Two New Acylated Flavonol Glycosides from the Seeds of Lepidium sativum

Abstract
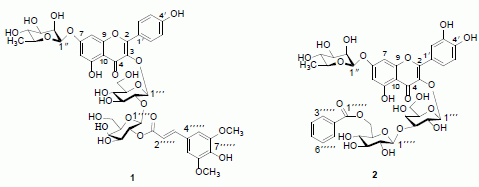
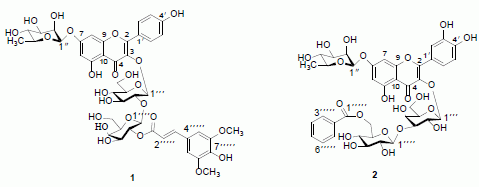
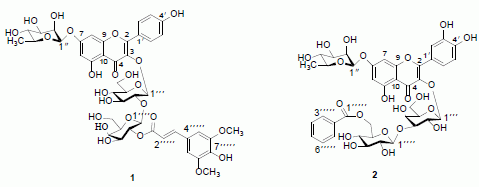
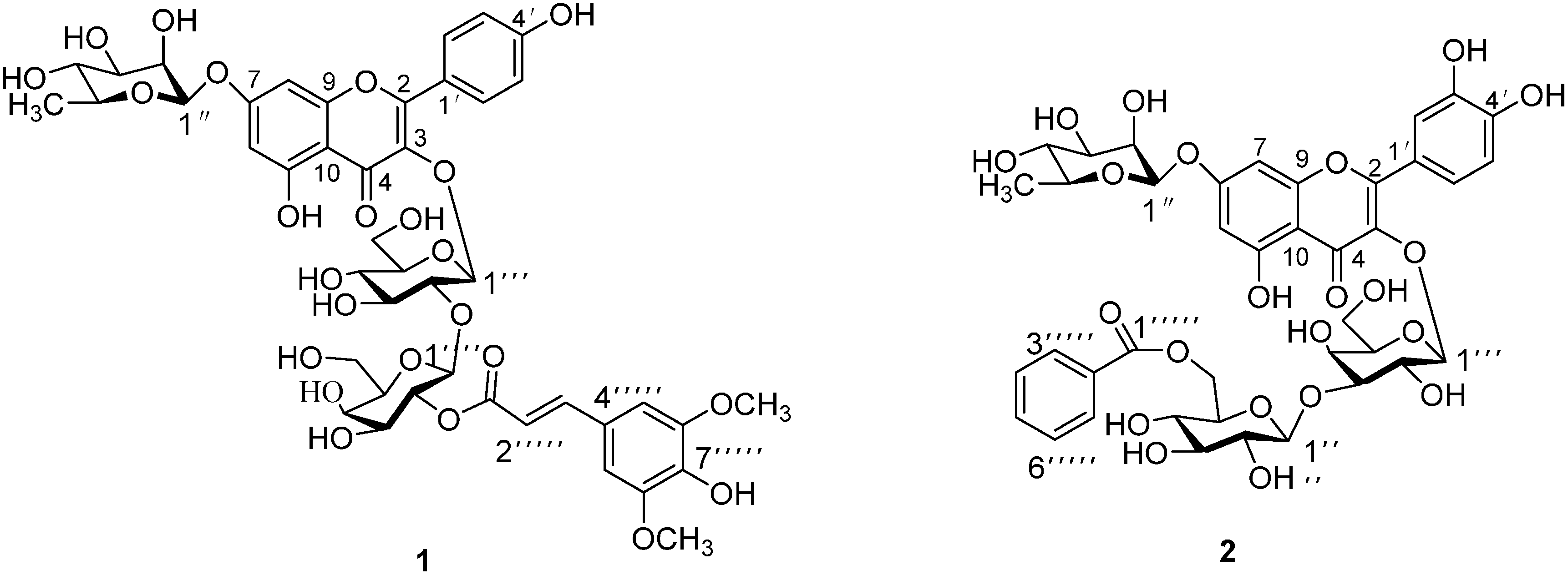
:
1. Introduction
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | ||||
|---|---|---|---|---|---|---|
| δC | δH (mult, J in Hz) | HMBC | δC | δH (mult, J in Hz) | HMBC | |
| 2 | 155.7 | 155.7 | ||||
| 3 | 133.1 | 133.3 | ||||
| 4 | 177.4 | 177.4 | ||||
| 5 | 160.7 | OH (12.66) | 160.9 | 12.65 (OH) | ||
| 6 | 99.3 | 6.32(d, 2.4) | C-5, C-7, C-8 | 99.2 | 6.38 (d, 1.8) | C-5, 8, 10 |
| 7 | 161.5 | 161.4 | ||||
| 8 | 93.8 | 6.63(d, 2.4) | C-7, C-9, C-10 | 94.1 | 6.69 (d, 1.8) | C-6, 7, 9, 10 |
| 9 | 155.7 | 155.6 | ||||
| 10 | 105.5 | 105.5 | ||||
| 1ꞌ | 120.9 | 122.2 | ||||
| 2ꞌ | 130.9 | 8.01 (d, 9) | C-2, C-4ꞌ | 115.8 | 7.50 (d, 1.8) | C-1ꞌ, 3ꞌ, 4ꞌ, 2 |
| 3ꞌ | 115.4 | 6.89 (d, 9 ) | C-4ꞌ | 144.9 | ||
| 4ꞌ | 156.0 | OH (s, 10.18) | 148.8 | |||
| 5ꞌ | 115.4 | 6.89 (d, 9) | C-4ꞌ | 115.2 | 6.82 (d, 8.4) | C-3ꞌ, 4ꞌ, 6ꞌ |
| 6ꞌ | 130.9 | 8.01 (d, 9) | C-2ꞌ | 120.7 | 7.65 (dd, 8.4, 1.8) | C-2ꞌ, 4ꞌ, 5ꞌ, 2 |
| 1ꞌꞌ | 98.5 | 5.50 (d, 1.2) | C-7, 3ꞌꞌ | 98.4 | 5.54 (s) | C-7, 2ꞌꞌ |
| 2ꞌꞌ | 69.8 | 3.87 (br. m) | C-3ꞌꞌ, 4ꞌꞌ | 69.8 | 3.86 (br. s) | C-5ꞌꞌ |
| 3ꞌꞌ | 70.2 | 3.63 (m) | 70.3 | 3.65 (dd, 3.0, 9.0 ) | ||
| 4ꞌꞌ | 71.6 | 3.31 (m) | C-3ꞌꞌ | 71.6 | 3.33 (m) | C-2ꞌꞌ, 6ꞌꞌ |
| 5ꞌꞌ | 70.0 | 3.43 (m) | C-4ꞌꞌ, 6ꞌꞌ | 70.1 | 3.45 (m) | C-3ꞌꞌ, 4ꞌꞌ |
| 6ꞌꞌ | 18.0 | 1.13 (3H, d, 6.0) | C-4ꞌꞌ, 5ꞌꞌ | 17.9 | 1.14 (d, 6.0) | C-4ꞌꞌ, 5ꞌꞌ |
| 1ꞌꞌꞌ | 97.0 | 5.81 (d, J = 7.8) | C-3, 2ꞌꞌꞌ, 5ꞌꞌꞌ | 98.5 | 5.59 (d, 7.8 ) | C-3 |
| 2ꞌꞌꞌ | 77.4 | 3.77 (dd, 9.6, 7.8) | C-5ꞌꞌꞌ, 1ꞌꞌꞌ | 72.9 | 3.61 (dd, 9.0, 7.8) | C-3ꞌꞌꞌ |
| 3ꞌꞌꞌ | 75.6 | 3.34 (m) | C-2ꞌꞌꞌ | 81.9 | 3.76 (dd, 9.0, 4.0) | C-1ꞌꞌꞌꞌ, C-1ꞌꞌꞌ, C-2ꞌꞌꞌ |
| 4ꞌꞌꞌ | 68.1 | 3.65 (m) | C-5ꞌꞌꞌ | 67.5 | 3.68 (dd, 4.0, 3.0) | C-ꞌꞌꞌ, 3ꞌꞌꞌ |
| 5ꞌꞌꞌ | 72.0 | 3.62 (m) | C-6ꞌꞌꞌ | 75.8 | 3.30 (m) | C-1ꞌꞌꞌ, 4ꞌꞌꞌ, 6ꞌꞌꞌ |
| 6ꞌꞌꞌ | 61.0 | 3.72 (m) 3.51 (m) | 59.9 | 3.23 (m), 3.38 (m) | C-4ꞌꞌꞌ, 5ꞌꞌꞌ | |
| 1ꞌꞌꞌꞌ | 98.3 | 5.08 (d, 7.8) | C-2ꞌꞌꞌ | 105.0 | 4.59 (d, 7.8 ) | C-3ꞌꞌꞌ, 2ꞌꞌꞌꞌ, 5ꞌꞌꞌꞌ |
| 2ꞌꞌꞌꞌ | 73.5 | 4.69 (dd, 9.0, 7.8) | C-1ꞌꞌꞌꞌ, 3ꞌꞌꞌꞌ, 1ꞌꞌꞌꞌꞌ | 74.8 | 3.15 (dd, 9.0, 7.8 ) | C-1ꞌꞌꞌꞌ, 5ꞌꞌꞌꞌ |
| 3ꞌꞌꞌꞌ | 74.4 | 3.45 (m) | C-2ꞌꞌꞌꞌ, 4ꞌꞌꞌꞌ | 74.0 | 3.56 (dd, 9.0, 7.2) | C-4ꞌꞌꞌꞌ |
| 4ꞌꞌꞌꞌ | 70.4 | 3.23 (m) | C-3ꞌꞌꞌꞌ, 5ꞌꞌꞌꞌ | 70.0 | 3.23 (m) | C-5ꞌꞌꞌꞌ |
| 5ꞌꞌꞌꞌ | 76.6 | 3.28 (m) | C-4ꞌꞌꞌꞌ | 76.0 | 3.30 | C-4ꞌꞌꞌꞌ |
| 6ꞌꞌꞌꞌ | 59.9 | 3.23 (m) 3.36 (m) | C-3ꞌꞌꞌꞌ, 5ꞌꞌꞌꞌ | 64.2 | 4.30 (dd, 6.0, 12.0) 4.40 (br. d, 12.0) | C-1ꞌꞌꞌꞌꞌ, C-3ꞌꞌꞌꞌ C-1ꞌꞌꞌꞌꞌ, 3ꞌꞌꞌꞌ |
| 1ꞌꞌꞌꞌꞌ | 165.7 | 165.4 | ||||
| 2ꞌꞌꞌꞌꞌ | 115.2 | 6.31 (d, 16.2) | C-4ꞌꞌꞌꞌꞌ, C-1ꞌꞌꞌꞌꞌ | 129.4 | ||
| 3ꞌꞌꞌꞌꞌ | 144.5 | 7.37 (d, 16.2) | C-1ꞌꞌꞌꞌꞌ, 2ꞌꞌꞌꞌꞌ, 4ꞌꞌꞌꞌꞌ, 5ꞌꞌꞌꞌꞌ, 9ꞌꞌꞌꞌꞌ | 128.7 | 7.69 (d, 7.2) | C-1ꞌꞌꞌꞌꞌ, 5ꞌꞌꞌꞌꞌ |
| 4ꞌꞌꞌꞌꞌ | 124.3 | 128.1 | 7.20 (t, 7.2) | C-1ꞌꞌꞌꞌꞌ, 3ꞌꞌꞌꞌꞌ, 7ꞌꞌꞌꞌꞌ, 5ꞌꞌꞌꞌꞌ | ||
| 5ꞌꞌꞌꞌꞌ | 105.4 | 6.70 (s) | C-3ꞌꞌꞌꞌꞌ, 4ꞌꞌꞌꞌꞌ 6ꞌꞌꞌꞌꞌ, 7ꞌꞌꞌꞌꞌ, 8ꞌꞌꞌꞌꞌ | 132.7 | 7.38 (t, 7.2) | C-3ꞌꞌꞌꞌꞌ, 7ꞌꞌꞌꞌꞌ |
| 6ꞌꞌꞌꞌꞌ | 147.7 | 128.1 | 7.20 (t, 7.2 ) | C-1ꞌꞌꞌꞌꞌ, 3ꞌꞌꞌꞌꞌ, 7ꞌꞌꞌꞌꞌ, 5ꞌꞌꞌꞌꞌ | ||
| 7ꞌꞌꞌꞌꞌ | 137.9 | 8.75 (OH) | 128.7 | 7.69 (d, 7.2) | C-1ꞌꞌꞌꞌꞌ, 5ꞌꞌꞌꞌꞌ | |
| 8ꞌꞌꞌꞌꞌ | 147.7 | |||||
| 9ꞌꞌꞌꞌꞌ | 105.4 | 6.70 (s) | C-3ꞌꞌꞌꞌꞌ, 4ꞌꞌꞌꞌꞌ, 6ꞌꞌꞌꞌꞌ, 7ꞌꞌꞌꞌꞌ, 8ꞌꞌꞌꞌꞌ | |||
| CH3O | 55.8 | 3.72 (s) × 2 | C-5ꞌꞌꞌꞌꞌ, 6ꞌꞌꞌꞌꞌ, 8ꞌꞌꞌꞌꞌ, 9ꞌꞌꞌꞌꞌ | |||

3. Experimental
3.1. General Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Spectroscopic Data
 = −127.7° (c = 0.065, MeOH); UV λmax (MeOH) nm: 224, 268, and 331 nm; IR νmax (KBr) cm−1: 3259, 1660, 1595, 1520. 1H- and 13C-NMR data, see Table 1; HR-ESI-MS m/z 985.2630 [M+Na]+, (calc for C44H50O24Na, 985.2589). = −82.8° (c = 0.064, MeOH); UV (MeOH) λmax 203, 257, and 358 nm; IR νmax (KBr) cm−1: 3254, 1656, 1590, 1513. 1H- and 13C-NMR data, see Table 1; HR-ESI-MS m/z 899.2222 [M+Na]+, (calc. for C40H44O22Na: 899.2221).
= −127.7° (c = 0.065, MeOH); UV λmax (MeOH) nm: 224, 268, and 331 nm; IR νmax (KBr) cm−1: 3259, 1660, 1595, 1520. 1H- and 13C-NMR data, see Table 1; HR-ESI-MS m/z 985.2630 [M+Na]+, (calc for C44H50O24Na, 985.2589). = −82.8° (c = 0.064, MeOH); UV (MeOH) λmax 203, 257, and 358 nm; IR νmax (KBr) cm−1: 3254, 1656, 1590, 1513. 1H- and 13C-NMR data, see Table 1; HR-ESI-MS m/z 899.2222 [M+Na]+, (calc. for C40H44O22Na: 899.2221).3.5. Determination of Sugar Components
3.6. NO Inhibition Assay
3.7. α-Glucosidase Inhibition Assay
4. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgements
Author Contributions
Conflicts of Interest
References
- Al-Yahya, M.A.; Mossa, J.S.; Ageel, A.M.; Rafatullah, S. Pharmacological and safety evaluation studies on Lepidium sativum L., Seeds. Phytomedicine 1994, 1, 155–159. [Google Scholar] [CrossRef]
- Eddouks, M.; Maghrani, M.; Zeggwagh, N.A.; Michel, J.B. Study of the hypoglycaemic activity of Lepidium sativum L. aqueous extract in normal and diabetic rats. J. Ethnopharmacol. 2005, 97, 391–395. [Google Scholar] [CrossRef]
- Maghrani, M.; Zeggwagh, N.A.; Michel, J.B.; Eddouks, M. Antihypertensive effect of Lepidium sativum L. in spontaneously hypertensive rats. J. Ethnopharmacol. 2005, 100, 193–197. [Google Scholar] [CrossRef]
- Agarwal, J.; Verma, D.L. Antioxidant activity-guided fractionation of aqueous extracts from Lepidium sativum and identification of active flavonol glycosides. Acad. Arena 2011, 3, 14–18. [Google Scholar]
- Najeeb Ur, R.; Mehmood, M.H.; Alkharfy, K.M.; Gilani, A.H. Prokinetic and laxative activities of Lepidium sativum seed extract with species and tissue selective gut stimulatory actions. J. Ethnopharmacol. 2011, 134, 878–883. [Google Scholar] [CrossRef]
- Maier, U.H.; Gundlach, H.; Zenk, M.H. Seven imidazole alkaloids from Lepidium sativum. Phytochemistry 1998, 49, 1791–1795. [Google Scholar] [CrossRef]
- Agarwal, J.; Verma, D.L. Antioxidative activity and flavonoid composition from Lepidium sativum. Nat. Sci. 2011, 9, 21–25. [Google Scholar]
- Saba; Mughal, M.H.; Ali, M.; Iqbal, M.; Srivastava, P.S. A steryl ester from Lepidium sativum. Phytochemistry 1999, 50, 1375–1377. [Google Scholar] [CrossRef]
- Pande, S.D.; Ali, M.; Iqbal, M.; Srivastava, P.S. Three new phytoconstituents from Lepidium sativum. Die Pharmazie 1999, 54, 851–853. [Google Scholar]
- Wang, X.M.; Wan, C.P.; Zhou, S.R.; Qiu, Y. Two new flavonol glycosides from Sarcopyramis bodinieri var. delicate. Molecules 2008, 13, 1399–1405. [Google Scholar] [CrossRef]
- Vitalini, S.; Braca, A.; Passarella, D.; Fico, G. New flavonol glycosides from Aconitum burnatii Gayer and Aconitum variegatum L. Fitoterapia 2010, 81, 940–947. [Google Scholar] [CrossRef]
- Agrawal, P.K. NMR spectroscopy in the structural elucidation of oligosaccharides and glycosides. Phytochemistry 1992, 31, 3307–3330. [Google Scholar] [CrossRef]
- Hirayama, C.; Ono, H.; Meng, Y.; Shimada, T.; Daimon, T. Flavonoids from the cocoon of Rondotia menciana. Phytochemistry 2013, 94, 108–112. [Google Scholar] [CrossRef]
- Mousallami, A.M.; Afifi, M.S.; Hussein, S.A. Acylated flavonol diglucosides from Lotus polyphyllos. Phytochemistry 2002, 60, 807–811. [Google Scholar] [CrossRef]
- Wei, X.H.; Yang, S.J.; Liang, N.; Hu, D.Y.; Jin, L.H.; Xue, W.; Yang, S. Chemical constituents of Caesalpinia decapetala (Roth) Alston. Molecules 2013, 18, 1325–1336. [Google Scholar] [CrossRef]
- Zhang, H.M.; Wang, C.F.; Shen, S.M.; Wang, G.L.; Liu, P.; Liu, Z.M.; Wang, Y.Y.; Du, S.S.; Liu, Z.L.; Deng, Z.W. Antioxidant phenolic compounds from Pu-erh tea. Molecules 2012, 17, 14037–14045. [Google Scholar] [CrossRef]
- Liu, R.; Feng, L.; Sun, A.; Kong, L. Preparative isolation and purification of coumarins from Cnidium monnieri (L.) Cusson by high-speed counter-current chromatography. J. Chromatorg. A 2004, 1055, 71–76. [Google Scholar] [CrossRef]
- Yuan, H.E.; Zhou, X.D.; Meng, L.J.; Qin, F.M.; Zhou, G.X. Chemical constituents from Commelina communis. China J. Chin. Mater. Med. 2013, 38, 3304–3308. [Google Scholar]
- Yu, Q.; Matsunami, K.; Otsuka, H.; Takeda, Y. Staphylionosides A-K: Megastigmane glucosides from the leaves of Staphylea bumalda DC. Chem. Pharm. Bull. 2005, 53, 800–807. [Google Scholar] [CrossRef]
- Wang, S.; Wang, W.; Luo, M.; Zhao, X.; Zu, Y.; Zho, Y. Regulating effect of compound glycyrrhizin on LPS-induced inflammatory factor release from murine RAW 264.7 cells. Prog. Pharm. Sci. 2012, 36, 465–470. [Google Scholar]
- Zhang, J.; Zhao, S.; Yin, P.; Yan, L.; Han, J.; Shi, L.; Zhou, X.; Liu, Y.; Ma, C. Alpha-glucosidase inhibitory activity of polyphenols from the burs of Castanea mollissima Blume. Molecules 2014, 19, 8373–8386. [Google Scholar] [CrossRef]
- Gao, X.; He, J.; Wu, X.D.; Peng, L.Y.; Dong, L.B.; Deng, X.; Li, Y.; Cheng, X.; Zhao, Q.S. Further lignans from Saururus chinensis. Planta Med. 2013, 79, 1720–1723. [Google Scholar] [CrossRef]
- Hua, J.; Qi, J.; Yu, B.Y. Iridoid and phenylpropanoid glycosides from Scrophularia ningpoensis Hemsl. and their alpha-glucosidase inhibitory activities. Fitoterapia 2014, 93, 67–73. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–8 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fan, Q.-L.; Zhu, Y.-D.; Huang, W.-H.; Qi, Y.; Guo, B.-L. Two New Acylated Flavonol Glycosides from the Seeds of Lepidium sativum. Molecules 2014, 19, 11341-11349. https://doi.org/10.3390/molecules190811341
Fan Q-L, Zhu Y-D, Huang W-H, Qi Y, Guo B-L. Two New Acylated Flavonol Glycosides from the Seeds of Lepidium sativum. Molecules. 2014; 19(8):11341-11349. https://doi.org/10.3390/molecules190811341
Chicago/Turabian StyleFan, Qing-Lu, Yin-Di Zhu, Wen-Hua Huang, Yun Qi, and Bao-Lin Guo. 2014. "Two New Acylated Flavonol Glycosides from the Seeds of Lepidium sativum" Molecules 19, no. 8: 11341-11349. https://doi.org/10.3390/molecules190811341
APA StyleFan, Q. -L., Zhu, Y. -D., Huang, W. -H., Qi, Y., & Guo, B. -L. (2014). Two New Acylated Flavonol Glycosides from the Seeds of Lepidium sativum. Molecules, 19(8), 11341-11349. https://doi.org/10.3390/molecules190811341