Inorganic Materials as Supports for Covalent Enzyme Immobilization: Methods and Mechanisms



Abstract
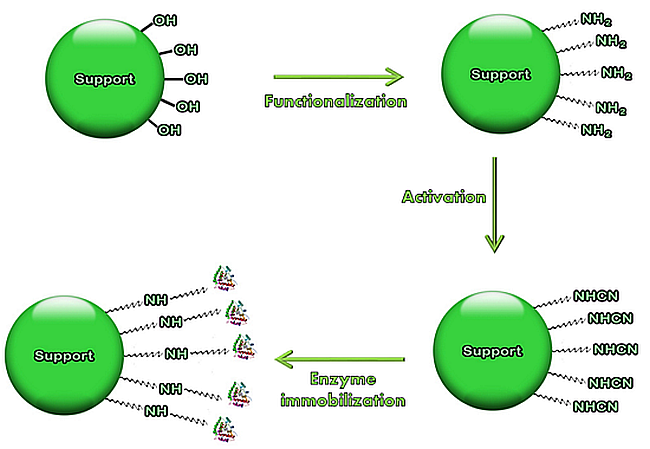
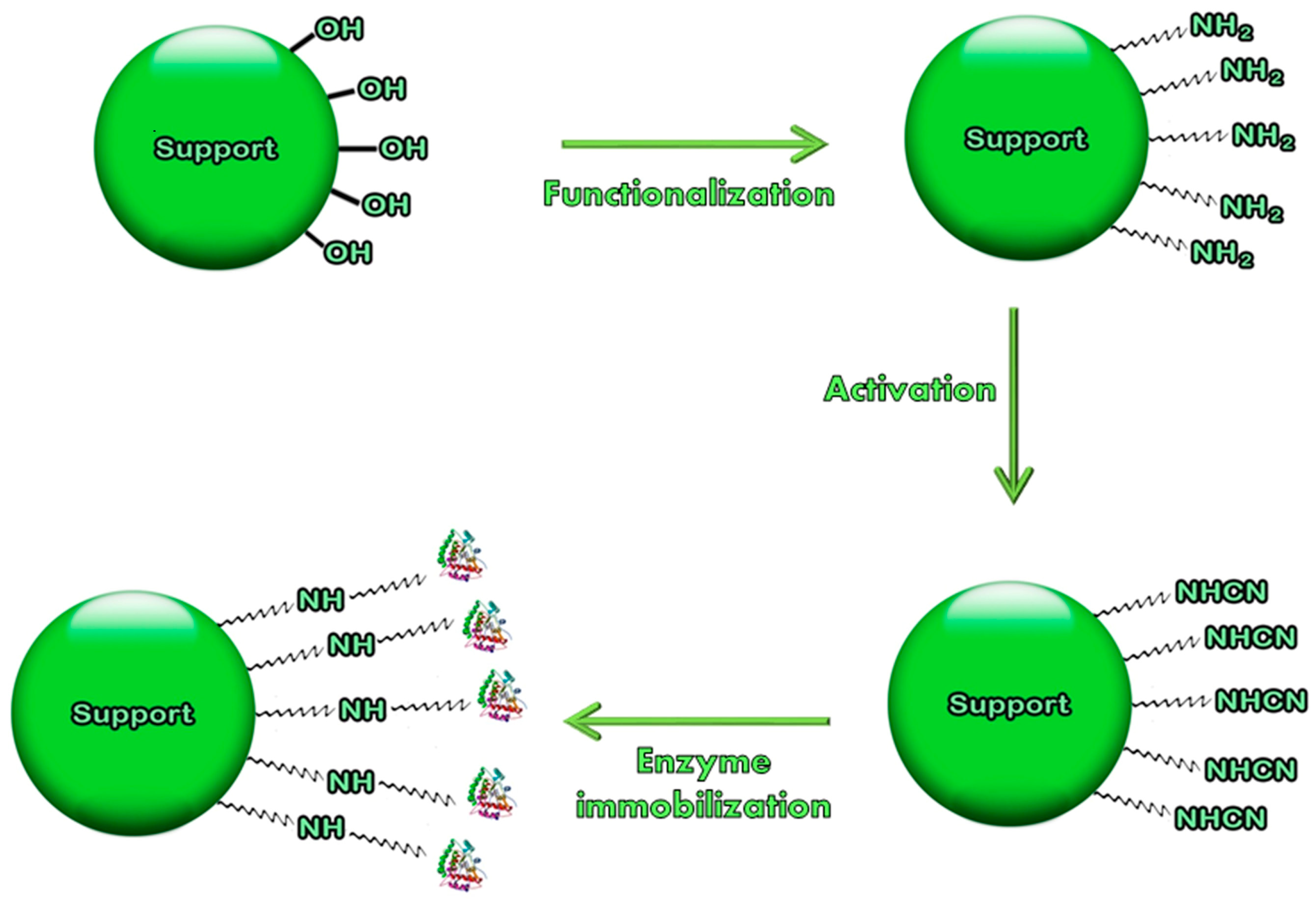
:
1. Introduction

- The support should be relatively inexpensive and environmentally harmless, minimizing the economic impact of the process.
- The support should be able to load a significant amount of enzyme per unit of weight. Accordingly, porosity could be a beneficial feature, but the diameter of the pores has to remain within a proper range (wider than the average protein diameter), since smaller pores merely exclude the protein, and too large ones will cause a significant drop in the surface area. In both cases the loading capacity is adversely affected [3,9,29]. High surface area and proper particle size should be also considered [30,31].
- Hydrophobicity of the surface should be usually minimized, since it favors undesired protein adsorption and denaturation [24,31]. Contrary behavior has been described only for well-known hydrophobic enzymes, such as lipases [8,32]. Generally speaking, the support should present the optimal micro-environment to enhance the catalytic features of the immobilized enzymes [8].
- Functionalization and activation require reactive chemical functions on the surface of the support. These groups should present minimal steric hindrance (especially for multi-point attachment [7]) and high superficial density.
- After immobilization, the support should be however totally inert under the enzymatic operational conditions, not interfering with the desired reaction.
- Interferences by unspecific protein/support interactions (i.e., adsorption, ion exchange) should be minimized, except in the case of specifically desired multifunctional immobilization [33].
- Microbial resistance is mandatory for a commercially viable enzyme.
- Chemical durability should be also considered. For instance, pH values far from neutrality could significantly affect the stability of inorganic structures [35].
2. Inorganic Supports
2.1. Silica-Based Supports
2.1.1. General
2.1.2. Silica Surface Chemistry

2.1.3. Siliceous Porous Materials
2.1.4. Controlled Pore Glass (CPG)
2.1.5. Fumed Silica
2.1.6. Silica-based Nanoparticles
2.2. Ceramics
2.3. Titania and Zirconia
2.4. Alumina
2.5. Magnetic Supports
2.6. Other Inorganic Supports
3. Advantages and Drawbacks of Covalent Enzyme Immobilization
- (i)
- Encapsulation and entrapment do not involve chemical bonds between the support and the protein, which is in fact simply included in the 3D network of the support, making impossible its diffusion away from the carrier. Accordingly, minimal modification of the native structure is involved, but leakage of enzyme is often observed [2]. Besides, mass transfer issues can often occur, involving both substrates and products.
- (ii)
- Adsorption and electrostatic interaction are often overlapping phenomena due to non-specific weak interactions, still not completely clarified [27,138]. However, the simplicity of this approach and the low modification of protein surfaces are responsible for the wide diffusion of such techniques [46]. Unfortunately, the non-specificity of the interactions could lead to unexpected leakage resulting from changes in several operational parameters (pH, temperature, and ionic strength particularly), thus suggesting the application of physically adsorbed enzymes mainly in hydrophobic environments [30].
- (iii)
- Cross-linked enzymes (CLEs) (such as cross-linked enzyme crystals CLECs, or aggregates CLEAs) involve the formation of covalent bonds among protein molecules using bifunctional reagents [6,9], often avoiding the use of any carrier. Glutaraldehyde and bis(imidoesters) are the most used bifunctional cross-linking agents. The covalent nature of the interaction is reflected in the minimal leakage and boosted operational stability of the enzymes (also under harsh conditions) [30], whereas the negative side is the possible chemical modification of the protein surface. Substrate/product diffusion rates can be also affected, and use of toxic reagents under complicated reaction conditions are often necessary [30].
- (iv)
- Affinity interaction between ligand-grafted carrier and protein can represent a valid alternative [139,140], since it could allow high-strength bonding (and so minimal leakage), without affecting a protein’s native structure [141]. Unfortunately, this approach requires the presence of specific chemical functions on the protein and a different carrier grafting for each protein, often rendering its broad diffusion for industrial enzymes uneconomical.
- (v)
- Covalent attachment tops the other approaches concerning the strength of the interactions, typically minimizing protein leakage. Several aminoacid side chains can form covalent bonds with activated inorganic supports. Particularly, the widespread lysine ε-NH2. Massive structural modifications of the immobilized proteins are accordingly likely to occur. Even when this is excluded, the simple bad orientation of the active site could affect the proper interaction between enzymes and substrates [24]. All these phenomena could thus affect catalytic activity.

| Method of Immobilization | Advantages | Disadvantages |
|---|---|---|
| Encapsulation/entrapment |
|
|
| Enzyme cross-linking |
|
|
| Adsorption |
|
|
| Electrostatic interaction |
|
|
| Affinity |
|
|
| Covalent binding |
|
|
4. Functionalization of Inorganic Supports
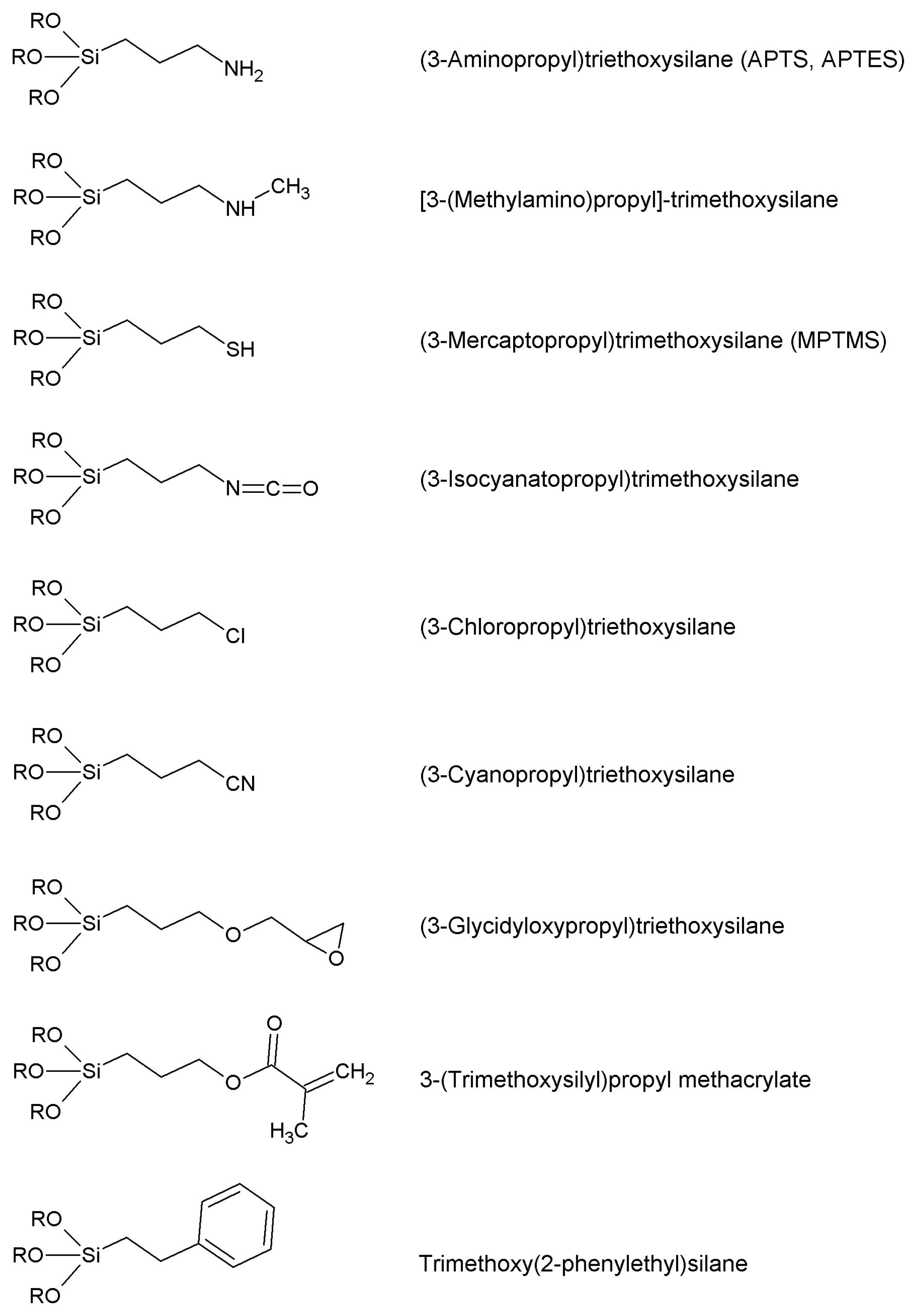
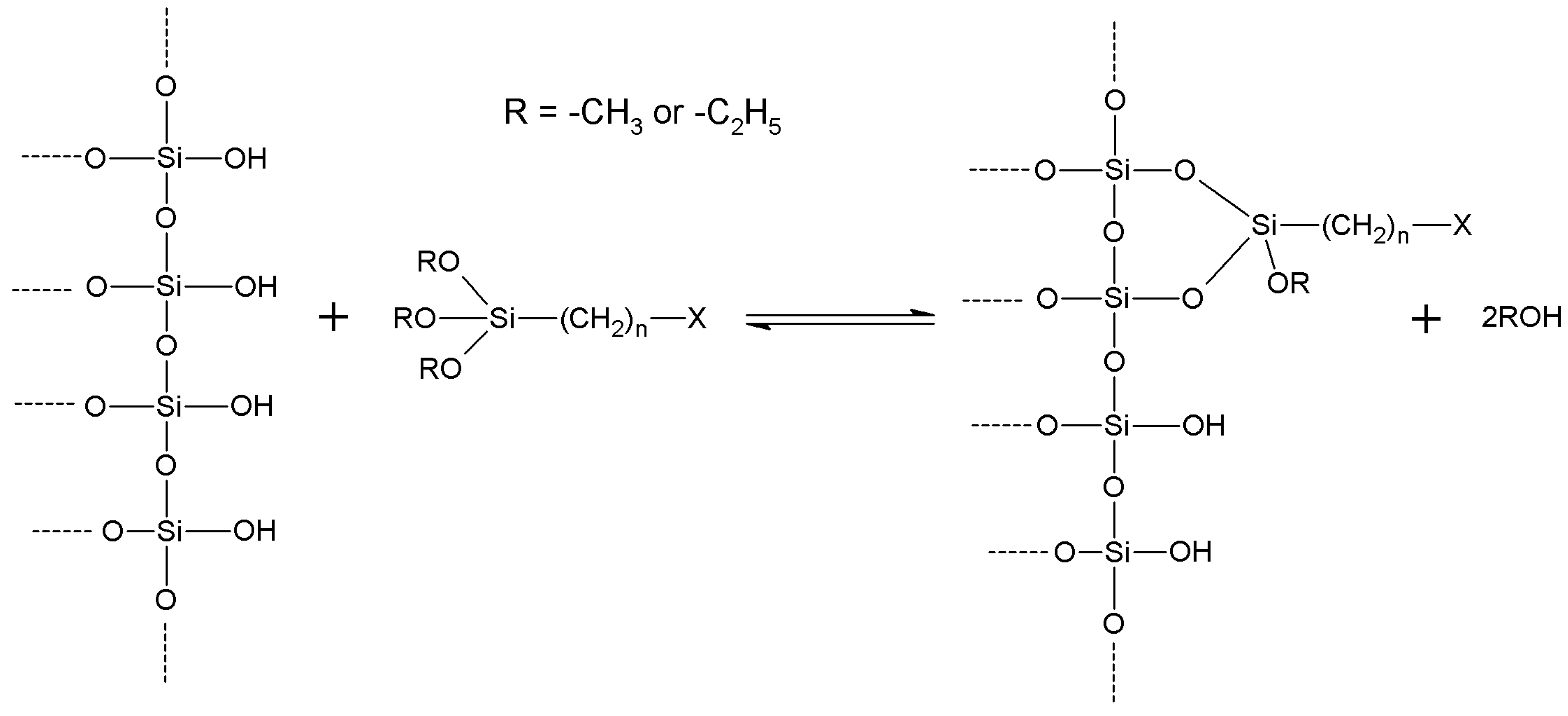
4.1. Silanization: General
- (i)
- Grafting: the plain support is treated under suitable conditions with a chosen organosilane, forming some sort of covalently bound coating. This coating is formed by the organic functions of the starting silane;
- (ii)
- Co-condensation: support particles such those described in § 2, are synthesized by means of sol-gel procedures, starting from a proper mixture of tetraethyl (or tetramethyl) orthosilicate and the chosen trialkoxyorganosilane. Tetraalkyl orthosilicates can be replaced by other alkoxides such as tetraethoxytitanium or so on. The growing particles incorporate the added organosilane and a very regular distribution of the organic functions is usually the result of such one-pot synthesis. However, excessive proportions of the organosilane adversely affect the structure of the obtained particles, and disordered structures should be expected in many cases when a high degree of organic functionalization is required. Also, organic functions that remain deeply incorporated within the very silica backbone are useless with respect to further derivatization/activation. Moreover, hydrolysis rate of the chosen organosilane can be significantly different form that of the alkyl orthosilicate: therefore, preparations showing heterogeneous distribution of the organic functions could arise.
- (iii)
- Use of the so-called silsesquioxanes (general empirical formula R2Si2O3), oligomers derived from hydrolysis—under proper experimental conditions—of organosilanes with general formula X3SiR, where X is an easily hydrolysable function such as Cl– or RO– [145]. Bridged organosilanes produce particular silsesquioxanes that could be incorporated within the particle structure by means of a sol-gel method, and later subjected to ammonolysis (with gaseous ammonia) at high temperatures to break one head of the Si–C bonds bridges while inserting –NH2 groups on to the organic moieties [146]. The method is promising but requires specialty instrumentation for high-temperature ammonolysis; certain bridged disilanes caused the collapse of the mesoporous structures when subjected to ammonolysis. On the whole, the use of silsesquioxanes (that could also be obtained as polymers of undefined degree of polymerization) is not always well distinguishable from co-condensation.

4.2. Grafting the Chosen Functional Group


4.3. Catechols as Derivatizing Agents
4.4. The Phosphate/Phosphonate Route
4.5. Gold Activation
5. Support Activation and Enzyme Immobilization Techniques
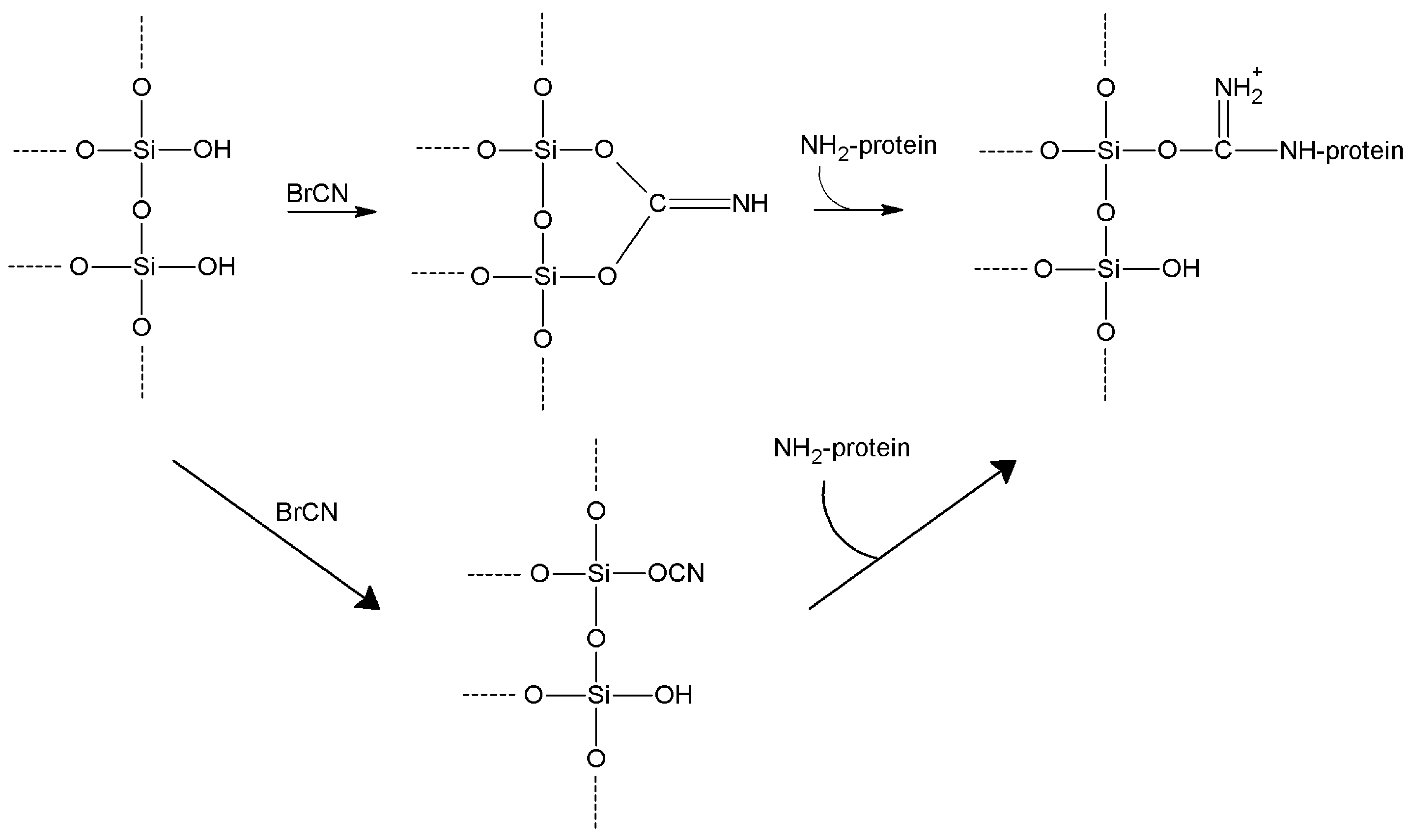
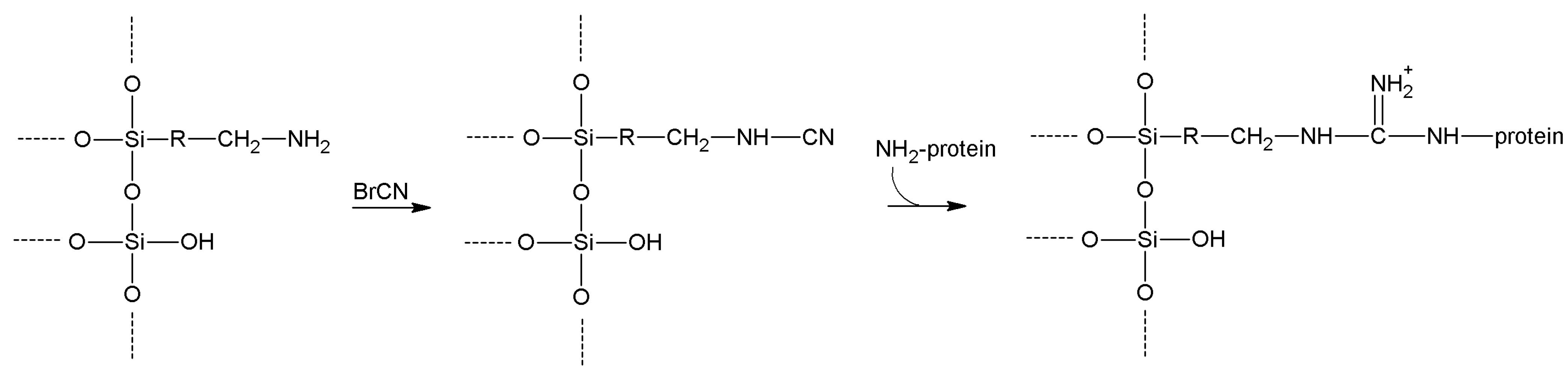
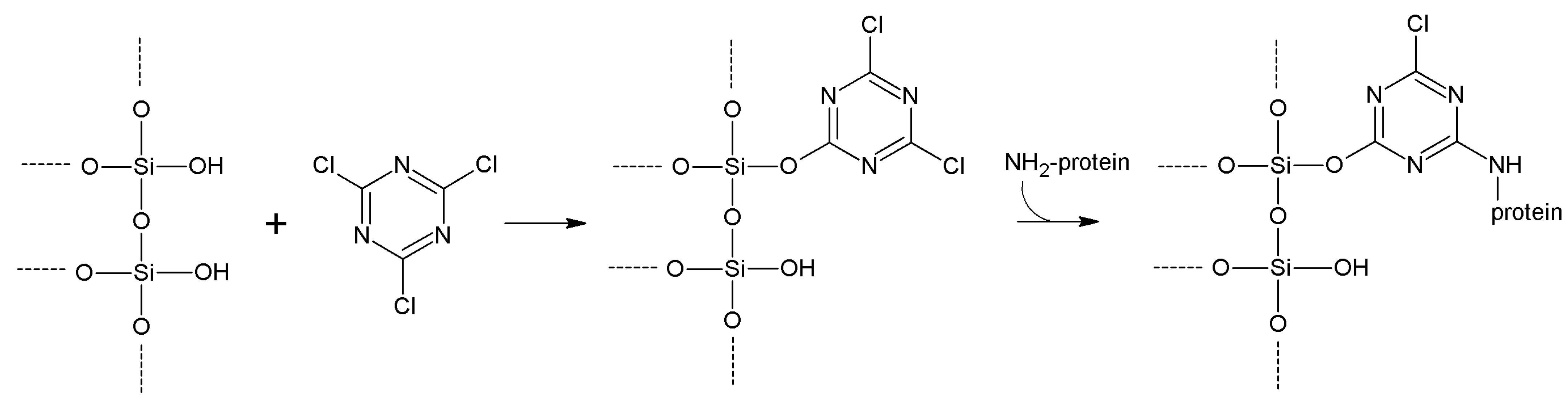
5.1. Cyanogen and Cyanuric Halides
| Activation Method | Support Reactive Group | Protein Reactive Group | Type of Bond | Bond Stability | Cost of the Reagents | Molecular Spacer |
|---|---|---|---|---|---|---|
| Cyanogen bromide | -OH
-NH2 | -NH2 | Isourea or imido-carbonate | Low | Moderate | Very short |
| Cyanuric chloride | -OH
-NH2 | -NH2 | Secondary amine | High | Low | Medium length |
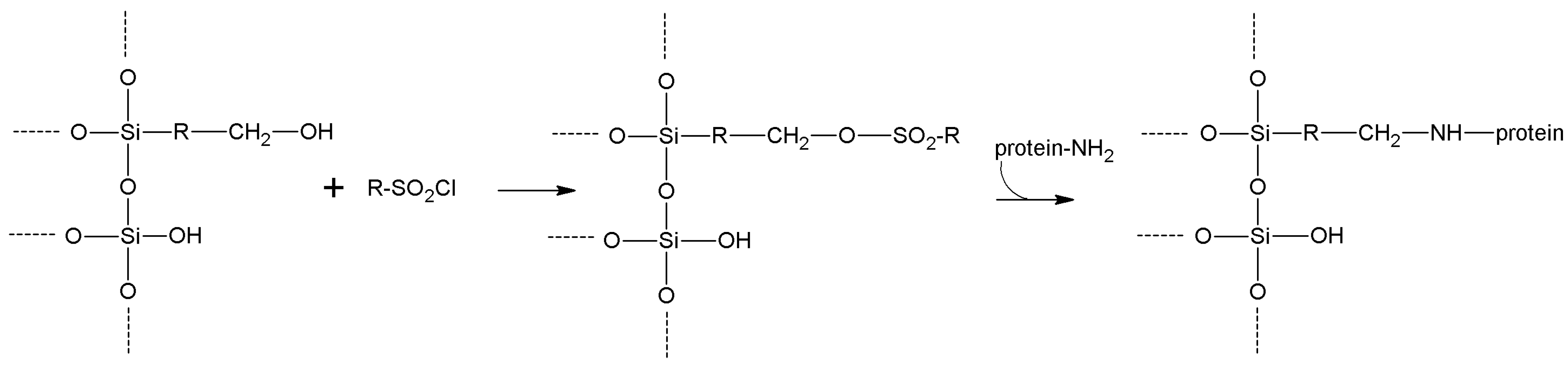
| Sulfonyl halides | -OH | -NH2 -SH | Secondary amine or thioether | High | Moderate/high | None |
| Acyl halides | -OH | -NH2 | Carbamate | High | Moderate/high | Very short |
| Thionyl chloride | -COOH | -NH2 -SH | Amide/thioester | High | Low | None |
| Metal halides | -OH | -SH | Metal bridge | Moderate | Moderate | Very short |
| Glutaraldehyde | -NH2 | -NH2 | Secondary amine | High | Low | Long |
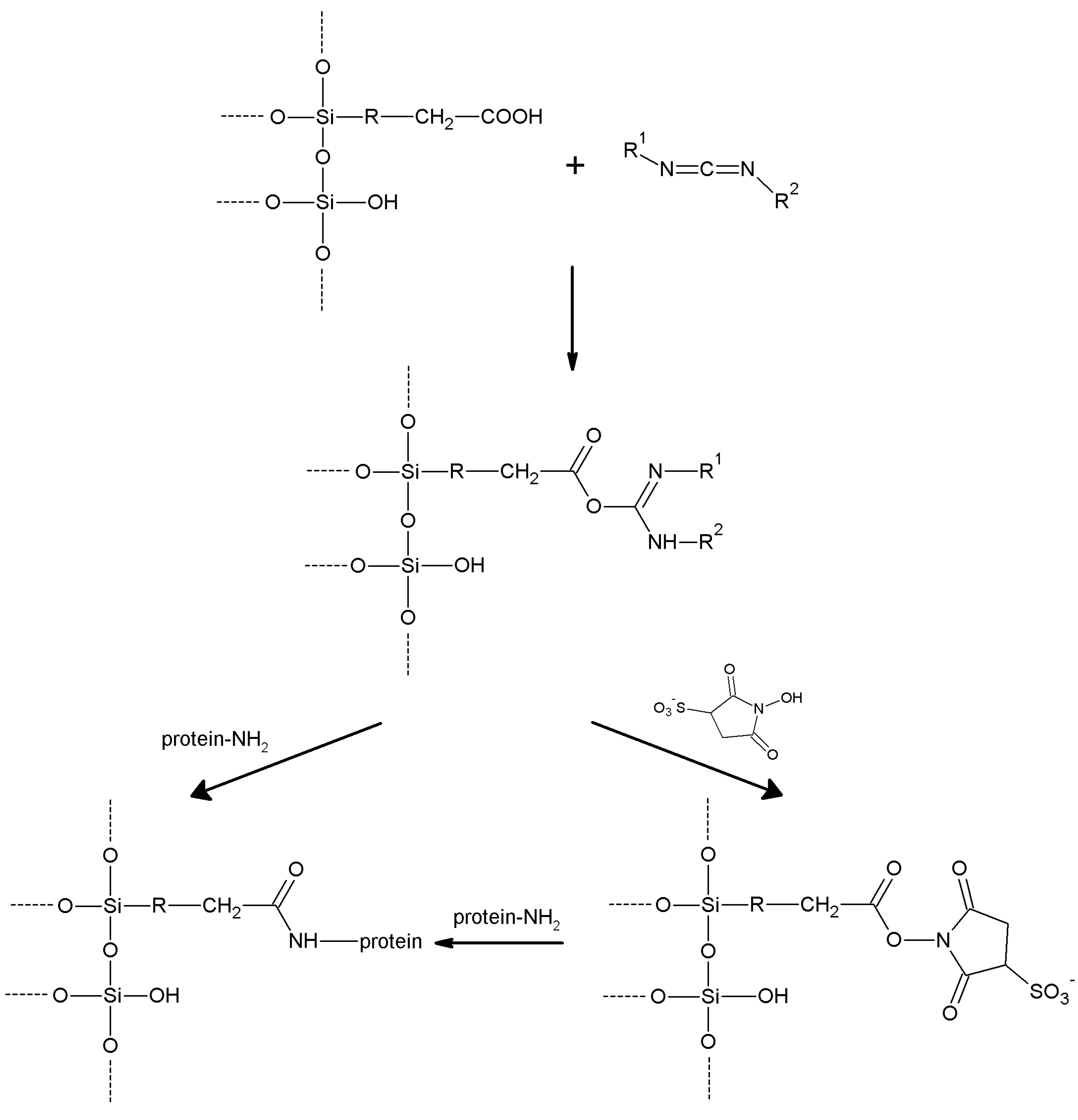
| Carbodiimides | -COOH/
-NH2 | -NH2/
-COOH | Amide | High | High | None |
| Divinylsulfone | -OH
-NH- | -SH
-NH2 | Ether/Secondary amine/thioether | Good (at neutral pH) | Moderate | Medium length |
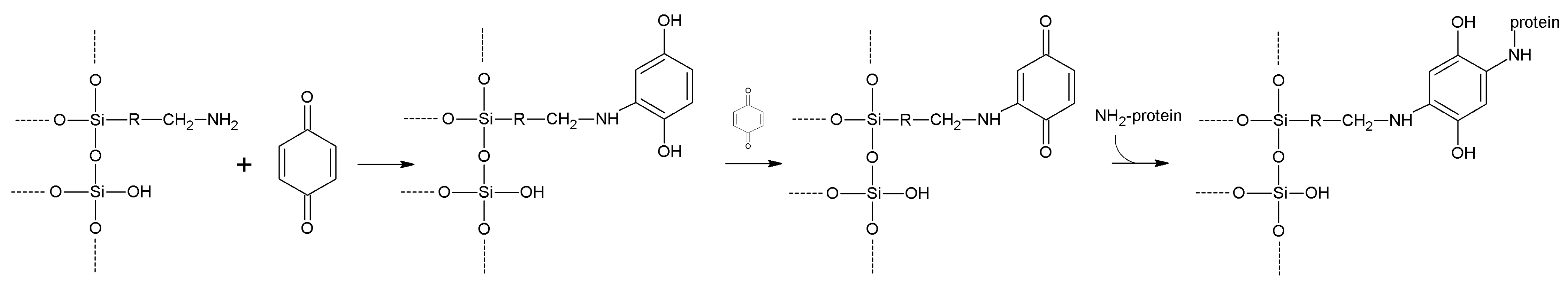
| Benzoquinone | -OH
-NH2 | -NH2 -SH | Anilinyl | High | Low | Medium length |
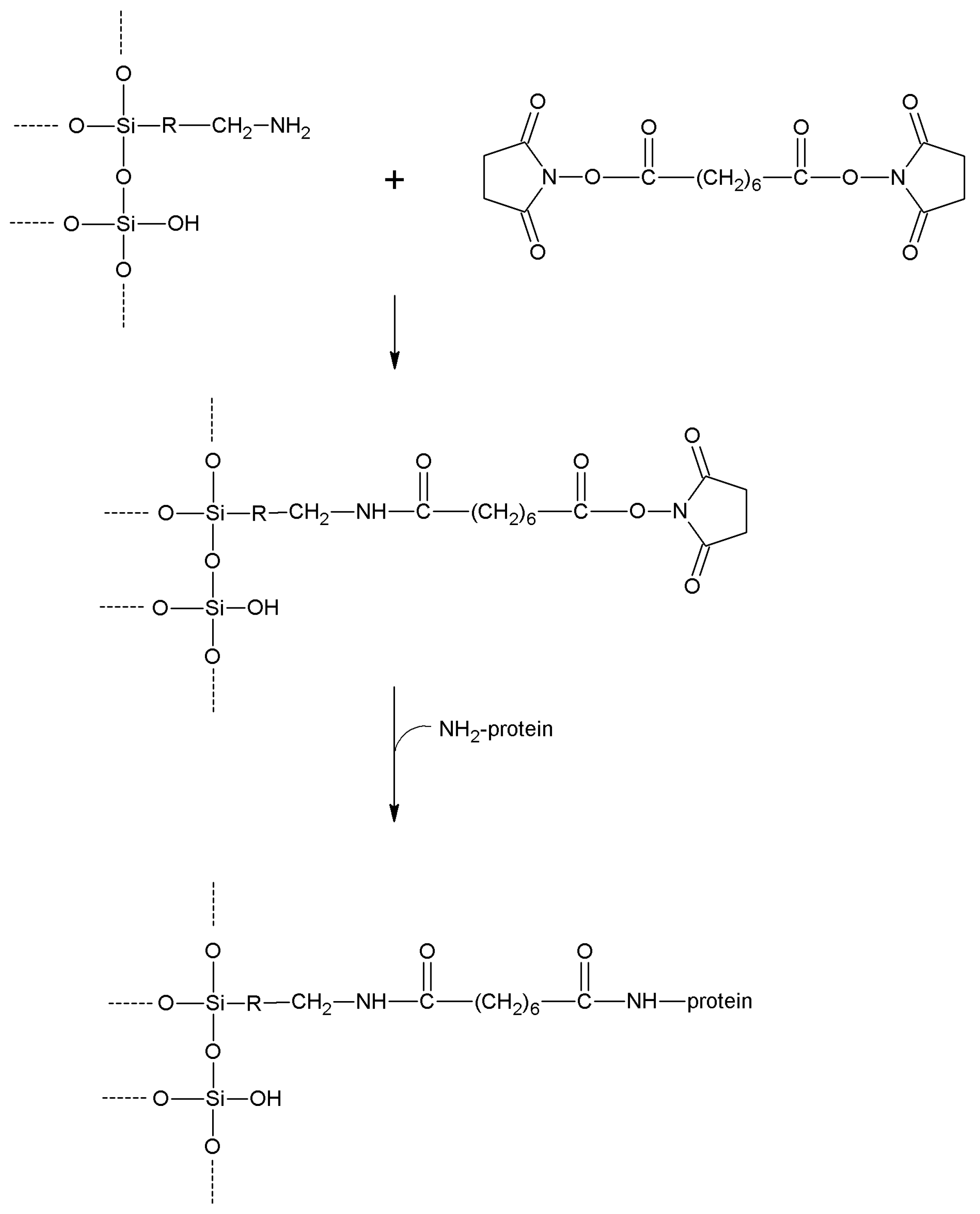
| Disuccinimidyl suberate | -NH2 | -NH2 | Amide | High | High | Long |
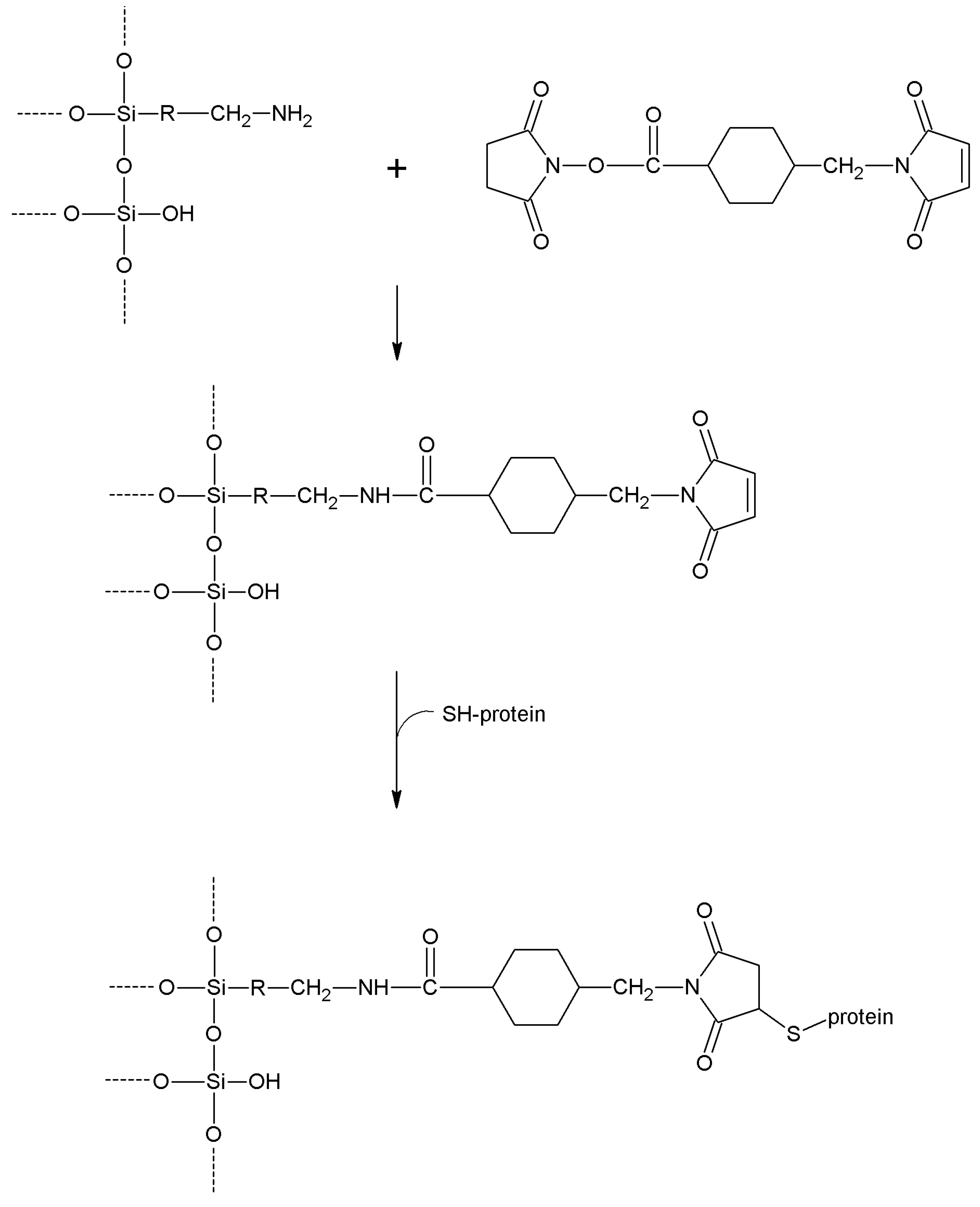
| Succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate | -NH2 | -SH | Amide
Thioether | High | High | Long |
| 2-2'- and 4,4'-Dipyridyldisulfide | -SH | -SH | Disulfide | Moderate | High | Very short |
| 1,6-Bismaleimidohexane | -SH | -SH | Thioether | High | High | Long |
| Carbonyl diimidazole | -OH | -NH2 | Carbamate | Moderate | Low | Very short |
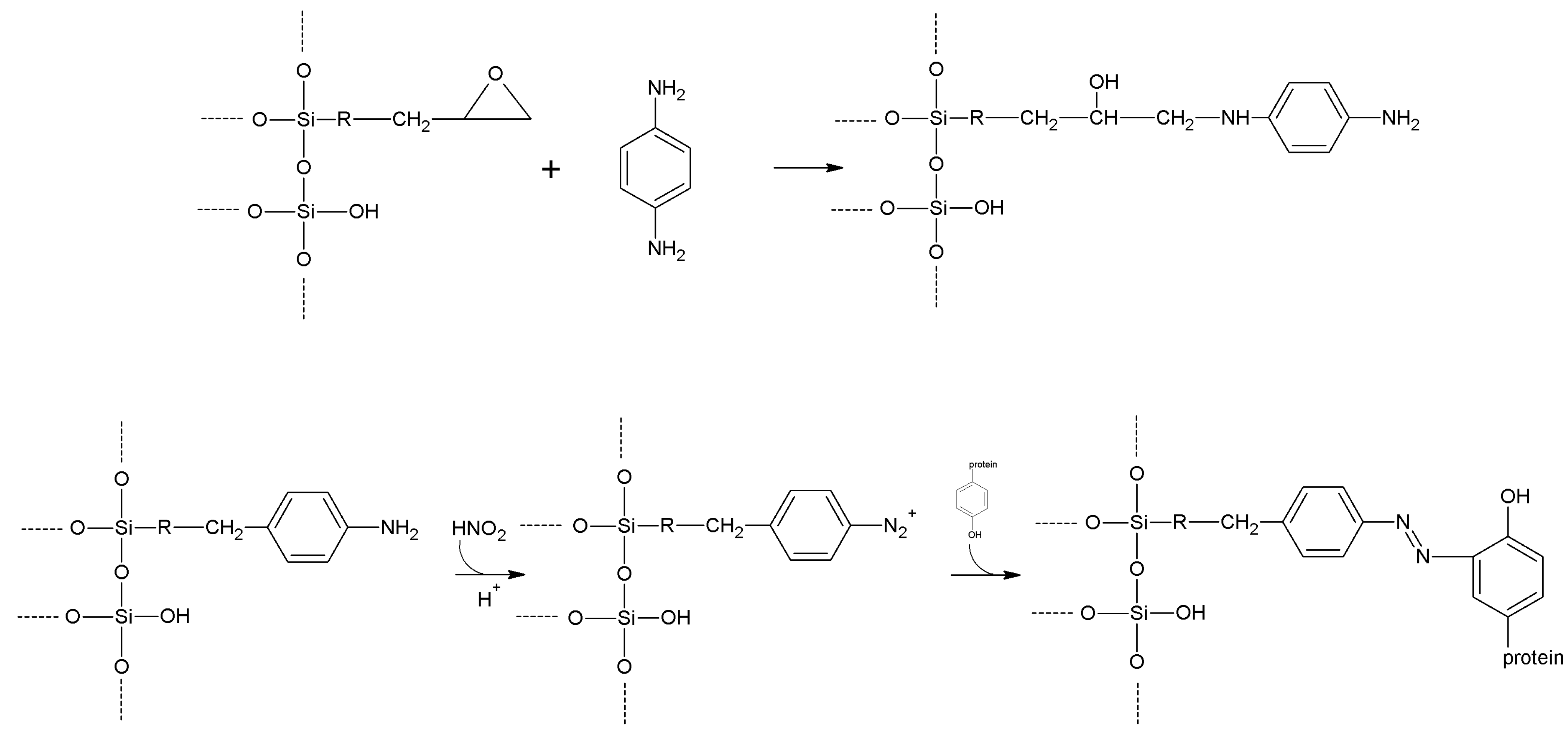
| Diazotization | Aromatic-NH2 | Aromatic -OH | Azo bond | High | Moderate | Medium |
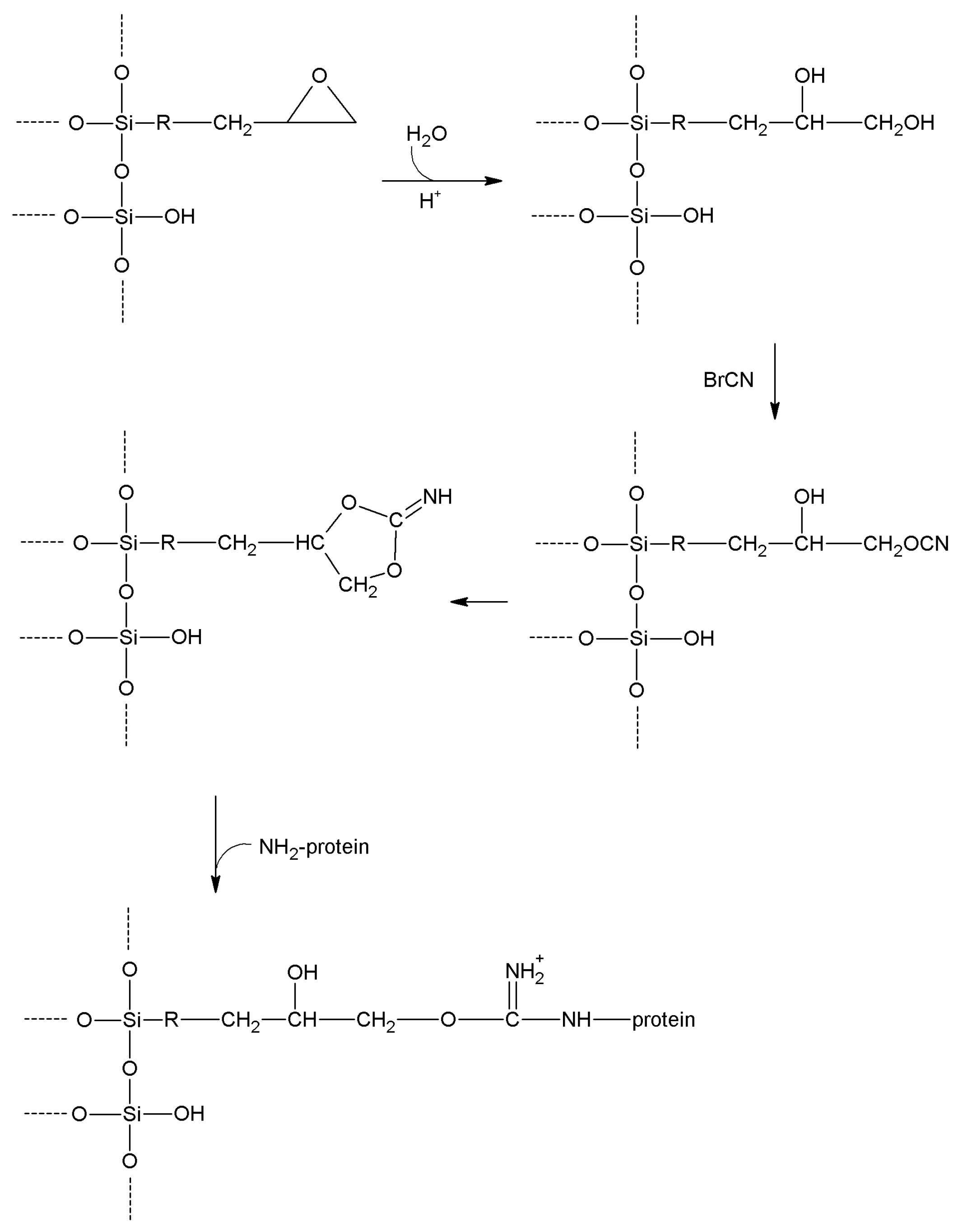
| Epichlorohyridin | -OH
-NH2 | -NH2 | Secondary amine | High | Low | Short |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




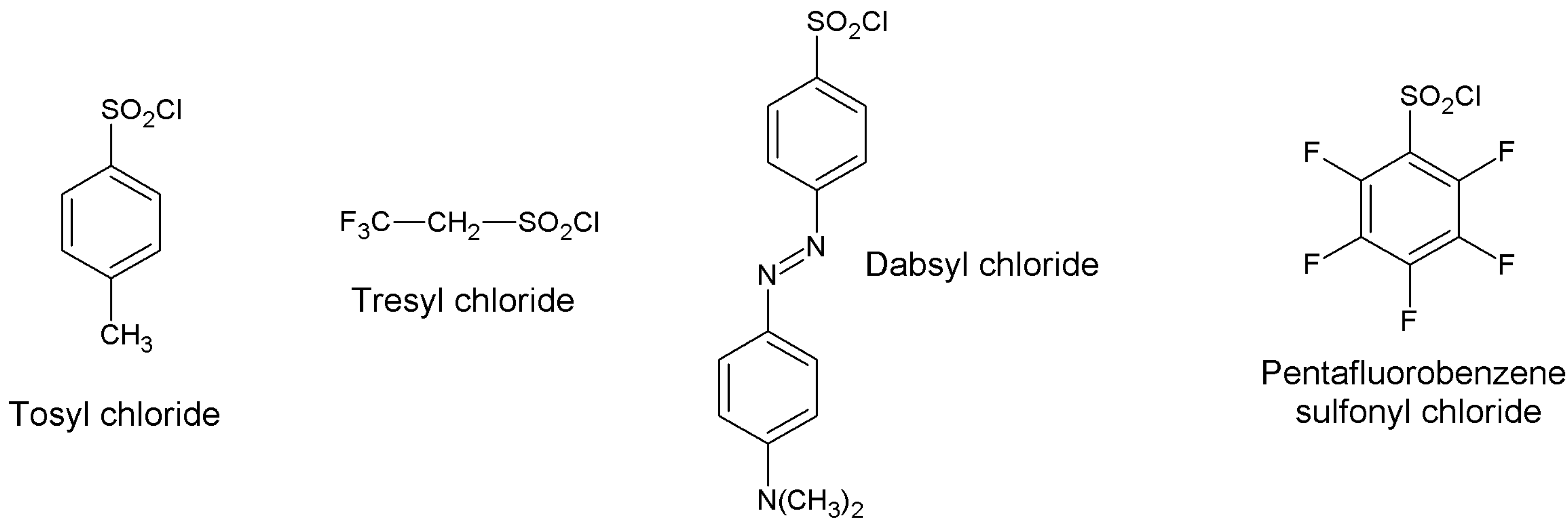
5.2. Sulfonyl Halides


5.3. Other Acyl Halides and Analogues



5.4. Metal Halides

5.5. Glutaraldehyde

5.6. Carbodiimides- and Active-Esters-Based Methods

5.7. Other Bifunctional Agents






5.8. Activation of Thiol-Functionalized Supports


5.9. Other Activating Methods



5.10. Direct Protein Coupling (without Activation)
6. Conclusions
Conflicts of Interest
References
- Minteer, S.D. Enzyme stabilization and immobilization. In Methods and Protocols; Series: Methods in Molecular Biology; A product of Humana Press: New York, NY, USA, 2011. [Google Scholar]
- Sassolas, A.; Blum, L.J.; Leca-Bouvier, B.D. Immobilization strategies to develop enzymatic biosensors. Biotechnol. Adv. 2012, 30, 489–511. [Google Scholar] [CrossRef]
- Magner, E. Immobilisation of enzymes on mesoporous silicate materials. Chem. Soc. Rev. 2013, 42, 6213–6222. [Google Scholar] [CrossRef]
- Hartmann, M. Ordered mesoporous materials for bioadsorption and biocatalysis. Chem. Mater. 2005, 17, 4577–4593. [Google Scholar] [CrossRef]
- Zhao, X.S.; Bao, X.Y.; Guo, W.; Lee, F.Y. Immobilizing catalysts on porous materials. Mater. Today 2006, 9, 32–39. [Google Scholar] [CrossRef]
- Sheldon, R.A. Enzyme immobilization: The quest for optimum performance. Adv. Synth. Catal. 2007, 349, 1289–1307. [Google Scholar] [CrossRef]
- Mateo, C.; Palomo, J.M.; Fernandez-Lorente, G.; Guisan, J.M.; Fernandez-Lafuente, R. Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzym. Microb. Technol. 2007, 40, 1451–1463. [Google Scholar] [CrossRef]
- Rodrigues, R.C.; Ortiz, C.; Berenguer-Murcia, A.; Torres, R.; Fernández-Lafuente, R. Modifying enzyme activity and selectivity by immobilization. Chem. Soc. Rev. 2013, 42, 6290–6307. [Google Scholar] [CrossRef]
- Tran, D.N.; Balkus, K.J. Perspective of recent progress in immobilization of enzymes. ACS Catal. 2011, 1, 956–968. [Google Scholar] [CrossRef]
- Jung, D.; Streb, C.; Hartmann, M. Oxidation of indole using chloroperoxidase and glucose oxidase immobilized on sba-15 as tandem biocatalyst. Microporous Mesoporous Mater. 2008, 113, 523–529. [Google Scholar] [CrossRef]
- Cass, A.E.G.; Cooper, J.M. Biosensors: A Practical Approach; Oxford University Press: Oxford, UK, 2004. [Google Scholar]
- Agüí, L.; Yáñez-Sedeño, P.; Pingarrón, J.M. Role of carbon nanotubes in electroanalytical chemistry. A review. Anal. Chim. Acta 2008, 622, 11–47. [Google Scholar]
- Cipolatti, E.P.; Silva, M.J.A.; Klein, M.; Feddern, V.; Feltes, M.M.C.; Oliveira, J.V.; Ninow, J.L.; de Oliveira, D. Current status and trends in enzymatic nanoimmobilization. J. Mol. Catal. B Enzym. 2014, 99, 56–67. [Google Scholar]
- Sasaki, T.; Kajino, T.; Li, B.; Sugiyama, H.; Takahashi, H. New pulp biobleaching system involving manganese peroxidase immobilized in a silica support with controlled pore sizes. Appl. Environ. Microbiol. 2001, 67, 2208–2212. [Google Scholar]
- Hartmann, M.; Jung, D. Biocatalysis with enzymes immobilized on mesoporous hosts: The status quo and future trends. J. Mater. Chem. 2010, 20, 844–857. [Google Scholar]
- Cesarini, S.; Pastor, F.I.J.; Diaz, P. Improvement of P. Aeruginosa 42A2 lipase preparations for FAMEs production, both in immobilized and soluble form. J. Mol. Catal. B Enzym. 2014, 99, 1–7. [Google Scholar] [CrossRef]
- Kobayashi, A.; Nonaka, H.; Funaoka, M. Comparison of softwood and hardwood lignocresol-immobilized cellulases. Nihon Enerugi Gakkaishi J. Jpn. Inst. Energy 2012, 91, 992–997. [Google Scholar]
- Liu, Y.; Hua, X. Production of biodiesel using a nanoscaled immobilized lipase as the catalyst. Catal. Lett. 2014, 144, 248–251. [Google Scholar]
- Salis, A.; Casula, M.F.; Bhattacharyya, M.S.; Pinna, M.; Solinas, V.; Monduzzi, M. Physical and chemical lipase adsorption on sba-15: Effect of different interactions on enzyme loading and catalytic performance. ChemCatChem 2010, 2, 322–329. [Google Scholar] [CrossRef]
- Salis, A.; Pinna, M.; Monduzzi, M.; Solinas, V. Comparison among immobilised lipases on macroporous polypropylene toward biodiesel synthesis. J. Mol. Catal. B Enzym. 2008, 54, 19–26. [Google Scholar] [CrossRef]
- Tran, C.T.H.; Nosworthy, N.J.; Kondyurin, A.; McKenzie, D.R.; Bilek, M.M.M. Celb and β-glucosidase immobilization for carboxymethyl cellulose hydrolysis. RSC Adv. 2013, 3, 23604–23611. [Google Scholar]
- Laveille, P.; Falcimaigne, A.; Chamouleau, F.; Renard, G.; Drone, J.; Fajula, F.; Pulvin, S.; Thomas, D.; Bailly, C.; Galarneau, A. Hemoglobin immobilized on mesoporous silica as effective material for the removal of polycyclic aromatic hydrocarbons pollutants from water. New J. Chem. 2010, 34, 2153–2165. [Google Scholar] [CrossRef]
- Riva, S. Laccases: Blue enzymes for green chemistry. Trends Biotechnol. 2006, 24, 219–226. [Google Scholar] [CrossRef]
- Zhou, Z.; Hartmann, M. Recent progress in biocatalysis with enzymes immobilized on mesoporous hosts. Top. Catal. 2012, 55, 1081–1100. [Google Scholar] [CrossRef]
- Falk, M.; Andoralov, V.; Blum, Z.; Sotres, J.; Suyatin, D.B.; Ruzgas, T.; Arnebrant, T.; Shleev, S. Biofuel cell as a power source for electronic contact lenses. Biosens. Bioelectron. 2012, 37, 38–45. [Google Scholar] [CrossRef]
- Krishnan, S.; Armstrong, F.A. Order-of-magnitude enhancement of an enzymatic hydrogen-air fuel cell based on pyrenyl carbon nanostructures. Chem. Sci. 2012, 3, 1015–1023. [Google Scholar] [CrossRef]
- Hudson, S.; Cooney, J.; Magner, E. Proteins in mesoporous silicates. Angew. Chem. Int. Ed. 2008, 47, 8582–8594. [Google Scholar] [CrossRef]
- Ispas, C.; Sokolov, I.; Andreescu, S. Enzyme-functionalized mesoporous silica for bioanalytical applications. Anal. Bioanal. Chem. 2009, 393, 543–554. [Google Scholar] [CrossRef]
- Weetall, H.H. Covalent coupling methods for inorganic support materials. In Methods in Enzymology; Klaus, M., Ed.; Academic Press: Waltham, MA, USA, 1976; Volume 44, pp. 134–148. [Google Scholar]
- Hartmann, M.; Kostrov, X. Immobilization of enzymes on porous silicas-benefits and challenges. Chem. Soc. Rev. 2013, 42, 6277–6289. [Google Scholar] [CrossRef]
- Cao, L. Carrier-bound Immobilized Enzymes: Principles, Application and Design; John Wiley & Sons: New York, NY, USA, 2006. [Google Scholar]
- Sörensen, M.H.; Ng, J.B.S.; Bergström, L.; Alberius, P.C.A. Improved enzymatic activity of thermomyces lanuginosus lipase immobilized in a hydrophobic particulate mesoporous carrier. J. Colloid Interface Sci. 2010, 343, 359–365. [Google Scholar] [CrossRef]
- Barbosa, O.; Torres, R.; Ortiz, C.; Berenguer-Murcia, A.; Rodrigues, R.C.; Fernandez-Lafuente, R. Heterofunctional supports in enzyme immobilization: From traditional immobilization protocols to opportunities in tuning enzyme properties. Biomacromolecules 2013, 14, 2433–2462. [Google Scholar] [CrossRef]
- Buchholz, K.; Klein, J. Characterization of immobilized biocatalysts. In Methods in Enzymology; Klaus, M., Ed.; Academic Press: Waltham, MA, USA, 1987; Volume 135, pp. 3–30. [Google Scholar]
- Weetall, H.H. Preparation of immobilized proteins covalently coupled through silane coupling agents to inorganic supports. Appl. Biochem. Biotechnol. 1993, 41, 157–188. [Google Scholar] [CrossRef]
- Lilly, M.D. Enzymes immobilized to cellulose. In Methods in Enzymology; Klaus, M., Ed.; Academic Press: Waltham, MA, USA, 1976; Volume 44, pp. 46–53. [Google Scholar]
- Porath, J.; Axén, R. Immobilization of enzymes to agar, agarose, and sephadex support. In Methods in Enzymology; Klaus, M., Ed.; Academic Press: Waltham, MA, USA, 1976; Volume 44, pp. 19–45. [Google Scholar]
- Epton, R.; Hibbert, B.L.; Thomas, T.H. Enzymes covalently bound to polyacrylic and polymethacrylic copolymers. In Methods in Enzymology; Klaus, M., Ed.; Academic Press: Waltham, MA, USA, 1976; Volume 44, pp. 84–107. [Google Scholar]
- Mosbach, R.; Koch-Schmidt, A.-C.; Mosbach, K. Immobilization of enzymes to various acrylic copolymer. In Methods in Enzymology; Klaus, M., Ed.; Academic Press: Waltham, MA, USA, 1976; Volume 44, pp. 53–65. [Google Scholar]
- Nan, C.; Zhang, Y.; Zhang, G.; Dong, C.; Shuang, S.; Choi, M.M.F. Activation of nylon net and its application to a biosensor for determination of glucose in human serum. Enzym. Microb. Technol. 2009, 44, 249–253. [Google Scholar] [CrossRef]
- Hornby, W.E.; Goldstein, L. Immobilization of enzymes on nylon. In Methods in Enzymology; Klaus, M., Ed.; Academic Press: Waltham, MA, USA, 1976; Volume 44, pp. 118–134. [Google Scholar]
- Hoffmann, F.; Cornelius, M.; Morell, J.; Fröba, M. Silica-based mesoporous organic-inorganic hybrid materials. Angew. Chem. Int. Ed. 2006, 45, 3216–3251. [Google Scholar] [CrossRef]
- Gustafsson, H.; Johansson, E.M.; Barrabino, A.; Odén, M.; Holmberg, K. Immobilization of lipase from mucor miehei and rhizopus oryzae into mesoporous silica-the effect of varied particle size and morphology. Colloids Surf. B. Biointerfaces 2012, 100, 22–30. [Google Scholar] [CrossRef]
- Philippot, E.; Palmier, D.; Pintard, M.; Goiffon, A. A general survey of quartz and quartz-like materials: Packing distortions, temperature, and pressure effects. J. Solid State Chem. 1996, 123, 1–13. [Google Scholar]
- Iler, R.K. The Chemistry of Silica; John Wiley and Sons: New York, NY, USA, 1979. [Google Scholar]
- Rimola, A.; Costa, D.; Sodupe, M.; Lambert, J.-F.; Ugliengo, P. Silica surface features and their role in the adsorption of biomolecules: Computational modeling and experiments. Chem. Rev. 2013, 113, 4216–4313. [Google Scholar] [CrossRef]
- Jesionowski, T.; Zdarta, J.; Krajewska, B. Enzyme immobilization by adsorption: A review. Adsorption 2014, 20, 801–821. [Google Scholar] [CrossRef]
- Chao, E.C.T.; Shoemaker, E.M.; Madsen, B.M. First natural occurrence of coesite. Science 1960, 132, 220–222. [Google Scholar]
- Ross, N.L.; Jin-Fu, S.; Hazen, R.M.; Gasparik, T. High-pressure crystal chemistry of stishovite. Am. Mineral. 1990, 75, 739–747. [Google Scholar]
- Weiss, A.; Weiss, A. Über siliciumchalkogenide. Vi. Zur kenntnis der faserigen siliciumdioxyd-modifikation. Z. Anorg. Allg. Chem. 1954, 276, 95–112. [Google Scholar] [CrossRef]
- Hamann, D.R. Energies of strained silica rings. Phys. Rev. B 1997, 55, 14784–14793. [Google Scholar] [CrossRef]
- Zhuravlev, L.T. The surface chemistry of amorphous silica. Zhuravlev model. Colloids Surf. Physicochem. Eng. Asp. 2000, 173, 1–38. [Google Scholar] [CrossRef]
- Morrow, B.A.; Cody, I.A.; Lee, L.S.M. Infrared studies of reactions on oxide surfaces. 7. Mechanism of the adsorption of water and ammonia on dehydroxylated silica. J. Phys. Chem. 1976, 80, 2761–2767. [Google Scholar] [CrossRef]
- Mondal, B.; Ghosh, D.; Das, A.K. Thermochemistry for silicic acid formation reaction: Prediction of new reaction pathway. Chem. Phys. Lett. 2009, 478, 115–119. [Google Scholar] [CrossRef]
- Cotton, F.A.; Wilkinson, G.; Murillo, C.A.; Bochmann, M. Advanced Inorganic Chemistry; Wiley-Interscience: Hoboken, NJ, USA, 1999. [Google Scholar]
- West, R.; Whatley, L.S.; Lake, K.J. Hydrogen bonding studies. V. The relative basicities of ethers, alkoxysilanes and siloxanes and the nature of the silicon-oxygen bond1,2. J. Am. Chem. Soc. 1961, 83, 761–764. [Google Scholar] [CrossRef]
- Davydov, V.Y. Adsorption on Silica Surfaces; Papirer, E., Ed.; CRC Press, Taylor & Francis Group: Santa Barbara, CA, USA, 2000; Volume 90, p. 63. [Google Scholar]
- Knight, C.T.G.; Balec, R.J.; Kinrade, S.D. The structure of silicate anions in aqueous alkaline solutions. Angew. Chem. Int. Ed. 2007, 46, 8148–8152. [Google Scholar] [CrossRef]
- Maciel, G.E.; Sindorf, D.W. Silicon-29 nmr study of the surface of silica gel by cross polarization and magic-angle spinning. J. Am. Chem. Soc. 1980, 102, 7606–7607. [Google Scholar]
- Messing, R.A. Adsorption and inorganic bridge formations. In Methods in Enzymology; Klaus, M., Ed.; Academic Press: Waltham, MA, USA, 1976; Volume 44, pp. 148–169. [Google Scholar]
- Rouquerol, J.; Avnir, D.; Everett, D.H.; Fairbridge, C.; Haynes, M.; Pernicone, N.; Ramsay, J.D.F.; Sing, K.S.W.; Unger, K.K. Guidelines for the characterization of porous solids. Stud. Surf. Sci. Catal. 1994, 87, 1–9. [Google Scholar] [CrossRef]
- Xu, R.; Pang, W.; Yu, J.; Huo, Q.; Chen, J. Chemistry of Zeolites and Related Porous Materials: Synthesis and Structure; Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Weetall, H.H. Analitical Uses of Immobilized Biological Compounds for Detection, Medical and Industrial Uses; Guilbault, G.G., Mascini, M., Eds.; Reidel Publishing Co.: Boston, MA, USA, 1988. [Google Scholar]
- Beau, R.; Duchene, J.; Page, M.L. Porous Silica Particles Containing a Crystallized Phase and Method. U.S. Patent 3493341, 2 March 1970. [Google Scholar]
- Yanagisawa, T.; Shimizu, T.; Kuroda, K.; Kato, C. The preparation of alkyltrimethylammonium-kanemite complexes and their conversion to microporous materials. Bull. Chem. Soc. Jpn. 1990, 63, 988–992. [Google Scholar] [CrossRef]
- Inagaki, S.; Fukushima, Y.; Kuroda, K. Synthesis of highly ordered mesoporous materials from a layered polysilicate. J. Chem. Soc. Chem. Commun. 1993. [Google Scholar] [CrossRef]
- Monnier, A.; Schüth, F.; Huo, Q.; Kumar, D.; Margolese, D.; Maxwell, R.S.; Stucky, G.D.; Krishnamurty, M.; Petroff, P.; Firouzi, A.; et al. Cooperative formation of inorganic-organic interfaces in the synthesis of silicate mesostructures. Science 1993, 261, 1299–1303. [Google Scholar]
- Beck, J.S.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E.; Kresge, C.T.; Schmitt, K.D.; Chu, C.T.W.; Olson, D.H.; Sheppard, E.W.; McCullen, S.B.; et al. A new family of mesoporous molecular sieves prepared with liquid crystal templates. J. Am. Chem. Soc. 1992, 114, 10834–10843. [Google Scholar] [CrossRef]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 1992, 359, 710–712. [Google Scholar] [CrossRef]
- Trewyn, B.G.; Slowing, I.I.; Giri, S.; Chen, H.T.; Lin, V.S.Y. Synthesis and functionalization of a mesoporous silica nanoparticle based on the sol-gel process and applications in controlled release. Acc. Chem. Res. 2007, 40, 846–853. [Google Scholar] [CrossRef]
- Zhao, D.; Feng, J.; Huo, Q.; Melosh, N.; Fredrickson, G.H.; Chmelka, B.F.; Stucky, G.D. Triblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom pores. Science 1998, 279, 548–552. [Google Scholar] [CrossRef]
- Zhao, D.; Huo, Q.; Feng, J.; Chmelka, B.F.; Stucky, G.D. Nonionic triblock and star diblock copolymer and oligomeric sufactant syntheses of highly ordered, hydrothermally stable, mesoporous silica structures. J. Am. Chem. Soc. 1998, 120, 6024–6036. [Google Scholar] [CrossRef]
- Meléndez-Ortiz, H.I.; Mercado-Silva, A.; García-Cerda, L.A.; Castruita, G.; Perera-Mercado, Y.A. Hydrothermal synthesis of mesoporous silica mcm-41 using commercial sodium silicate. J. Mex. Chem. Soc. 2013, 57, 73–79. [Google Scholar]
- Vivero-Escoto, J.L.; Trewyn, B.G.; Lin, S.Y.V. Mesoporous silica nanoparticles: Synthesis and applications. In Annual Review of Nano Research; World Scientific: Singapore, Singapore, 2009; Volume 3, pp. 191–231. [Google Scholar]
- Bagshaw, S.A.; Prouzet, E.; Pinnavaia, T.J. Templating of mesoporous molecular sieves by nonionic polyethylene oxide surfactants. Science 1995, 269, 1242–1244. [Google Scholar] [CrossRef]
- Schmidt-Winkel, P.; Lukens, W.W., Jr.; Zhao, D.; Yang, P.; Chmelka, B.F.; Stucky, G.D. Mesocellular siliceous foams with uniformly sized cells and windows. J. Am. Chem. Soc. 1999, 121, 254–255. [Google Scholar] [CrossRef]
- Mazurin, O.V.; Porai-Koshits, E.A. Phase Separation in Glass; North-Holland: New York, NY, USA, 1985; Volume 89, pp. 1246–1247. [Google Scholar]
- Weetall, H.H.; Filbert, A.M. porous glass for affinity chromatography applications. Methods Enzymol. 1974, 34, 59–72. [Google Scholar] [CrossRef]
- Alptekin, O.; Tükel, S.S.; Yildirim, D.; Alagöz, D. Characterization and properties of catalase immobilized onto controlled pore glass and its application in batch and plug-flow type reactors. J. Mol. Catal. B Enzym. 2009, 58, 124–131. [Google Scholar] [CrossRef]
- Girelli, A.M.; Mattei, E.; Messina, A.; Papaleo, D. Immobilization of mushroom tyrosinase on controlled pore glass: Effect of chemical modification. Sens. Actuators B Chem. 2007, 125, 48–54. [Google Scholar] [CrossRef]
- Cruz, J.C.; Pfromm, P.H.; Tomich, J.M.; Rezac, M.E. Conformational changes and catalytic competency of hydrolases adsorbing on fumed silica nanoparticles: I. Tertiary structure. Colloids Surf. B Biointerfaces 2010, 79, 97–104. [Google Scholar] [CrossRef]
- Gun’ko, V.M.; Turov, V.V.; Zarko, V.I.; Dudnik, V.V.; Tischenko, V.A.; Kazakova, O.A.; Voronin, E.F.; Siltchenko, S.S.; Barvinchenko, V.N.; Chuiko, A.A. Aqueous suspensions of fumed silica and adsorption of proteins. J. Colloid Interface Sci. 1997, 192, 166–178. [Google Scholar] [CrossRef]
- Hench, L.L.; West, J.K. The sol-gel process. Chem. Rev. 1990, 90, 33–72. [Google Scholar] [CrossRef]
- Jafarzadeh, M.; Rahman, I.A.; Sipaut, C.S. Synthesis of silica nanoparticles by modified sol-gel process: The effect of mixing modes of the reactants and drying techniques. J. Sol-Gel Sci. Technol. 2009, 50, 328–336. [Google Scholar] [CrossRef]
- Stöber, W.; Fink, A.; Bohn, E. Controlled growth of monodisperse silica spheres in the micron size range. J. Colloid Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Mittal, K.L. Silanes and other Coupling Agents; CRC Press: Boca Raton, FL, USA, 2007; Volume 4. [Google Scholar]
- Pizzi, A.; Mittal, K.L. Handbook of Adhesive Technology,Revised and Expanded; CRC Press: Boca Raton, FL, USA, 2003. [Google Scholar]
- Karimi, M.; Chaudhury, I.; Jianjun, C.; Safari, M.; Sadeghi, R.; Habibi-Rezaei, M.; Kokini, J. Immobilization of endo-inulinase on non-porous amino functionalized silica nanoparticles. J. Mol. Catal. B Enzym. 2014, 104, 48–55. [Google Scholar] [CrossRef]
- Amirnejat, S.; Movahedi, F.; Masrouri, H.; Mohadesi, M.; Kassaee, M.Z. Silica nanoparticles immobilized benzoylthiourea ferrous complex as an efficient and reusable catalyst for one-pot synthesis of benzopyranopyrimidines. J. Mol. Catal. A Chem. 2013, 378, 135–141. [Google Scholar] [CrossRef]
- Wu, X.; Wu, M.; Zhao, J.X. Recent development of silica nanoparticles as delivery vectors for cancer imaging and therapy. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 297–312. [Google Scholar] [CrossRef]
- Li, H.; He, J.; Zhao, Y.; Wu, D.; Cai, Y.; Wei, Q.; Yang, M. Immobilization of glucose oxidase and platinum on mesoporous silica nanoparticles for the fabrication of glucose biosensor. Electrochim. Acta 2011, 56, 2960–2965. [Google Scholar] [CrossRef]
- Ansari, S.A.; Husain, Q. Potential applications of enzymes immobilized on/in nano materials: A review. Biotechnol. Adv. 2012, 30, 512–523. [Google Scholar] [CrossRef]
- Yang, H.; Wei, W.; Liu, S. Monodispersed silica nanoparticles as carrier for co-immobilization of bi-enzyme and its application for glucose biosensing. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 125, 183–188. [Google Scholar] [CrossRef]
- Soleimani, M.; Khani, A.; Najafzadeh, K. amylase immobilization on the silica nanoparticles for cleaning performance towards starch soils in laundry detergents. J. Mol. Catal. B Enzym. 2012, 74, 1–5. [Google Scholar] [CrossRef]
- Zhang, S.; Lu, Y.; Ye, X. Catalytic behavior of carbonic anhydrase enzyme immobilized onto nonporous silica nanoparticles for enhancing co2 absorption into a carbonate solution. Int. J. Greenh. Gas Control 2013, 13, 17–25. [Google Scholar] [CrossRef]
- Deer, W.A. An Introduction to the Rock-forming Minerals; Deer, W.A., Howie, R.A., Zussman, J., Eds.; Longman Scientific & Technical: Harlow, England; Wiley: New York, NY, USA, 1992. [Google Scholar]
- Anthony, J.W.; Bideaux, R.A.; Bladh, K.W.; Nichols, M.C. Montmorillonite; Mineralogical Society of America: Chantilly, VA, USA, 2011. [Google Scholar]
- Guo, Y.; Zhang, Y.; Huang, H.; Meng, K.; Hu, K.; Hu, P.; Wang, X.; Zhang, Z.; Meng, X. Novel glass ceramic foams materials based on red mud. Ceram. Int. 2014, 40, 6677–6683. [Google Scholar]
- Osma, J.F.; Toca-Herrera, J.L.; Rodríguez-Couto, S. Biodegradation of a simulated textile effluent by immobilised-coated laccase in laboratory-scale reactors. Appl. Catal. A Gen. 2010, 373, 147–153. [Google Scholar] [CrossRef]
- Dimitrov, M.; Guncheva, M.; Zhiryakova, D.; Lazarova, T.; Lalev, G.; Tsoncheva, T. Nanostructured tin dioxide—A promising multipurpose support material for catalytic and biocatalytic applications. Chem. Eng. J. 2014, 252, 55–63. [Google Scholar] [CrossRef]
- Jean, G.; Sciamanna, V.; Demuynck, M.; Cambier, F.; Gonon, M. Macroporous ceramics: Novel route using partial sintering of alumina-powder agglomerates obtained by spray-drying. Ceram. Int. 2014, 40, 10197–10203. [Google Scholar] [CrossRef]
- Pontón, P.I.; d’Almeida, J.R.; Marinkovic, B.A.; Savić, S.M.; Mancic, L.; Rey, N.A.; Morgado, E., Jr.; Rizzo, F.C. The effects of the chemical composition of titanate nanotubes and solvent type on 3-aminopropyltriethoxysilane grafting efficiency. Appl. Surf. Sci. 2014, 301, 315–322. [Google Scholar] [CrossRef]
- Bertuoli, P.T.; Piazza, D.; Scienza, L.C.; Zattera, A.J. Preparation and characterization of montmorillonite modified with 3-aminopropyltriethoxysilane. Appl. Clay Sci. 2014, 87, 46–51. [Google Scholar] [CrossRef]
- Ohji, T. Chapter 11.2.2—Porous ceramic materials. In Handbook of Advanced Ceramics, 2nd ed.; Somiya, S., Ed.; Academic Press: Oxford, UK, 2013; pp. 1131–1148. [Google Scholar]
- Treccani, L.; Yvonne Klein, T.; Meder, F.; Pardun, K.; Rezwan, K. Functionalized ceramics for biomedical, biotechnological and environmental applications. Acta Biomater. 2013, 9, 7115–7150. [Google Scholar] [CrossRef]
- De Lathouder, K.M.; Lozano-Castelló, D.; Linares-Solano, A.; Wallin, S.A.; Kapteijn, F.; Moulijn, J.A. Carbon-ceramic composites for enzyme immobilization. Microporous Mesoporous Mater. 2007, 99, 216–223. [Google Scholar]
- De Lathouder, K.M.; van Benthem, D.T.J.; Wallin, S.A.; Mateo, C.; Lafuente, R.F.; Guisan, J.M.; Kapteijn, F.; Moulijn, J.A. Polyethyleneimine (pei) functionalized ceramic monoliths as enzyme carriers: Preparation and performance. J. Mol. Catal. B Enzym. 2008, 50, 20–27. [Google Scholar] [CrossRef]
- Albertini, A.V.P.; Cadena, P.G.; Silva, J.L.; Nascimento, G.A.; Reis, A.L.S.; Freire, V.N.; Santos, R.P.; Martins, J.L.; Cavada, B.S.; Neto, P.J.R.; et al. Performance of invertase immobilized on glass-ceramic supports in batch bioreactor. Chem. Eng. J. 2012, 187, 341–350. [Google Scholar] [CrossRef]
- Wang, W.; Li, Z.; Liu, W.; Wu, J. Horseradish peroxidase immobilized on the silane-modified ceramics for the catalytic oxidation of simulated oily water. Sep. Purif. Technol. 2012, 89, 206–211. [Google Scholar] [CrossRef]
- Verné, E.; Ferraris, S.; Vitale-Brovarone, C.; Spriano, S.; Bianchi, C.L.; Naldoni, A.; Morra, M.; Cassinelli, C. Alkaline phosphatase grafting on bioactive glasses and glass ceramics. Acta Biomater. 2010, 6, 229–240. [Google Scholar] [CrossRef]
- Hadjiivanov, K.I.; Klissurski, D.G. Surface chemistry of titania (anatase) and titania-supported catalysts. Chem. Soc. Rev. 1996, 25, 61–69. [Google Scholar] [CrossRef]
- Kim, K.D.; Kim, S.H.; Kim, H.T. Applying the taguchi method to the optimization for the synthesis of tio2 nanoparticles by hydrolysis of teot in micelles. Colloids Surf. Physicochem. Eng. Asp. 2005, 254, 99–105. [Google Scholar] [CrossRef]
- Bessekhouad, Y.; Robert, D.; Weber, J.V. Synthesis of photocatalytic tio2 nanoparticles: Optimization of the preparation conditions. J. Photochem. Photobiol. A Chem. 2003, 157, 47–53. [Google Scholar] [CrossRef]
- Davydov, A.A. Infrared Spectroscopy of Adsorbed Species on the Surface of Transition Metal Oxides; Wiley: Chichester, Sussex, 1990. [Google Scholar]
- Dutoit, D.C.M.; Schneider, M.; Baiker, A. Titania-silica mixed oxides. I. Influence of sol-gel and drying conditions on structural properties. J. Catal. 1995, 153, 165–176. [Google Scholar] [CrossRef]
- Chen, X.; Mao, S.S. Titanium dioxide nanomaterials: Synthesis, properties, modifications and applications. Chem. Rev. 2007, 107, 2891–2959. [Google Scholar] [CrossRef]
- Radoičić, M.B.; Janković, I.A.; Despotović, V.N.; Šojić, D.V.; Savić, T.D.; Šaponjić, Z.V.; Abramović, B.F.; Čomor, M.I. The role of surface defect sites of titania nanoparticles in the photocatalysis: Aging and modification. Appl. Catal. B Environ. 2013, 138–139, 122–127. [Google Scholar] [CrossRef]
- Nawrocki, J.; Dunlap, C.J.; Carr, P.W.; Blackwell, J.A. New materials for biotechnology: Chromatographic stationary phases based on zirconia. Biotechnol. Prog. 1994, 10, 561–573. [Google Scholar] [CrossRef]
- Reshmi, R.; Sanjay, G.; Sugunan, S. Immobilization of α-amylase on zirconia: A heterogeneous biocatalyst for starch hydrolysis. Catal. Commun. 2007, 8, 393–399. [Google Scholar] [CrossRef]
- Maciver, D.S.; Tobin, H.H.; Barth, R.T. Catalytic aluminas i. Surface chemistry of eta and gamma alumina. J. Catal. 1963, 2, 485–497. [Google Scholar] [CrossRef]
- McHardy, W.; Thomson, A. Conditions for the formation of bayerite and gibbsite. Mineral. Mag. 1971, 38, 358–368. [Google Scholar]
- Ferreira, A.R.; Küçükbenli, E.; de Gironcoli, S.; Souza, W.F.; Chiaro, S.S.X.; Konstantinova, E.; Leitão, A.A. Structural models of activated γ-alumina surfaces revisited: Thermodynamics, nmr and ir spectroscopies from ab initio calculations. Chem. Phys. 2013, 423, 62–72. [Google Scholar] [CrossRef]
- Yang, Z.; Si, S.; Zhang, C. Study on the activity and stability of urease immobilized onto nanoporous alumina membranes. Microporous Mesoporous Mater. 2008, 111, 359–366. [Google Scholar] [CrossRef]
- Vallés, D.; Furtado, S.; Villadóniga, C.; Cantera, A.M.B. Adsorption onto alumina and stabilization of cysteine proteinases from crude extract of solanum granuloso-leprosum fruits. Process Biochem. 2011, 46, 592–598. [Google Scholar] [CrossRef]
- Fadda, M.; Rescigno, A.; Rinaldi, A.; Sanjust, E. Covalent coupling of concanavalin a to commercial alumina. Biotechnol. Appl. Biochem. 1992, 16, 221–227. [Google Scholar]
- Zhou, Y.; Wang, S.; Ding, B.; Yang, Z. Modification of magnetite nanoparticles via surface-initiated atom transfer radical polymerization (atrp). Chem. Eng. J. 2008, 138, 578–585. [Google Scholar] [CrossRef]
- Zhu, Y.; Wu, Q. Synthesis of magnetite nanoparticles by precipitation with forced mixing. J. Nanopart. Res. 1999, 1, 393–396. [Google Scholar] [CrossRef]
- Nejati, K.; Zabihi, R. Preparation and magnetic properties of nano size nickel ferrite particles using hydrothermal method. Chem. Cent. J. 2012, 6. [Google Scholar] [CrossRef]
- Yalçiner, F.; Çevik, E.; Şenel, M.; Baykal, A. Development of an amperometric hydrogen peroxide biosensor based on the immobilization of horseradish peroxidase onto nickel ferrite nanoparticle-chitosan composite. Nano-Micro Lett. 2011, 3, 91–98. [Google Scholar]
- Maaz, K.; Mumtaz, A.; Hasanain, S.K.; Ceylan, A. Synthesis and magnetic properties of cobalt ferrite (cofe2o4) nanoparticles prepared by wet chemical route. J. Magn. Magn. Mater. 2007, 308, 289–295. [Google Scholar] [CrossRef]
- Halling, P.J.; Dunnill, P. Magnetic supports for immobilized enzymes and bioaffinity adsorbents. Enzym. Microb. Technol. 1980, 2, 2–10. [Google Scholar] [CrossRef]
- Subramanian, A.; Kennel, S.J.; Oden, P.I.; Jacobson, K.B.; Woodward, J.; Doktycz, M.J. Comparison of techniques for enzyme immobilization on silicon supports. Enzym. Microb. Technol. 1999, 24, 26–34. [Google Scholar] [CrossRef]
- Guncheva, M.; Dimitrov, M.; Napoly, F.; Draye, M.; Andrioletti, B. Novel hybrid materials on the basis of nanostructured tin dioxide and a lipase from rhizopus delemar with improved enantioselectivity. J. Mol. Catal. B Enzym. 2014, 102, 72–80. [Google Scholar] [CrossRef]
- Frasconi, M.; Mazzei, F.; Ferri, T. Protein immobilization at gold-thiol surfaces and potential for biosensing. Anal. Bioanal. Chem. 2010, 398, 1545–1564. [Google Scholar] [CrossRef]
- Jadhav, S.A. Functional self-assembled monolayers (sams) of organic compounds on gold nanoparticles. J. Mater. Chem. 2012, 22, 5894–5899. [Google Scholar] [CrossRef]
- Hayakawa, T.; Yoshinari, M.; Nemoto, K. Direct attachment of fibronectin to tresyl chloride-activated titanium. J. Biomed. Mater. Res. Part A 2003, 67, 684–688. [Google Scholar] [CrossRef]
- Cuetos, A.; Rioz-Martínez, A.; Valenzuela, M.L.; Lavandera, I.; de Gonzalo, G.; Carriedo, G.A.; Gotor, V. Immobilized redox enzymatic catalysts: Baeyer-villiger monooxygenases supported on polyphosphazenes. J. Mol. Catal. B Enzym. 2012, 74, 178–183. [Google Scholar] [CrossRef]
- Ramsden, J.J. Puzzles and paradoxes in protein adsorption. Chem. Soc. Rev. 1995, 24, 73–78. [Google Scholar] [CrossRef]
- Bucur, B.; Danet, A.F.; Marty, J.L. Cholinesterase immobilisation on the surface of screen-printed electrodes based on concanavalin a affinity. Anal. Chim. Acta 2005, 530, 1–6. [Google Scholar] [CrossRef]
- Mattiasson, B. Affinity immobilization. Methods Enzymol. 1988, 137, 647–656. [Google Scholar] [CrossRef]
- Sollai, F.; Noli, B.; Floris, G.; Sanjust, E. Irreversible affinity immobilization of lentil seedling amine oxidase with activity retention. Environ. Eng. Manag. J. 2007, 6, 31–35. [Google Scholar]
- Fernandez-Lafuente, R. Stabilization of multimeric enzymes: Strategies to prevent subunit dissociation. Enzym. Microb. Technol. 2009, 45, 405–418. [Google Scholar] [CrossRef]
- Garcia-Galan, C.; Dos Santos, J.C.S.; Barbosa, O.; Torres, R.; Pereira, E.B.; Corberan, V.C.; Gonçalves, L.R.B.; Fernandez-Lafuente, R. Tuning of lecitase features via solid-phase chemical modification: Effect of the immobilization protocol. Process Biochem. 2014, 49, 604–616. [Google Scholar] [CrossRef]
- Vansant, E.F.; van der Voort, P.; Vrancken, K.C. Characterization and Chemical Modification of the Silica Surface; Elsevier: Amsterdam, The Netherlands, 1995; Volume 93. [Google Scholar]
- Li, G.; Wang, L.; Ni, H.; Pittman, C., Jr. Polyhedral oligomeric silsesquioxane (poss) polymers and copolymers: A review. J. Inorg. Organomet. Polym. 2001, 11, 123–154. [Google Scholar] [CrossRef]
- Asefa, T.; Kruk, M.; Coombs, N.; Grondey, H.; MacLachlan, M.J.; Jaroniec, M.; Ozin, G.A. Novel route to periodic mesoporous aminosilicas, pmas: Ammonolysis of periodic mesoporous organosilicas. J. Am. Chem. Soc. 2003, 125, 11662–11673. [Google Scholar]
- Pasternack, R.M.; Rivillon Amy, S.; Chabal, Y.J. Attachment of 3-(aminopropyl)triethoxysilane on silicon oxide surfaces: Dependence on solution temperature. Langmuir 2008, 24, 12963–12971. [Google Scholar] [CrossRef]
- Acres, R.G.; Ellis, A.V.; Alvino, J.; Lenahan, C.E.; Khodakov, D.A.; Metha, G.F.; Andersson, G.G. Molecular structure of 3-aminopropyltriethoxysilane layers formed on silanol-terminated silicon surfaces. J. Phys. Chem. C 2012, 116, 6289–6297. [Google Scholar]
- Sanz-Moral, L.M.; Rueda, M.; Nieto, A.; Novak, Z.; Martín, Á. Gradual hydrophobic surface functionalization of dry silica aerogels by reaction with silane precursors dissolved in supercritical carbon dioxide. J. Supercrit. Fluids 2013, 84, 74–79. [Google Scholar] [CrossRef]
- McKittrick, M.W.; Jones, C.W. Toward single-site, immobilized molecular catalysts: Site-isolated ti ethylene polymerization catalysts supported on porous silica. J. Am. Chem. Soc. 2004, 126, 3052–3053. [Google Scholar] [CrossRef]
- Cuoq, F.; Masion, A.; Labille, J.; Rose, J.; Ziarelli, F.; Prelot, B.; Bottero, J.-Y. Preparation of amino-functionalized silica in aqueous conditions. Appl. Surf. Sci. 2013, 266, 155–160. [Google Scholar] [CrossRef]
- Inman, J.K. [3] covalent linkage of functional groups, ligands, and proteins to polyacrylamide beads. Methods Enzymol. 1974, 34, 30–58. [Google Scholar] [CrossRef]
- Kaiser, E.; Colescott, R.L.; Bossinger, C.D.; Cook, P.I. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal. Biochem. 1970, 34, 595–598. [Google Scholar] [CrossRef]
- Juntapram, K.; Praphairaksit, N.; Siraleartmukul, K.; Muangsin, N. Synthesis and characterization of chitosan-homocysteine thiolactone as a mucoadhesive polymer. Carbohydr. Polym. 2012, 87, 2399–2408. [Google Scholar] [CrossRef]
- Zucca, P.; Mocci, G.; Rescigno, A.; Sanjust, E. 5, 10, 15, 20-tetrakis (4-sulfonato-phenyl) porphine-mn (iii) immobilized on imidazole-activated silica as a novel lignin-peroxidase-like biomimetic catalyst. J. Mol. Catal. A Chem. 2007, 278, 220–227. [Google Scholar] [CrossRef]
- Zucca, P.; Rescigno, A.; Pintus, M.; Rinaldi, A.C.; Sanjust, E. Degradation of textile dyes using immobilized lignin peroxidase-like metalloporphines under mild experimental conditions. Chem. Cent. J. 2012, 6, 1–8. [Google Scholar] [CrossRef]
- Zucca, P.; Vinci, C.; Rescigno, A.; Dumitriu, E.; Sanjust, E. Is the bleaching of phenosafranine by hydrogen peroxide oxidation catalyzed by silica-supported 5, 10, 15, 20-tetrakis-(sulfonatophenyl) porphine-mn (iii) really biomimetic? Mol. Catal. A Chem. 2010, 321, 27–33. [Google Scholar] [CrossRef]
- Zucca, P.; Vinci, C.; Sollai, F.; Rescigno, A.; Sanjust, E. Degradation of alizarin red s under mild experimental conditions by immobilized 5, 10, 15, 20-tetrakis (4-sulfonatophenyl) porphine–mn (iii) as a biomimetic peroxidase-like catalyst. J. Mol. Catal. A Chem. 2008, 288, 97–102. [Google Scholar] [CrossRef]
- Hwang, S.; Lee, K.-T.; Park, J.-W.; Min, B.-R.; Haam, S.; Ahn, I.-S.; Jung, J.-K. Stability analysis of bacillus stearothermophilus l1 lipase immobilized on surface-modified silica gels. Biochem. Eng. J. 2004, 17, 85–90. [Google Scholar] [CrossRef]
- Wu, H.; Liang, Y.; Shi, J.; Wang, X.; Yang, D.; Jiang, Z. Enhanced stability of catalase covalently immobilized on functionalized titania submicrospheres. Mater. Sci. Eng. C 2013, 33, 1438–1445. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, C.; Liang, Y.; Shi, J.; Wang, X.; Jiang, Z. Catechol modification and covalent immobilization of catalase on titania submicrospheres. J. Mol. Catal. B Enzym. 2013, 92, 44–50. [Google Scholar] [CrossRef]
- Sanjust, E.; Bannister, J.; Petruzzelli, D.; Cocco, D. Nadh oxidase activity of the fad-alumina derivative. Ital. J. Biochem. 1991, 40, 177–178. [Google Scholar]
- Pawsey, S.; Yach, K.; Reven, L. Self-assembly of carboxyalkylphosphonic acids on metal oxide powders. Langmuir 2002, 18, 5205–5212. [Google Scholar] [CrossRef]
- Coletti-Previero, M.A.; Previero, A. Alumina-phosphate complexes for immobilization of biomolecules. Anal. Biochem. 1989, 180, 1–10. [Google Scholar] [CrossRef]
- Clausen, A.M.; Subramanian, A.; Carr, P.W. Purification of monoclonal antibodies from cell culture supernatants using a modified zirconia based cation-exchange support. J. Chromatogr. 1999, 831, 63–72. [Google Scholar]
- Srere, P.A.; Uyeda, K. Functional groups on enzymes suitable for binding to matrices. In Methods in Enzymology; Klaus, M., Ed.; Academic Press: Waltham, MA, USA, 1976; Volume 44, pp. 11–19. [Google Scholar]
- Morales-Sanfrutos, J.; Lopez-Jaramillo, J.; Ortega-Muñoz, M.; Megia-Fernandez, A.; Perez-Balderas, F.; Hernandez-Mateo, F.; Santoyo-Gonzalez, F. Vinyl sulfone: A versatile function for simple bioconjugation and immobilization. Org. Biomol. Chem. 2010, 8, 667–675. [Google Scholar] [CrossRef]
- Rodrigues, R.C.; Godoy, C.A.; Volpato, G.; Ayub, M.A.Z.; Fernandez-Lafuente, R.; Guisan, J.M. Immobilization-stabilization of the lipase from thermomyces lanuginosus: Critical role of chemical amination. Process Biochem. 2009, 44, 963–968. [Google Scholar] [CrossRef]
- López-Gallego, F.; Montes, T.; Fuentes, M.; Alonso, N.; Grazu, V.; Betancor, L.; Guisán, J.M.; Fernández-Lafuente, R. Improved stabilization of chemically aminated enzymes via multipoint covalent attachment on glyoxyl supports. J. Biotechnol. 2005, 116, 1–10. [Google Scholar] [CrossRef]
- Kohn, J.; Wilchek, M. A new approach (cyano-transfer) for cyanogen bromide activation of sepharose at neutral ph, which yields activated resins, free of interfering nitrogen derivatives. Biochem. Biophys. Res. Commun. 1982, 107, 878–884. [Google Scholar] [CrossRef]
- Kohn, J.; Wilchek, M. Procedures for the analysis of cyanogen bromide-activated sepharose or sephadex by quantitative determination of cyanate esters and imidocarbonates. Anal. Biochem. 1981, 115, 375–382. [Google Scholar] [CrossRef]
- Scouten, W.H. A survey of enzyme coupling techniques. In Methods in Enzymology; Academic Press: Waltham, MA, USA, 1987; Volume 135, pp. 30–65. [Google Scholar]
- Kohn, J.; Wilchek, M. Mechanism of activation of sepharose and sephadex by cyanogen bromide. Enzym. Microb. Technol. 1982, 4, 161–163. [Google Scholar] [CrossRef]
- Wilchek, M.; Oka, T.; Topper, Y.J. Structure of a soluble super active insulin is revealed by the nature of the complex between cyanogen bromide activated sepharose and amines (n substituted isoureas/n1 n2 disubstituted guanidines/insulin sepharose/insulin insensitivity/super active peptide hormones). Proc. Natl. Acad. Sci. USA 1975, 72, 1055–1058. [Google Scholar] [CrossRef]
- Nilsson, K.; Larsson, P.-O. High-performance liquid affinity chromatography on silica-bound alcohol dehydrogenase. Anal. Biochem. 1983, 134, 60–72. [Google Scholar] [CrossRef]
- Schnapp, J.; Shalitin, Y. Immobilization of enzymes by covalent binding to amine supports via cyanogen bromide activation. Biochem. Biophys. Res. Commun. 1976, 70, 8–14. [Google Scholar] [CrossRef]
- Kohn, J.; Wilchek, M. 1-cyano-4-dimethylamino pyridinium tetrafluoroborate as a cyanylating agent for the covalent attachment of ligand to polysaccharide resins. FEBS Lett. 1983, 154, 209–210. [Google Scholar] [CrossRef]
- Sun, S.; Wang, C.; Zhang, Y.; Liu, C.; Song, Y.; Jia, J. Immobilization of β-galactosidase on chitosan by 2,4,6-trichloro-1,3,5-triazine cethod. Available online: http://www.chemistrymag.org/cji/2007/099041pe.htm (accessed on 25 August 2014).
- Alcántara, A.R.; Borreguero, I.; López-Belmonte, M.T.; Sinisterra, J.V. Covalent immobilization of crude and partially-purified upases onto inorganic supports: Stability and hyperactivation. Prog. Biotechnol. 1998, 15, 571–576. [Google Scholar] [CrossRef]
- Moreno, J.M.; Sinisterra, J.V. Immobilization of lipase from candida cylindracea on inorganic supports. J. Mol. Catal. 1994, 93, 357–369. [Google Scholar] [CrossRef]
- Nilsson, K.; Mosbach, K. Tresyl chloride-activated supports for enzyme immobilization. In Methods in Enzymology; Klaus, M., Ed.; Academic Press: Waltham, MA, USA, 1987; Volume 135, pp. 65–78. [Google Scholar]
- Crossland, R.; Wells, W.; Shiner, V., Jr. Sulfonate leaving groups, structure and reactivity. 2,2,2-Trifluoroethanesulfonate. J. Am. Chem. Soc. 1971, 93, 4217–4219. [Google Scholar] [CrossRef]
- Scouten, W.H.; van den Tweel, W.; Kranenburg, H.; Dekker, M. Colored sulfonyl chloride as an activating agent for hydroxylic matrices. In Methods in Enzymology; Klaus, M., Ed.; Academic Press: Waltham, MA, USA, 1987; Volume 135, pp. 79–84. [Google Scholar]
- Scouten, W.H.; Van den Tweel, W.; Delhaes, D. Sulfonyl chloride activation of hydroxylic materials. J. Chromatogr. Biomed. Appl. 1986, 376, 289–298. [Google Scholar] [CrossRef]
- Selvamurugan, C.; Lavanya, A.; Sivasankar, B. A comparative study on immobilization of urease on different matrices. J. Sci. Ind. Res. 2007, 66, 655–659. [Google Scholar]
- Kohn, J.; Lenger, R.; Wilchek, M. P-nitrophenylcyanate: An efficient, convenient, and nonhazardous substitute for cyanogen bromide as an activating agent for sepharose. Appl. Biochem. Biotechnol. 1983, 8, 227–235. [Google Scholar] [CrossRef]
- Miron, T.; Wilchek, M. Immobilization of proteins and ligands using chlorocarbonates. In Methodsin Enzymology; Klaus, M., Ed.; Academic Press: Waltham, MA, USA, 1987; Volume 135, pp. 84–90. [Google Scholar]
- Janolino, V.; Swaisgood, H. Immobilization of proteins on thionyl chloride-activated controlled-pore glass. In Immobilization of Enzymes and Cells; Bickerstaff, G., Ed.; Humana Press: New York, NY, USA, 1997; Volume 1, pp. 21–26. [Google Scholar]
- Jarzębski, A.B.; Szymańska, K.; Bryjak, J.; Mrowiec-Białoń, J. Covalent immobilization of trypsin on to siliceous mesostructured cellular foams to obtain effective biocatalysts. Catal. Today 2007, 124, 2–10. [Google Scholar] [CrossRef]
- Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Torres, R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Glutaraldehyde in bio-catalysts design: A useful crosslinker and a versatile tool in enzyme immobilization. RSC Adv. 2014, 4, 1583–1600. [Google Scholar]
- Migneault, I.; Dartiguenave, C.; Bertrand, M.J.; Waldron, K.C. Glutaraldehyde: Behavior in aqueous solution, reaction with proteins, and application to enzyme crosslinking. BioTechniques 2004, 37, 790–802. [Google Scholar]
- Barbosa, O.; Torres, R.; Ortiz, C.; Fernandez-Lafuente, R. Versatility of glutaraldehyde to immobilize lipases: Effect of the immobilization protocol on the properties of lipase b from candida antarctica. Process Biochem. 2012, 47, 1220–1227. [Google Scholar] [CrossRef]
- Ferreira, L.; Ramos, M.A.; Dordick, J.S.; Gil, M.H. Influence of different silica derivatives in the immobilization and stabilization of a bacillus licheniformis protease (subtilisin carlsberg). J. Mol. Catal. B Enzym. 2003, 21, 189–199. [Google Scholar] [CrossRef]
- Jung, D.; Streb, C.; Hartmann, M. Covalent anchoring of chloroperoxidase and glucose oxidase on the mesoporous molecular sieve sba-15. Int. J. Mol. Sci. 2010, 11, 762–778. [Google Scholar] [CrossRef]
- Forde, J.; Vakurov, A.; Gibson, T.D.; Millner, P.; Whelehan, M.; Marison, I.W.; Ó’Fágáin, C. Chemical modification and immobilisation of lipase b from candida antarctica onto mesoporous silicates. J. Mol. Catal. B Enzym. 2010, 66, 203–209. [Google Scholar] [CrossRef]
- Gao, Y.; Kyratzis, I. Covalent immobilization of proteins on carbon nanotubes using the cross-linker 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide—A critical assessment. Bioconjug. Chem. 2008, 19, 1945–1950. [Google Scholar] [CrossRef]
- Staros, J.V.; Wright, R.W.; Swingle, D.M. Enhancement of n-hydroxysulfosuccinimide of water-soluble carbodiimide-mediated coupling reactions. Anal. Biochem. 1986, 156, 220–222. [Google Scholar] [CrossRef]
- Sehgal, D.; Vijay, I.K. A method for the high efficiency of water-soluble carbodiimide-mediated amidation. Anal. Biochem. 1994, 218, 87–91. [Google Scholar] [CrossRef]
- Müller-Schulte, D.; Horster, F.A. Radiation grafted polyethylene as carrier for protein immobilization—2. Covalent immobilization of antibodies against thyroxine. Polym. Bull. 1982, 7, 395–399. [Google Scholar]
- Zhu, Y.T.; Ren, X.Y.; Liu, Y.M.; Wei, Y.; Qing, L.S.; Liao, X. Covalent immobilization of porcine pancreatic lipase on carboxyl-activated magnetic nanoparticles: Characterization and application for enzymatic inhibition assays. Mater. Sci. Eng. C 2014, 38, 278–285. [Google Scholar] [CrossRef]
- Poplawska, M.; Bystrzejewski, M.; Grudziński, I.P.; Cywińska, M.A.; Ostapko, J.; Cieszanowski, A. Immobilization of gamma globulins and polyclonal antibodies of class igg onto carbon-encapsulated iron nanoparticles functionalized with various surface linkers. Carbon 2014, 74, 180–194. [Google Scholar] [CrossRef]
- Cheng, F.; Shang, J.; Ratner, D.M. A versatile method for functionalizing surfaces with bioactive glycans. Bioconjug. Chem. 2010, 22, 50–57. [Google Scholar] [CrossRef]
- Morpurgo, M.; Veronese, F.M.; Kachensky, D.; Harris, J.M. Preparation of characterization of poly(ethylene glycol) vinyl sulfone. Bioconjug. Chem. 1996, 7, 363–368. [Google Scholar] [CrossRef]
- Santoyo-Gonzalez, F.; Hernandez-Mateo, F.; Lopez-Jaramillo, F.; Ortega-Muñoz, J. Compound of silica-vinylsulphone,synthesis and uses of the same. WO Patent 200910664, 2 April 2009. [Google Scholar]
- Zhou, C.; Zhu, S.; Wu, X.; Jiang, B.; Cen, T.; Shen, S. Post-immobilization of modified macromolecular reagents using assembled penicillin acylase for microenvironmental regulation of nanopores and enhancement of enzyme stability. Biotechnol. Bioprocess Eng. 2010, 15, 376–382. [Google Scholar] [CrossRef]
- Eldin, M.S.M.; Seuror, E.I.; Nasr, M.A.; El-Aassar, M.R.; Tieama, H.A. Affinity covalent immobilization of glucoamylase onto ρ-benzoquinone activated alginate beads: I. Beads preparation and characterization. Appl. Biochem. Biotechnol. 2011, 164, 10–22. [Google Scholar] [CrossRef]
- Maksimova, Y.G.; Rogozhnikova, T.A.; Ovechkina, G.V.; Maksimov, A.Y.; Demakov, V.A. Catalytic properties of a nitrile hydratase immobilized on activated chitosan. Appl. Biochem. Microbiol. 2012, 48, 434–439. [Google Scholar] [CrossRef]
- Mirsky, V.M.; Riepl, M.; Wolfbeis, O.S. Capacitive monitoring of protein immobilization and antigen-antibody reactions on monomolecular alkylthiol films on gold electrodes. Biosens. Bioelectron. 1997, 12, 977–989. [Google Scholar] [CrossRef]
- Aissaoui, N.; Landoulsi, J.; Bergaoui, L.; Boujday, S.; Lambert, J.-F. Catalytic activity and thermostability of enzymes immobilized on silanized surface: Influence of the crosslinking agent. Enzym. Microb. Technol. 2013, 52, 336–343. [Google Scholar] [CrossRef]
- Kim, H.S.; Kye, Y.S.; Hage, D.S. Development and evaluation of n-hydroxysuccinimide-activated silica for immobilizing human serum albumin in liquid chromatography columns. J. Chromatogr. 1049, 51–61. [Google Scholar]
- Horton, H.R.; Swaisgood, H.E. Covalent immobilization of proteins by techniques which permit subsequent release. Methods Enzymol. 1987, 135, 130–141. [Google Scholar] [CrossRef]
- Seto, H.; Takara, M.; Yamashita, C.; Murakami, T.; Hasegawa, T.; Hoshino, Y.; Miura, Y. Surface modification of siliceous materials using maleimidation and various functional polymers synthesized by reversible addition-fragmentation chain transfer polymerization. ACS Appl. Mater. Interfaces 2012, 4, 5125–5133. [Google Scholar] [CrossRef]
- Kim, J.; Cho, J.; Seidler, P.M.; Kurland, N.E.; Yadavalli, V.K. Investigations of chemical modifications of amino-terminated organic films on silicon substrates and controlled protein immobilization. Langmuir 2010, 26, 2599–2608. [Google Scholar] [CrossRef]
- Gilbert, H.F. Thiol/disulfide exchange equilibria and disulfidebond stability. In Methods in Enzymology; Lester, P., Ed.; Academic Press: Waltham, MA, USA, 1995; Volume 251, pp. 8–28. [Google Scholar]
- Hearn, M.T.W. 1,1'-Carbonyldiimidazole-mediated immobilization of enzymes and affinity ligands. Methods Enzymol. 1987, 135, 102–117. [Google Scholar] [CrossRef]
- Ho, J.; Al-Deen, F.M.N.; Al-Abboodi, A.; Selomulya, C.; Xiang, S.D.; Plebanski, M.; Forde, G.M. N,n'-carbonyldiimidazole-mediated functionalization of superparamagnetic nanoparticles as vaccine carrier. Colloids Surf. B Biointerfaces 2011, 83, 83–90. [Google Scholar] [CrossRef]
- Crowley, S.C.; Chan, K.C.; Walters, R.R. Optimization of protein immobilization on 1,1'-carbonyldiimidazole-activated diol-bonded silica. J. Chromatogr. 1986, 359, 359–368. [Google Scholar] [CrossRef]
- Curreli, N.; Oliva, S.; Rescigno, A.; Rinaldi, A.; Sollai, F.; Sanjust, E. Novel diazonium-functionalized support for immobilization experiments. J. Appl. Polym. Sci. 1997, 66, 1433–1438. [Google Scholar] [CrossRef]
- Xi, F.; Wu, J.; Jia, Z.; Lin, X. Preparation and characterization of trypsin immobilized on silica gel supported macroporous chitosan bead. Process Biochem. 2005, 40, 2833–2840. [Google Scholar] [CrossRef]
- Andrews, B.A.; Head, D.M.; Dunthorne, P.; Asenjo, J.A. Peg activation and ligand binding for the affinity partitioning of proteins in aqueous two-phase systems. Biotechnol. Tech. 1990, 4, 49–54. [Google Scholar] [CrossRef]
- Buthe, A.; Wu, S.; Wang, P. Nanoporous silica glass for the immobilization of interactive enzyme systems. Methods Mol. Biol. 2011, 679, 37–48. [Google Scholar] [CrossRef]
- Liu, X.; Chen, X.; Li, Y.; Cui, Y.; Zhu, H.; Zhu, W. Preparation of superparamagnetic sodium alginate nanoparticles for covalent immobilization of candida rugosa lipase. J. Nanopart. Res. 2012, 14. [Google Scholar] [CrossRef]
- Solná, R.; Skládal, P. Amperometric flow-injection determination of phenolic compounds using a biosensor with immobilized laccase, peroxidase and tyrosinase. Electroanalysis 2005, 17, 2137–2146. [Google Scholar] [CrossRef]
- Wang, A.; Du, F.; Wang, F.; Shen, Y.; Gao, W.; Zhang, P. Convenient one-step purification and immobilization of lipase using a genetically encoded aldehyde tag. Biochem. Eng. J. 2013, 73, 86–92. [Google Scholar] [CrossRef]
- Cardias, H.; Grininger, C.; Trevisan, H.; Guisan, J.; Giordano, R. Influence of activation on the multipoint immobilization of penicillin g acylase on macroporous silica. Braz. J. Chem. Eng. 1999, 16, 141–148. [Google Scholar] [CrossRef]
- Bolivar, J.M.; Mateo, C.; Grazu, V.; Carrascosa, A.V.; Pessela, B.C.; Guisan, J.M. Heterofunctional supports for the one-step purification, immobilization and stabilization of large multimeric enzymes: Amino-glyoxyl versus amino-epoxy supports. Process Biochem. 2010, 45, 1692–1698. [Google Scholar] [CrossRef]
- Mateo, C.; Abian, O.; Bernedo, M.; Cuenca, E.; Fuentes, M.; Fernandez-Lorente, G.; Palomo, J.M.; Grazu, V.; Pessela, B.C.C.; Giacomini, C.; et al. Some special features of glyoxyl supports to immobilize proteins. Enzym. Microb. Technol. 2005, 37, 456–462. [Google Scholar] [CrossRef]
- Mateo, C.; Grazú, V.; Pessela, B.C.C.; Montes, T.; Palomo, J.M.; Torres, R.; López-Gallego, F.; Fernández-Lafuente, R.; Guisán, J.M. Advances in the design of new epoxy supports for enzyme immobilization-stabilization. Biochem. Soc. Trans. 2007, 35, 1593–1601. [Google Scholar] [CrossRef]
- Mateo, C.; Abian, O.; Fernández-Lorente, G.; Pedroche, J.; Fernández-Lafuente, R.; Guisan, J.M.; Tam, A.; Daminati, M. Epoxy sepabeads: A novel epoxy support for stabilization of industrial enzymes via very intense multipoint covalent attachment. Biotechnol. Prog. 2002, 18, 629–634. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zucca, P.; Sanjust, E. Inorganic Materials as Supports for Covalent Enzyme Immobilization: Methods and Mechanisms. Molecules 2014, 19, 14139-14194. https://doi.org/10.3390/molecules190914139
Zucca P, Sanjust E. Inorganic Materials as Supports for Covalent Enzyme Immobilization: Methods and Mechanisms. Molecules. 2014; 19(9):14139-14194. https://doi.org/10.3390/molecules190914139
Chicago/Turabian StyleZucca, Paolo, and Enrico Sanjust. 2014. "Inorganic Materials as Supports for Covalent Enzyme Immobilization: Methods and Mechanisms" Molecules 19, no. 9: 14139-14194. https://doi.org/10.3390/molecules190914139
APA StyleZucca, P., & Sanjust, E. (2014). Inorganic Materials as Supports for Covalent Enzyme Immobilization: Methods and Mechanisms. Molecules, 19(9), 14139-14194. https://doi.org/10.3390/molecules190914139