Three-Dimensional Heterocycles: New Uracil-Based Structures Obtained by Nucleophilic Substitution at the sp2 Carbon of Bromoisoxazoline

Abstract
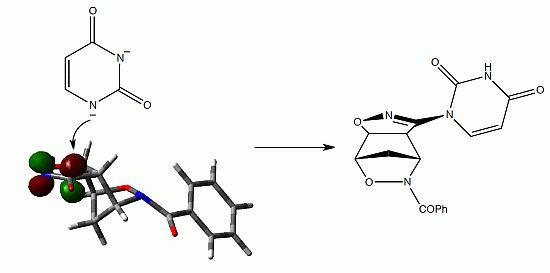
:
1. Introduction



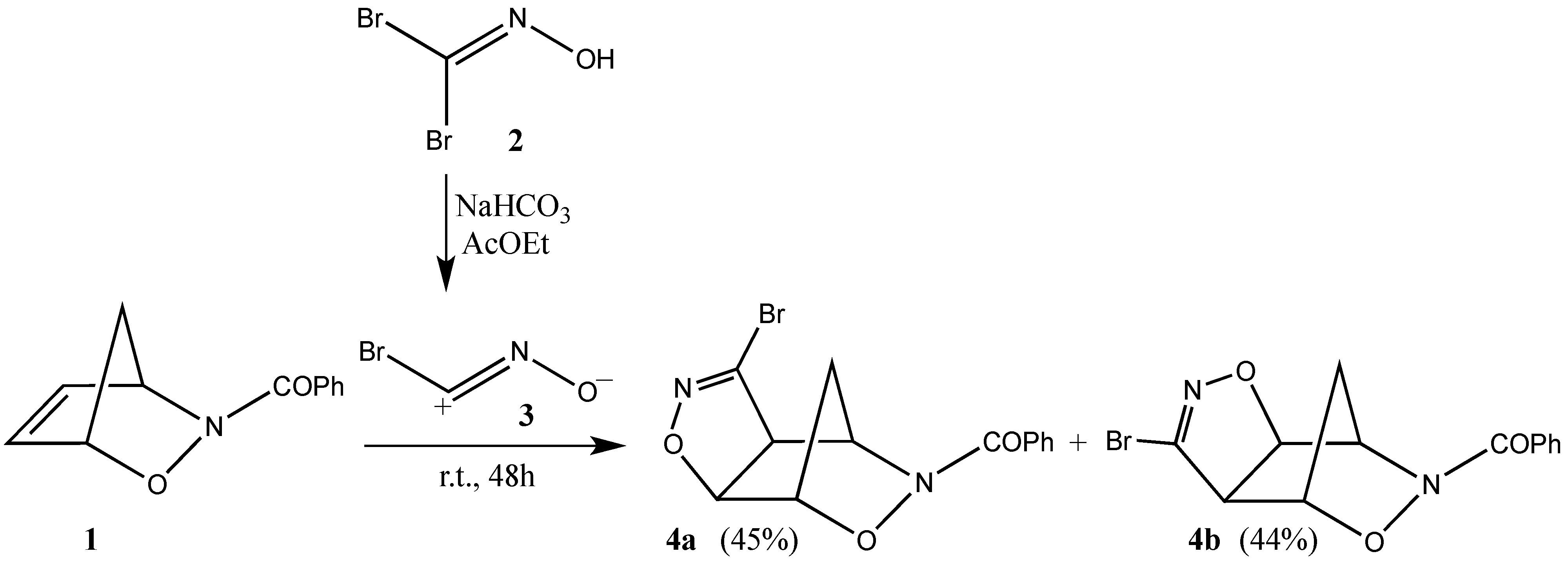
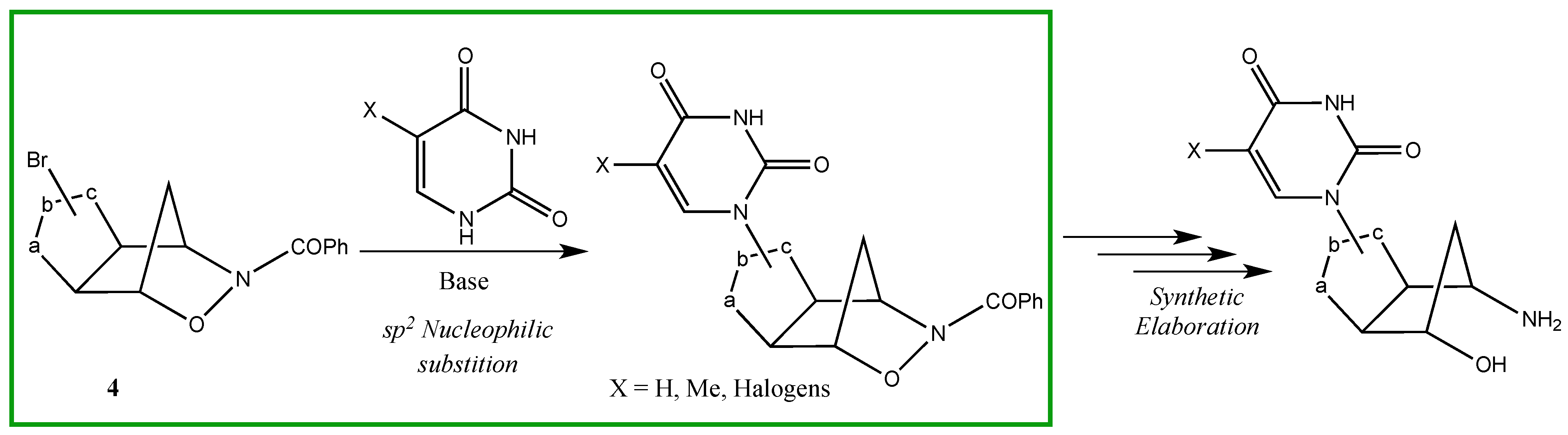
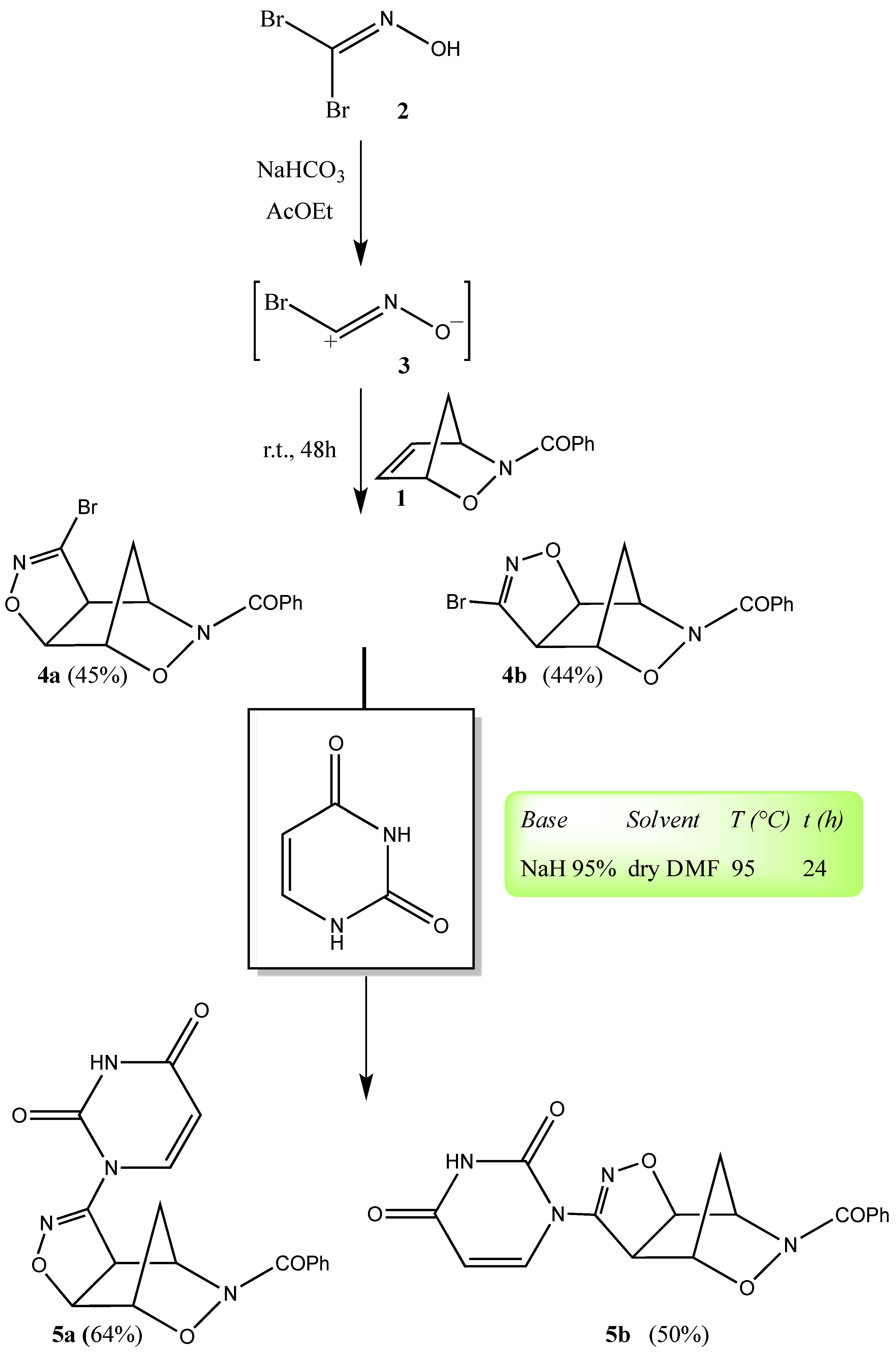
2. Results and Discussion



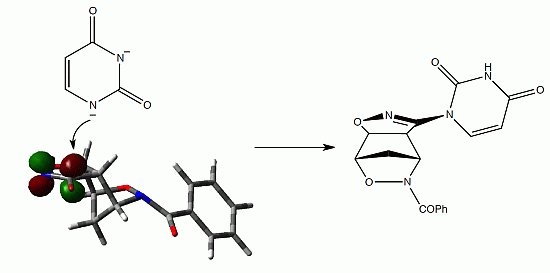{kind=link}
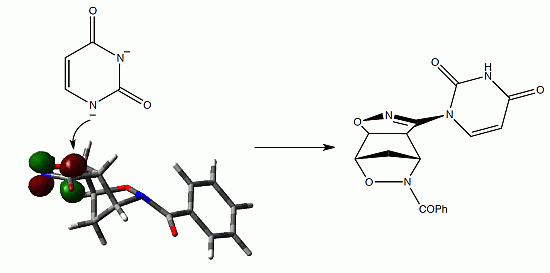{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
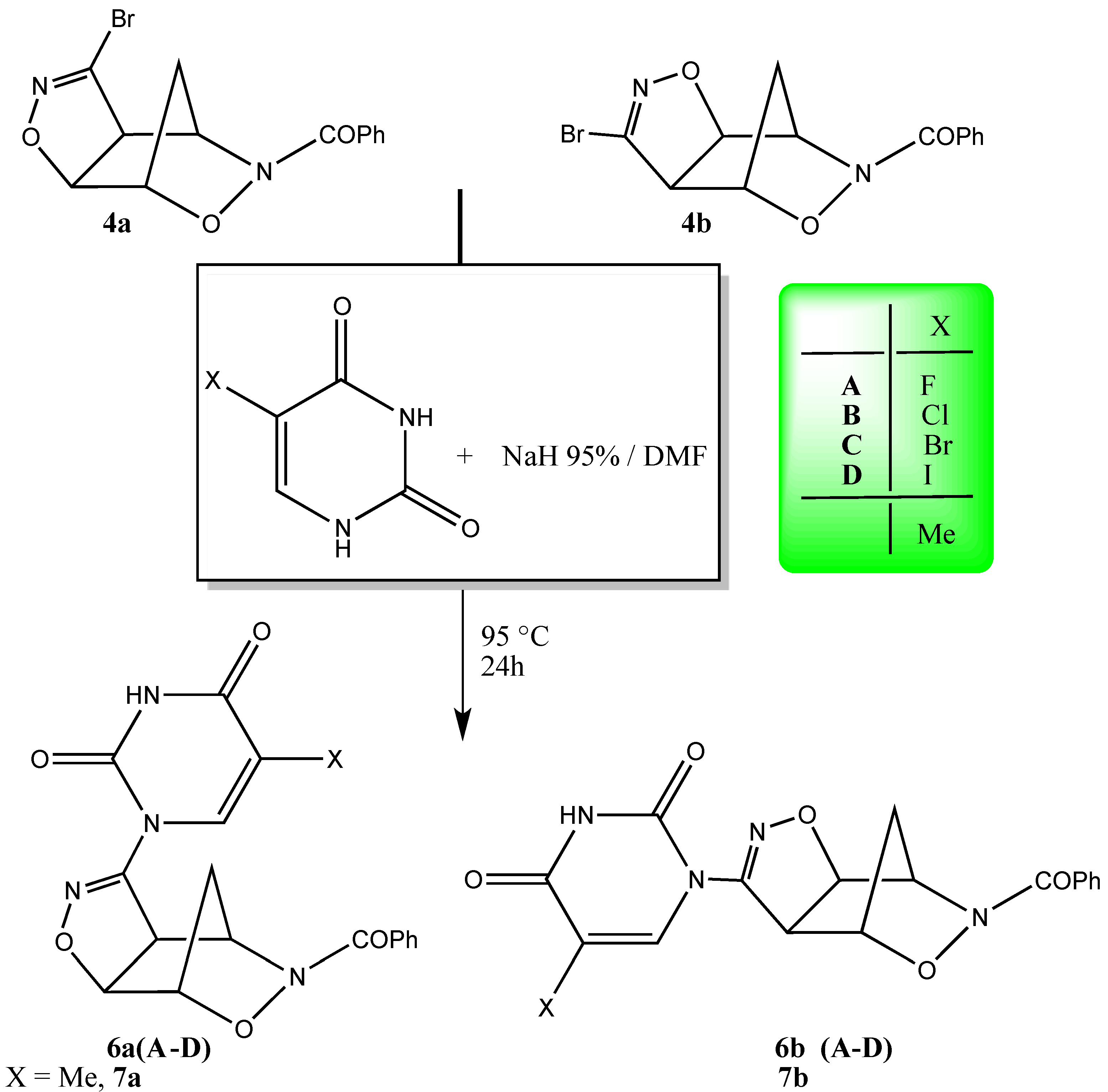{kind=link}
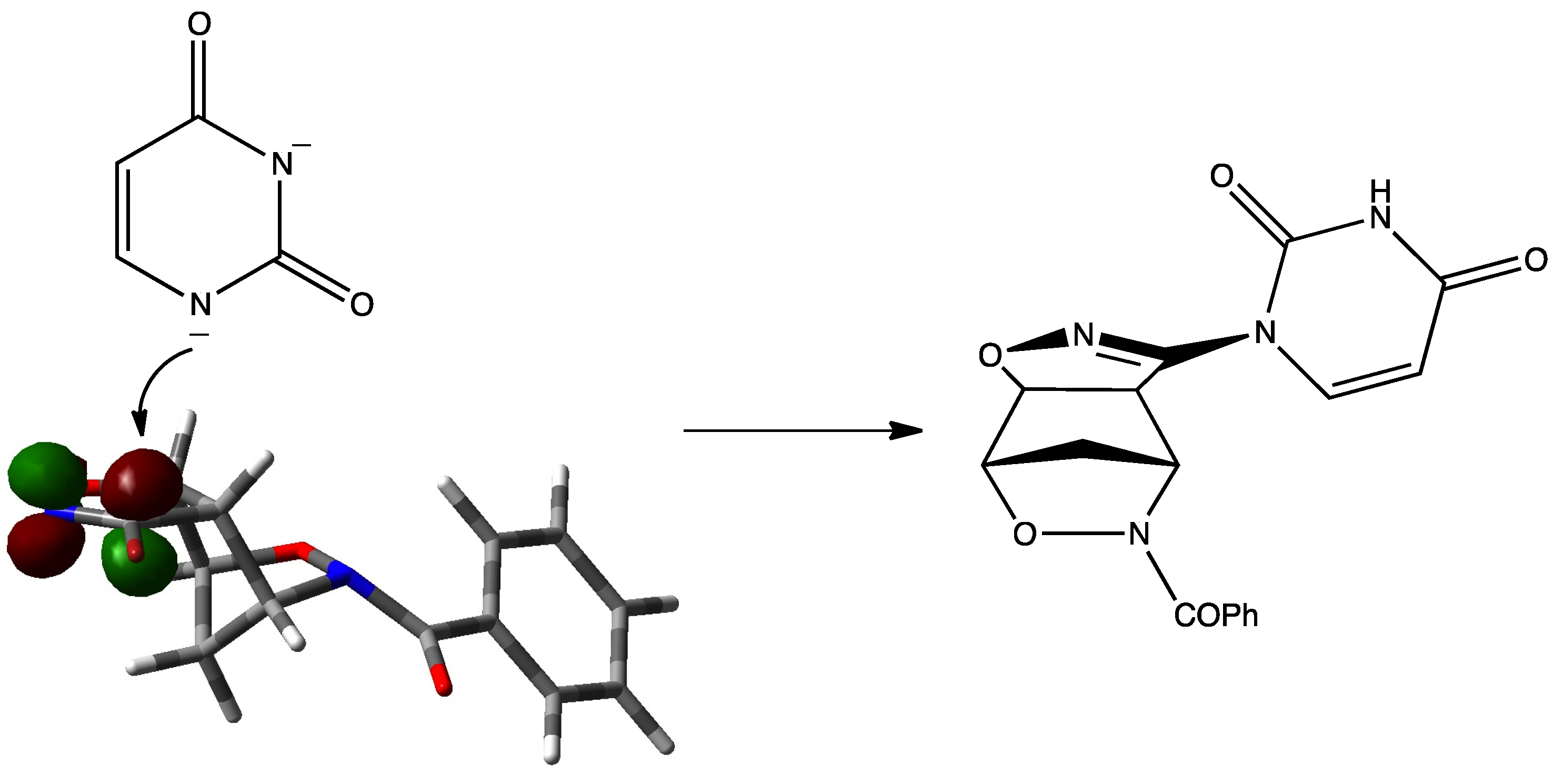{kind=link}
| Entry | Adduct 6a | Yield (%) | m.p. (°C) # | IR (cm−1) νNH | νC=O | 1H-NMR N-CH=CX | (δ, DMSO- d6) J (Hz) |
|---|---|---|---|---|---|---|---|
| 1 | A | 46 | 193–198 | 3394 | 1733 | 8.24 (d) | 7 |
| 2 | B | 53 | 180–189 | 3360 | 1734 | 8.23 (s) | |
| 3 | C | 56 | 96–100 | 3200 | 1734 | 8.27 (s) | |
| 4 | D | 57 | 110–115 | 3394 | 1632 | 8.23 (s) | |
| 6b | |||||||
| 5 | A | 64 | 95–100 | 3157 | 1723 | 8.24 (d) | 7 |
| 6 | B | 76 | 98–105 | 3178 | 1732 | 8.24 (s) | |
| 7 | C | 49 | 104–106 | 3121 | 1732 | 8.29 (s) | |
| 8 | D | 57 | 162–164 | 3160 | 1720 | 8.24 (s) | |
| 9 | 7a | 45 | 215–217 | 3176 | 1715 | 7.69 (d) | 1.81 (Me) |
| 10 | 7b | 55 | 220–222 | 3149 | 1710 | 7.69 (d) | 1.82 (Me) |


3. Experimental
3.1. General Information
3.2. Cycloaddition of Bromonitrile Oxide 3 to N-Benzoyl-2,3-oxazanorborn-5-ene (1)
3.3. General Procedure for the Synthesis of Compounds 5a,b, 6a,b (A–D) and 7a,b
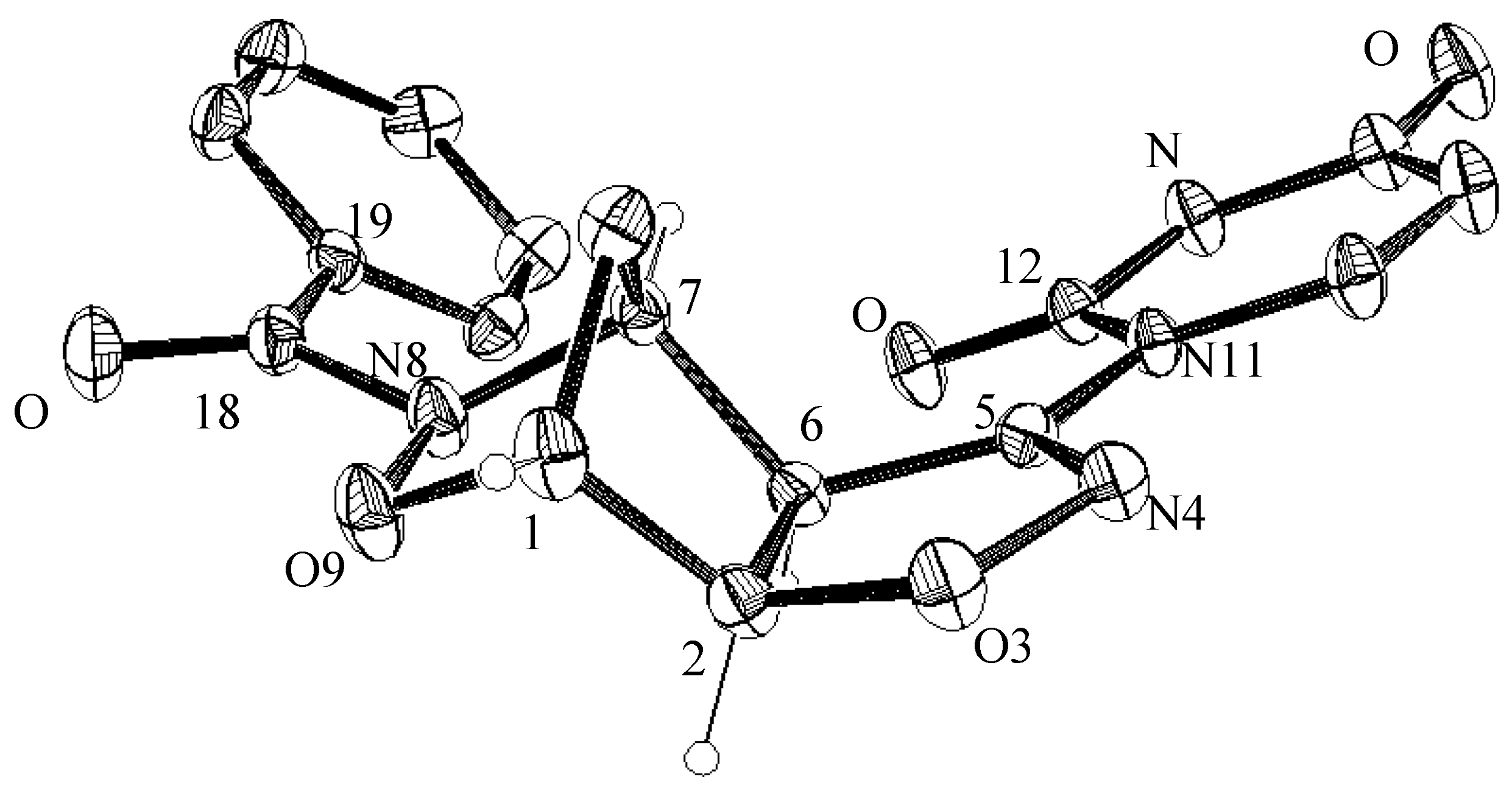
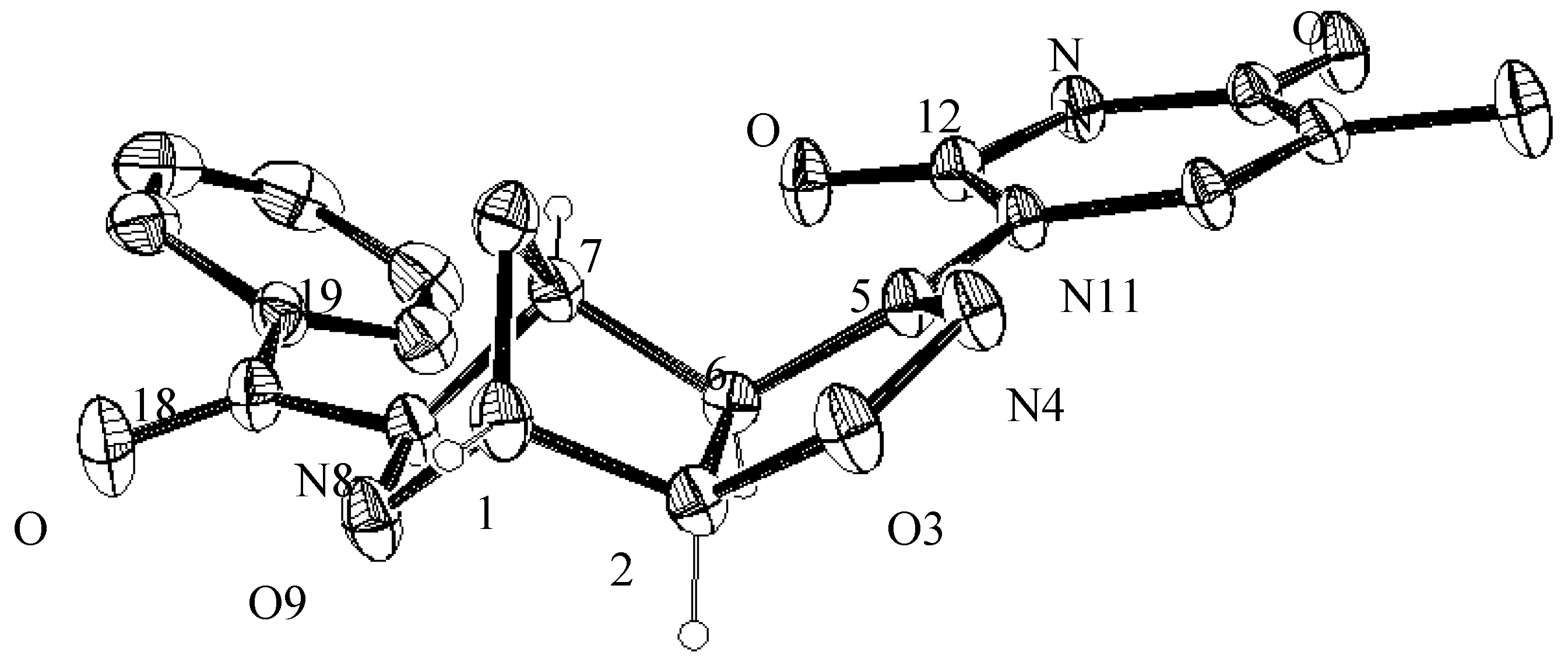
3.4. X-ray Crystallographic Analysis of Compounds 5a and 7a
| C1—C2 | 1.523(4) 1.507(6) | C5—C6 | 1.499(3) 1.498(5) |
| C1—O9 | 1.458(3) 1.456(5) | C5—N11 | 1.417(3) 1.408(4) |
| C1—C10 | 1.501(4) 1.497(6) | C6—C7 | 1.538(4) 1.533(5) |
| C2—O3 | 1.452(3) 1.453(4) | C7—N8 | 1.477(3) 1.487(5) |
| C2—C6 | 1.534(3) 1.540(5) | C7—C10 | 1.517(3) 1.520(6) |
| O3—N4 | 1.420(2) 1.402(4) | N8—O9 | 1.450(2) 1.455(4) |
| N4—C5 | 1.270(3) 1.272(4) | N8—C18 | 1.376(3) 1.377(5) |
| Bond Angles (°) | |||
| C2—C1—O9 | 104.8(2) 104.3(2) | C2—C6—C5 | 100.1(2) 99.8(3) |
| C2—C1—C10 | 103.9(2) 104.2(4) | C2—C6—C7 | 101.9(2) 101.7(3) |
| O9—C1—C10 | 103.3(2) 103.8(4) | C6—C7—N8 | 104.0(2) 104.2(3) |
| C1—C2—C6 | 102.6(2) 102.9(3) | C6—C7—C10 | 103.2(2) 102.7(4) |
| O3—C2—C6 | 105.8(2) 105.5(3) | N8—C7—C10 | 101.6(2) 101.2(3) |
| C2—O3—N4 | 109.4(2) 109.4(3) | C7—N8—O9 | 105.2(2) 105.3(3) |
| O3—N4—C5 | 109.1(2) 109.9(3) | C7—N8—C18 | 126.9(2) 124.1(3) |
| N4—C5—C6 | 115.6(2) 115.1(3) | O9—N8—C18 | 112.9(2) 110.0(3) |
| N4—C5—N11 | 118.1(2) 116.4(3) | C1—O9—N8 | 103.7(2) 103.4(3) |
| C6—C5—N11 | 126.4(2) 128.3(3) | C1—C10—C7 | 92.7(2) 92.9(4) |
| Empirical Formula | C17H14N4O5 |
|---|---|
| Formula weight | 354.32 |
| Crystal size, mm | 0.56 × 0.42 × 0.18 |
| Temperature, K | 293 |
| Crystal system | Orthorhombic |
| Space group | P bca |
| a, Å | 9.977(4) |
| b, Å | 16.774(3) |
| c, Å | 19.649(3) |
| α | 90 |
| β | 90 |
| γ | 90 |
| V, Å3 | 3288(1) |
| Z | 8 |
| Dcalcd, g·cm−3 | 1.431 |
| Absorption coeff., µ, mm−1 | 0.108 |
| Diffractometer/scan | Enraf–Nonius CAD–4, θ/2θ |
| λ, Å | 0.71073 |
| F(000) | 1472 |
| Range (°) for data | 2.0 < θ > 25 |
| Index ranges | 0 < h> 11, 0 < k> 19, 0 < l > 23 |
| No. of reflects. measd | 2893 |
| No. of unique reflects | 1824 |
| Correction applied | Lorentz–polarization |
| Refinement method | Full–matrix least–squares |
| No. of variables | 291 |
| Goodness–of–fit (2893) | 0.900 |
| R1 (I) > 2 σ (I), (1824) | 0.0403 |
| R1 (2883) | 0.0785 |
| (∆ρ) max, min, eÅ−3 | 0.124, −0.151 |
| Empirical Formula | C20H22N4O6 |
|---|---|
| Formula weight | 414.417 |
| Crystal size, mm | 0.50 × 0.385 × 0.14 |
| Temperature, K | 293 |
| Crystal system | Monoclinic |
| Space group | P 21/n |
| a, Å | 11.991(3) |
| b, Å | 11.4437(8) |
| c, Å | 14.63(1) |
| α | 90.0 |
| β | 101.34(4) |
| γ | 90.0 |
| V, Å3 | 1968.3(14) |
| Z | 4 |
| Dcalcd, g·cm−3 | 1.3985 |
| Absorption coeff., µ, mm−1 | 0.1051 |
| Diffractometer/scan | Enraf–Nonius CAD–4, θ/2 θ |
| Radiation | MoKα |
| λ, Å | 0.71073 |
| F(000) | 872 |
| Range (°) for data | 2.0 < θ > 25.0 |
| Index ranges | −11 < h > 11, 0 < k > 10, 0 < l > 14 |
| No. of reflects. measd | 1828 |
| No. of unique reflects | 1266 |
| Correction applied | Lorentz–polarization |
| Refinement method | Full–matrix least–squares |
| No. of variables | 359 |
| Goodness–of–fit (1828) | 1.024 |
| R1 (I) > 2 σ (I), (1266) | 0.0414 |
| R1 (1828) | 0.0728 |
| (∆ρ) max, min, eÅ−3 | 0.119, −0.129 |
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Savion, M.; Memeo, M.G.; Bovio, B.; Grazioso, G.; Legnani, L.; Quadrelli, P. Synthesis and molecular modeling of novel dihydroxycyclopentane-carbonitrile nor-nucleosides by bromonitrile oxide 1,3-dipolar cycloaddition. Tetrahedron 2012, 68, 1845–1852. [Google Scholar] [CrossRef]
- Quadrelli, P.; Bovio, B.; Piccinini, A.; Caramella, P.; de Sarlo, F.; Machetti, F. Conversion of a nitrosocarbonyl hetero Diels-Alder cycloadduct to useful isoxazoline-carbocyclic aminols. Tetrahedron 2009, 65, 10679–10686. [Google Scholar] [CrossRef]
- Bodnar, B.S.; Miller, M.J. The nitrosocarbonyl hetero-diels–alder reaction as a useful tool for organic syntheses. Angew. Chem. Int. Ed. 2011, 50, 5630–5647. [Google Scholar] [CrossRef]
- Scagnelli, L.; Memeo, M.G.; Carosso, S.; Bovio, B.; Quadrelli, P. Syntheses of New Carbanucleosides by Pericyclic Reactions. Eur. J. Org. Chem. 2013, 2013, 3835–3846. [Google Scholar] [CrossRef]
- Kirby, G.W. Electrophilic C-Nitroso-compomds. Chem. Soc. Rev. 1977, 6, 1–24. [Google Scholar] [CrossRef]
- Quadrelli, P.; Mella, M.; Gamba Invernizzi, A.; Caramella, P. The mild oxidation of nitrile oxides affords a convenient entry to nitrosocarbonyl intermediates, versatile tools in organic synthesis. Tetrahedron 1999, 55, 10497–10510. [Google Scholar] [CrossRef]
- Quadrelli, P.; Mella, M.; Paganoni, P.; Caramella, P. Cycloadditions of nitrile oxides to the highly reactive N-Acyl-2-oxa-3-azanorborn-5-enes afford versatile cycloadducts and a convenient entry to highly functionalized derivatives. Eur. J. Org. Chem. 2000, 14, 2613–2620. [Google Scholar]
- Quadrelli, P.; Scrocchi, R.; Caramella, P.; Rescifina, A.; Piperno, A. From cyclopentadiene to isoxazoline–carbocyclic nucleosides: A rapid access to biological molecules through nitrosocarbonyl chemistry. Tetrahedron 2004, 60, 3643–3651. [Google Scholar] [CrossRef]
- Quadrelli, P.; Mella, M.; Carosso, S.; Bovio, B.; Caramella, P. A straightforward synthesis of isoxazoline-based carbocyclic nucleosides from 1,3-cyclohexadiene through nitrosocarbonyl chemistry. Eur. J. Org. Chem. 2007, 36, 6003–6015. [Google Scholar]
- Moggio, Y.; Legnani, L.; Bovio, B.; Memeo, M.G.; Quadrelli, P. Synthesis of novel anthracene derivatives of isoxazolino-carbocyclic nucleoside analogues. Tetrahedron 2012, 68, 1384–1392. [Google Scholar] [CrossRef]
- Miyauchi, H.; Chiba, S.; Fukamizu, K.; Ando, K.; Narasaka, K. Synthesis of hetero- and carbocycles by nucleophilic substitution at sp2 carbon. Tetrahedron 2007, 63, 5940–5953. [Google Scholar] [CrossRef]
- North, M. Incorporation of conformationally constrained β-amino acids into peptides. J. Pept. Sci. 2000, 6, 301–313. [Google Scholar] [CrossRef]
- Memeo, M.G.; Bovio, B.; Quadrelli, P. RuO 4-catalyzed oxidation reactions of isoxazolino-2-azanorbornane derivatives: A short-cut synthesis of tricyclic lactams and peptidomimetic γ-amino acids. Tetrahedron 2011, 67, 1907–1914. [Google Scholar] [CrossRef]
- Caldirola, P.; Ciancaglione, M.; de Amici, M.; de Micheli, C. Conversion of isoxazolines to β-hydroxy esters. Synthesis of 2-deoxy-D-ribose. Tetrahedron Lett. 1986, 27, 4647–4650. [Google Scholar] [CrossRef]
- Pinto, A.; Conti, P.; Tamborini, L.; de Micheli, C. A novel simplified synthesis of acivicin. Tetrahedron 2009, 20, 508–511. [Google Scholar] [CrossRef]
- Freeman, J. Possible criteria for distinguishing between cyclic and acyclic activated complexes and among cyclic activated complexes in addition reactions. Chem. Rev. 1975, 75, 439–489. [Google Scholar] [CrossRef]
- Mehta, G.; Chandrasekhar, J. Electronic control of facial selection in additions to sterically unbiased ketones and olefins. Chem. Rev. 1999, 99, 1437–1467. [Google Scholar] [CrossRef]
- Huisgen, R.; Ooms, P.H.J.; Mingin, M.; Allinger, N.L. Exceptional reactivity of the bicyclo[2.2.1]heptene double bond. J. Am. Chem. Soc. 1980, 102, 3951–3953. [Google Scholar] [CrossRef]
- Rondan, N.G.; Paddon-Row, M.N.; Caramella, P.; Houk, K.N. Nonpanar alkenes and carbonyls: A molecular distortion which parallels addition stereoselectivity. J. Am. Chem. Soc. 1981, 103, 2436–2438. [Google Scholar] [CrossRef]
- Houk, K.N.; Rondan, N.G.; Brown, F.K.; Jorgensen, W.L.; Madura, J.D.; Spellmeyer, D.C. Electronic origins and consequences of pyramidalization of asymmetric alkenes in ground and triplet excited states. J. Am. Chem. Soc. 1983, 105, 5980–5988. [Google Scholar]
- Coutouli-Argyropoulou, E.; Pilanidou, P. An entry to new isoxazoline analogues of dideoxynucleosides by bromonitrile oxide 1,3-dipolar cycloaddition. Tetrahedron Lett. 2003, 44, 3755–3758. [Google Scholar] [CrossRef]
- Quadrelli, P.; Bovio, B.; Piccanello, A. Structure determination of 8-benzyl-5-phenyl-3-oxa-4,8-diazatricyclo[5.2.1.02,6]dec-4-ene and 1-(9-Ethoxy-5-phenyl-3-oxa-4,8-diaza-tricyclo[5.2.1.02,6]dec-4-en-8-yl)-ethanone: Their synthesis, chemical relationship and comparison with similar compounds. J. Chem. Crystallogr. 2012, 42, 43–66. [Google Scholar] [CrossRef]
- Cremer, D.; Pople, J. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Bernasconi, C.F.; Rappoport, Z. Recent advances in our mechanistic understanding of SNV reactions. Acc. Chem. Res. 2009, 42, 993–1003. [Google Scholar] [CrossRef]
- Fernandez, I.; Bickelhaupt, F.M.; Uggerud, E. Reactivity in nucleophilic vinylic substitution (SNV):SNVπ versus SNVσ mechanistic dichotomy. J. Org. Chem. 2013, 78, 8574–8484. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Calculated Structure of 4a at the B3LYP 6–31G* Level by Means of Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Sheldrick, G.M. SHELXL-93. Program for the Refinement of Crystal Structure; University of Göttingen: Göttingen, Germany, 1993. [Google Scholar]
- Johnson, C.K. ORTEP, Report ORNL-3793; Oak Ridge National Laboratory: Oak Ridge TN, USA, 1966. [Google Scholar]
- Wilson, A.J.C. Determination of absolute from relative X-ray intensity data. Nature 1942, 150, 152. [Google Scholar] [CrossRef]
- André, C.; Legrand, B.; Deng, C.; Didierjean, C.; Pickaert, G.; Martinez, J.; Averlant-Petit, M.C.; Amblard, M.; Calmes, M. (S)-ABOC: A rigid bicyclic β-amino acid as turn inducer. Org. Lett. 2012, 14, 960–963. [Google Scholar] [CrossRef]
- Gorska, K; Winssinger, N. Reactions templated by nucleic acids: More ways to translate oligonucleotide-based instructions into emerging function. Angew. Chem. Int. Ed. 2013, 52, 6820–6843. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2014 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Memeo, M.G.; Lapolla, F.; Bovio, B.; Quadrelli, P. Three-Dimensional Heterocycles: New Uracil-Based Structures Obtained by Nucleophilic Substitution at the sp2 Carbon of Bromoisoxazoline. Molecules 2014, 19, 8661-8678. https://doi.org/10.3390/molecules19068661
Memeo MG, Lapolla F, Bovio B, Quadrelli P. Three-Dimensional Heterocycles: New Uracil-Based Structures Obtained by Nucleophilic Substitution at the sp2 Carbon of Bromoisoxazoline. Molecules. 2014; 19(6):8661-8678. https://doi.org/10.3390/molecules19068661
Chicago/Turabian StyleMemeo, Misal Giuseppe, Francesco Lapolla, Bruna Bovio, and Paolo Quadrelli. 2014. "Three-Dimensional Heterocycles: New Uracil-Based Structures Obtained by Nucleophilic Substitution at the sp2 Carbon of Bromoisoxazoline" Molecules 19, no. 6: 8661-8678. https://doi.org/10.3390/molecules19068661
APA StyleMemeo, M. G., Lapolla, F., Bovio, B., & Quadrelli, P. (2014). Three-Dimensional Heterocycles: New Uracil-Based Structures Obtained by Nucleophilic Substitution at the sp2 Carbon of Bromoisoxazoline. Molecules, 19(6), 8661-8678. https://doi.org/10.3390/molecules19068661