Dynamics and Binding Energy Insights into the Bolinaquinone-Clathrin Terminal Domain Binding Site. Molecules. 2014; 19(5):6609-6622. https://doi.org/10.3390/molecules19056609
Chicago/Turabian StyleAbdel-Hamid, Mohammed K., and Adam McCluskey. 2014. "In Silico Docking, Molecular Dynamics and Binding Energy Insights into the Bolinaquinone-Clathrin Terminal Domain Binding Site" Molecules 19, no. 5: 6609-6622. https://doi.org/10.3390/molecules19056609
APA StyleAbdel-Hamid, M. K., & McCluskey, A. (2014). In Silico Docking, Molecular Dynamics and Binding Energy Insights into the Bolinaquinone-Clathrin Terminal Domain Binding Site. Molecules, 19(5), 6609-6622. https://doi.org/10.3390/molecules19056609