
Linking Protein Motion to Enzyme Catalysis

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
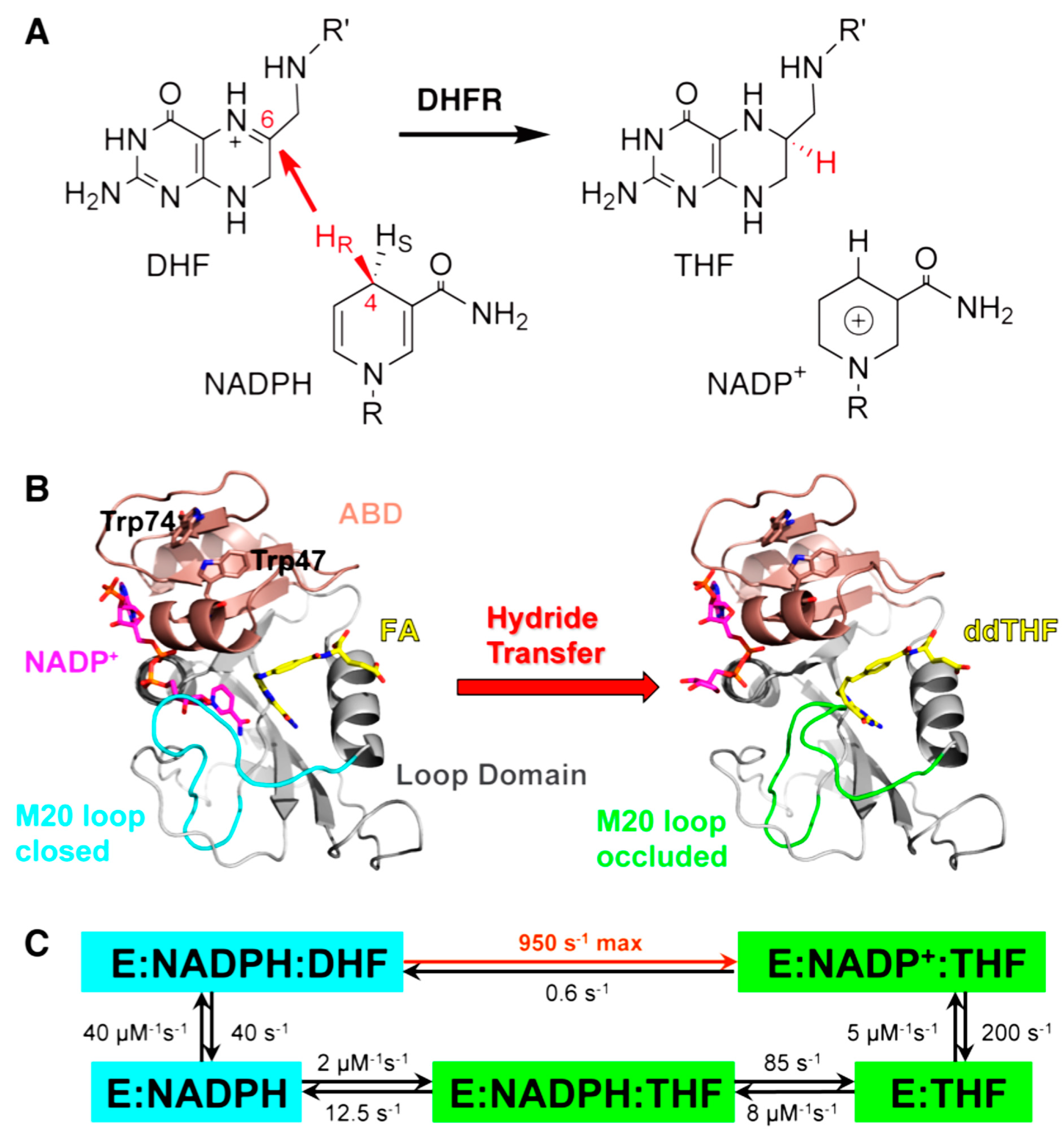
2. Dihydrofolate Reductase (DHFR)
2.1. Introduction

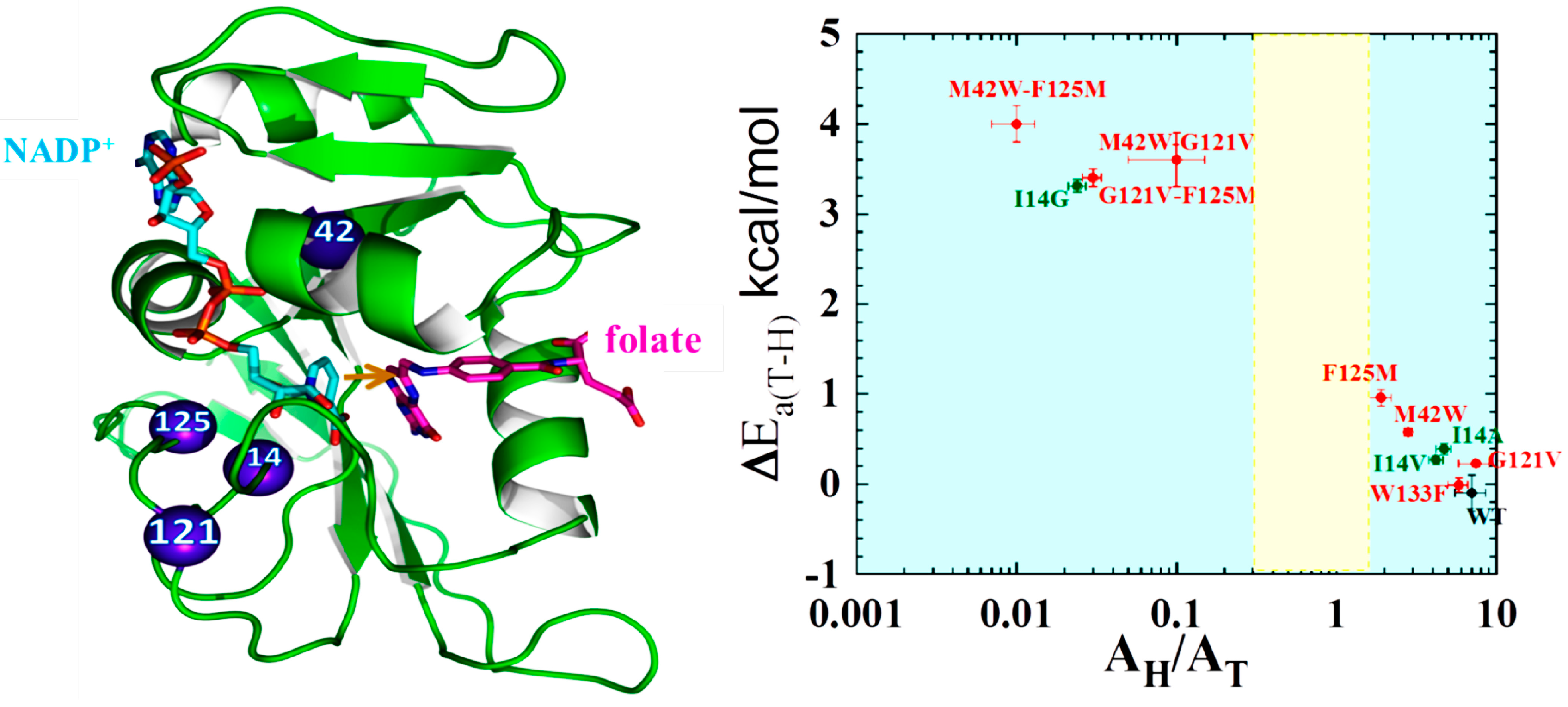
2.2. Effect of Remote Residues on the DHFR Catalyzed Chemistry

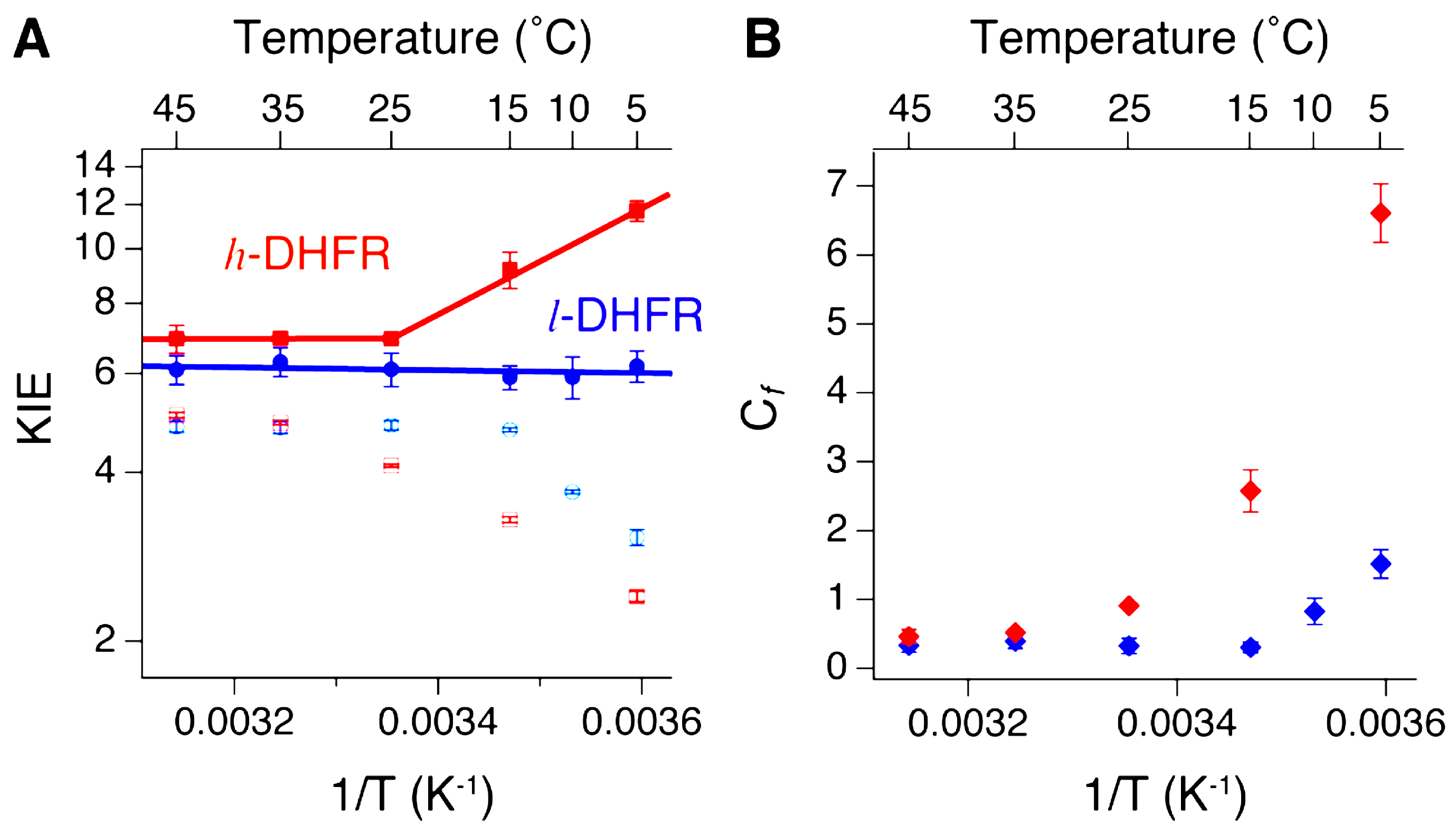
2.3. Heavy DHFR

3. Thymidylate Synthase (TSase)
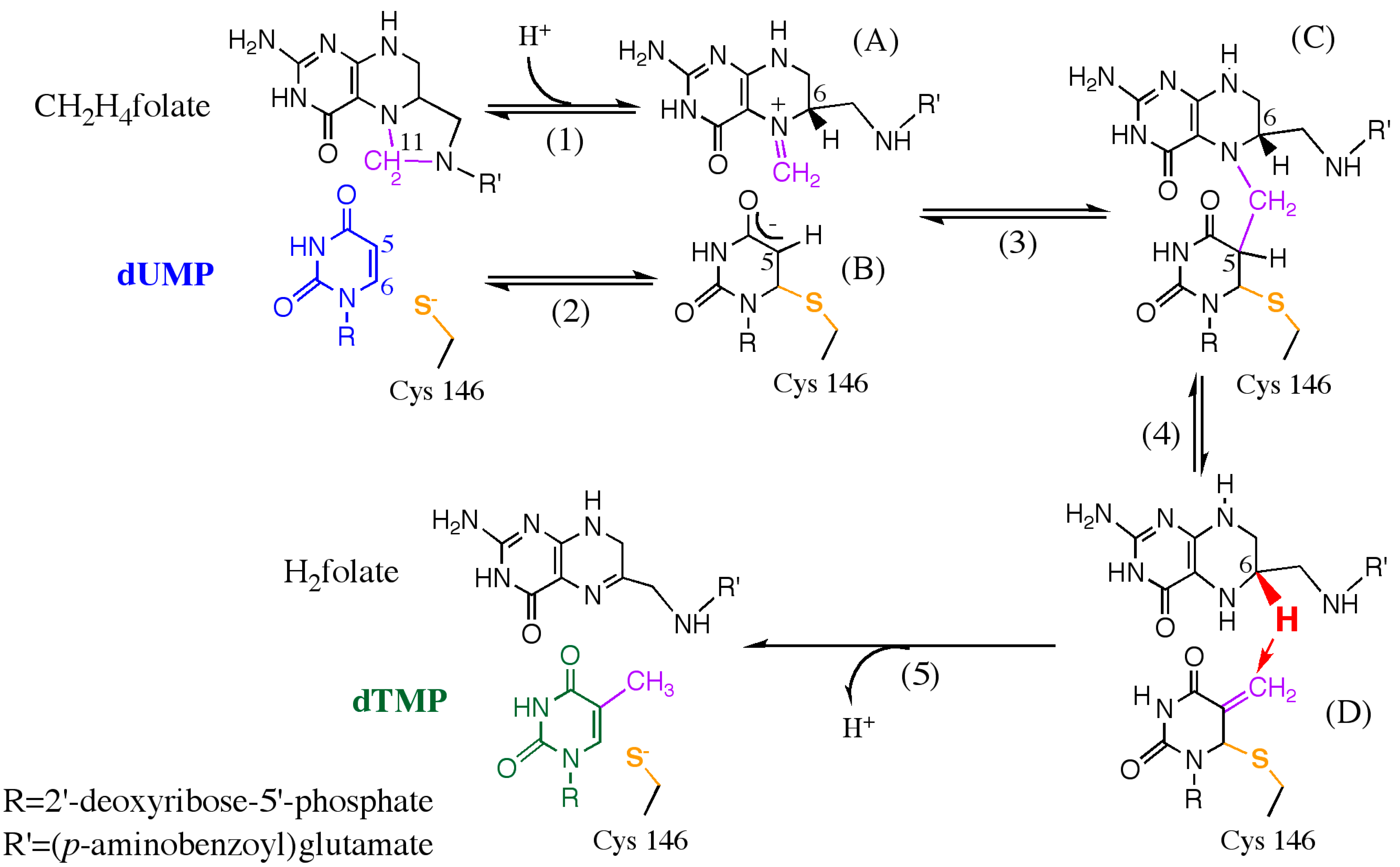
3.1. Introduction

3.2. Y209W TSase

3.3. Effect of Mg2+ on Motions of WT TSase

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wolfenden, R. Degrees of Difficulty of Water-Consuming Reactions in the Absence of Enzymes. Chem. Rev. 2006, 106, 3379–3396. [Google Scholar] [CrossRef] [PubMed]
- Benkovic, S.J.; Hammes-Schiffer, S. A Perspective on Enzyme Catalysis. Science 2003, 301, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Jäckel, C.; Kast, P.; Hilvert, D. Protein Design by Directed Evolution. Annu. Rev. Biochem. Biophys. 2008, 37, 153–173. [Google Scholar] [CrossRef]
- Fischer, E. Influence of configuration on the action of enzymes. Ber. Dtsch. Chem. Ges. 1894, 27, 2985–2993. [Google Scholar] [CrossRef]
- Pauling, L. Chemical Achievement and Hope for the Future. Am. Sci. 1948, 36, 51–58. [Google Scholar] [PubMed]
- Koshland, D.E. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Klinman, J.P.; Kohen, A. Hydrogen tunneling links protein dynamics to enzyme catalysis. Annu. Rev. Biochem. 2013, 82, 471–496. [Google Scholar] [CrossRef] [PubMed]
- Kohen, A. Role of Dynamics in Enzyme Catalysis: Substantial vs. Semantic Controversies. Acc. Chem. Res. 2014. [Google Scholar] [CrossRef]
- Wang, L.; Tharp, S.; Selzer, T.; Benkovic, S.J.; Kohen, A. Effects of a distal mutation on active site chemistry. Biochemistry 2006, 45, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Sen, A.; Francis, K.; Kohen, A. Extension and Limits of the Network of Coupled Motions Correlated to Hydride Transfer in Dihydrofolate Reductase. J. Am. Chem. Soc. 2014, 136, 2575–2582. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Abeysinghe, T.; Finer-Moore, J.S.; Stroud, R.M.; Kohen, A. A Remote Mutation Affects the Hydride Transfer by Disrupting Concerted Protein Motions in Thymidylate Synthase. J. Am. Chem. Soc. 2012, 134, 17722–17730. [Google Scholar] [CrossRef] [PubMed]
- Saen-Oon, S.; Ghanem, M.; Schramm, V.L.; Schwartz, S.D. Remote Mutations and Active Site Dynamics Correlate with Catalytic Properties of Purine Nucleoside Phosphorylase. Biophys. J. 2008, 94, 4078–4088. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, M.; Li, L.; Wing, C.; Schramm, V.L. Altered Thermodynamics from Remote Mutations Altering Human toward Bovine Purine Nucleoside Phosphorylase. Biochemistry 2008, 47, 2559–2564. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.P.; Tomchick, D.R.; Klinman, J.P. Enzyme structure and dynamics affect hydrogen tunneling: The impact of a remote side chain (I553) in soybean lipoxygenase-1. Proc. Natl. Acad. Sci. USA 2008, 105, 1146–1151. [Google Scholar] [CrossRef] [PubMed]
- Voet, D.; Voet, J.G. Biochemistry, 2nd ed.; Wiley: Hoboken, NJ, USA, 1995. [Google Scholar]
- Hammes-Schiffer, S.; Benkovic, S.J. Relating protein motion to catalysis. Annu. Rev. Biochem. 2006, 75, 519–541. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, M.R.; Kraut, J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: Crystallographic evidence. Biochemistry 1997, 36, 586–603. [Google Scholar] [CrossRef] [PubMed]
- Fierke, C.A.; Johnson, K.A.; Benkovic, S.J. Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry 1987, 26, 4085–4092. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Singh, P.; Czekster, C.M.; Kohen, A.; Schramm, V.L. Protein Mass-Modulated Effects in the Catalytic Mechanism of Dihydrofolate Reductase: Beyond Promoting Vibrations. J. Am. Chem. Soc. 2014, 136, 8333–8341. [Google Scholar] [CrossRef] [PubMed]
- Dametto, M.; Antoniou, D.; Schwartz, S.D. Barrier Crossing in Dihydrofolate Reductasedoes not involve a rate-promoting vibration. Mol. Phys. 2012, 110, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Arora, K.; Brooks, C.L., III. Functionally Important Conformations of the Met20 Loop in Dihydrofolate Reductase are Populated by Rapid Thermal Fluctuations. J. Am. Chem. Soc. 2009, 131, 5642–5647. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Warshel, A. Origin of the Temperature Dependence of Isotope Effects in Enzymatic Reactions: The Case of Dihydrofolate Reductase. J. Phys. Chem. B 2007, 111, 7852–7861. [Google Scholar] [CrossRef] [PubMed]
- Loveridge, E.J.; Behiry, E.M.; Guo, J.; Allemann, R.K. Evidence that a ‘dynamic knockout’ in Escherichia coli dihydrofolate reductase does not affect the chemical step of catalysis. Nat. Chem. 2012, 4, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.F.; Watney, J.B.; Hammes-Schiffer, S. Analysis of Electrostatics and Correlated Motions for Hydride Transfer in Dihydrofolate Reductase. J. Phys. Chem. B 2004, 108, 12231–12241. [Google Scholar] [CrossRef]
- Fan, Y.; Cembran, A.; Ma, S.; Gao, J. Connecting Protein Conformational Dynamics with Catalytic Function As Illustrated in Dihydrofolate Reductase. Biochemistry 2013, 52, 2036–2049. [Google Scholar] [CrossRef] [PubMed]
- Bystroff, C.; Kraut, J. Crystal structure of unliganded Escherichia coli dihydrofolate reductase. Ligand-induced conformational changes and cooperativity in binding. Biochemistry 1991, 30, 2227–2239. [Google Scholar]
- McElheny, D.; Schnell, J.R.; Lansing, J.C.; Dyson, H.J.; Wright, P.E. Defining the role of active-site loop fluctuations in dihydrofolate reductase catalysis. Proc. Natl. Acad. Sci. USA 2005, 102, 5032–5037. [Google Scholar] [CrossRef] [PubMed]
- Osborne, M.J.; Schnell, J.; Benkovic, S.J.; Dyson, H.J.; Wright, P.E. Backbone Dynamics in Dihydrofolate Reductase Complexes: Role of Loop Flexibility in the Catalytic Mechanism. Biochemistry 2001, 40, 9846–9859. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.T.; Taira, K.; Tu, C.P.D.; Benkovic, S.J. Probing the functional role of phenylalanine-31 of Escherichia coli dihydrofolate reductase by site-directed mutagenesis. Biochemistry 1987, 26, 4093–4100. [Google Scholar] [CrossRef] [PubMed]
- Falzone, C.J.; Wright, P.E.; Benkovic, S.J. Dynamics of a flexible loop in dihydrofolate reductase from Escherichia coli and its implication for catalysis. Biochemistry 1994, 33, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Falzone, C.J.; Wright, P.E.; Benkovic, S.J. Functional role of a mobile loop of Escherichia coli dihydrofolate reductase in transition-state stabilization. Biochemistry 1992, 31, 7826–7833. [Google Scholar] [CrossRef] [PubMed]
- Antikainen, N.M.; Smiley, R.D.; Benkovic, S.J.; Hammes, G.G. Conformation Coupled Enzyme Catalysis: Single-Molecule and Transient Kinetics Investigation of Dihydrofolate Reductase. Biochemistry 2005, 44, 16835–16843. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, L.; Fahmi, N.E.; Benkovic, S.J.; Hecht, S.M. Two Pyrenylalanines in Dihydrofolate Reductase Form an Excimer Enabling the Study of Protein Dynamics. J. Am. Chem. Soc. 2012, 134, 18883–18885. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D.; McElheny, D.; Dyson, H.J.; Wright, P.E. The dynamic energy landscape of dihydrofolate reductase catalysis. Science 2006, 313, 1638–1642. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.S.; Clarkson, M.W.; Degnan, S.C.; Erion, R.; Kern, D.; Alber, T. Hidden alternative structures of proline isomerase essential for catalysis. Nature 2009, 462, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Henzler-Wildman, K.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Loria, J.P.; Berlow, R.B.; Watt, E.D. Characterization of Enzyme Motions by Solution NMR Relaxation Dispersion. Acc. Chem. Res. 2008, 41, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Meadows, C.W.; Tsang, J.E.; Klinman, J.P. Picosecond-Resolved Fluorescence Studies of Substrate and Cofactor-Binding Domain Mutants in a Thermophilic Alcohol Dehydrogenase Uncover an Extended Network of Communication. J. Am. Chem. Soc. 2014, 136, 14821–14833. [Google Scholar] [CrossRef] [PubMed]
- Suel, G.M.; Lockless, S.W.; Wall, M.A.; Ranganathan, R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat. Struct. Mol. Biol. 2003, 10, 59–69. [Google Scholar] [CrossRef]
- Wong, K.F.; Selzer, T.; Benkovic, S.J.; Hammes-Schiffer, S. Impact of distal mutations on the network of coupled motions correlated to hydride transfer in dihydrofolate reductase. Proc. Natl. Acad. Sci. USA 2005, 102, 6807–6812. [Google Scholar] [CrossRef] [PubMed]
- Radkiewicz, J.L.; Brooks, C.L. Protein Dynamics in Enzymatic Catalysis: Exploration of Dihydrofolate Reductase. J. Am. Chem. Soc. 2000, 122, 255–231. [Google Scholar] [CrossRef]
- Agarwal, P.K.; Billeter, S.R.; Rajagopalan, P.T.; Benkovic, S.J.; Hammes-Schiffer, S. Network of coupled promoting motions in enzyme catalysis. Proc. Natl. Acad. Sci. USA 2002, 99, 2794–2799. [Google Scholar] [CrossRef] [PubMed]
- Rod, T.H.; Radkiewicz, J.L.; Brooks, C.L. Correlated motion and the effect of distal mutations in dihydrofolate reductase. Proc. Natl. Acad. Sci. USA 2003, 100, 6980–6985. [Google Scholar] [CrossRef] [PubMed]
- Hammes-Schiffer, S. Hydrogen Tunneling and Protein Motion in Enzyme Reactions. Acc. Chem. Res. 2005, 39, 93–100. [Google Scholar] [CrossRef]
- Roston, D.; Cheatum, C.M.; Kohen, A. Hydrogen Donor-Acceptor Fluctuations from Kinetic Isotope Effects: A Phenomenological Model. Biochemistry 2012, 51, 6860–6870. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, R.S.; Wang, L.; Markham, K.A.; Rajagopalan, P.T.; Benkovic, S.J.; Kohen, A. Tunneling and coupled motion in the Escherichia coli dihydrofolate reductase catalysis. J. Am. Chem. Soc. 2004, 126, 4778–4779. [Google Scholar] [CrossRef] [PubMed]
- Nagel, Z.D.; Klinman, J.P. A 21st century revisionist’s view at a turning point in enzymology. Nat. Chem. Biol. 2009, 5, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Klinman, J.P. Beyond Tunnelling Corrections: Full Tunnelling Models for Enzymatic C–H Activation Reactions. In Quantum Tunneling in Enzyme Catalyzed Reactions; Allemann, R., Scrutton, N., Eds.; Royal Society of Chemistry: London, UK, 2009; pp. 132–160. [Google Scholar]
- Kohen, A. Isotopes Effects in Chemistry and Biology; Taylor and Francis: Boca Raton, FL, 2006; pp. 743–764. [Google Scholar]
- Nagel, Z.D.; Klinman, J.P. Update 1 of: Tunneling and Dynamics in Enzymatic Hydride Transfer. Chem. Rev. 2010, 110, PR41–PR67. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Roston, D.; Kohen, A. Experimental and Theoretical Studies of Enzyme-Catalyzed Hydrogen-Transfer Reactions. Adv. Protein Chem. Struct. Biol. 2012, 87, 155–180. [Google Scholar] [PubMed]
- Stojković, V.; Perissinotti, L.L.; Lee, J.; Benkovic, S.J.; Kohen, A. The effect of active-site isoleucine to alanine mutation on the DHFR catalyzed hydride-transfer. Chem. Commun. 2010, 46, 8974–8976. [Google Scholar] [CrossRef]
- Stojković, V.; Perissinotti, L.L.; Willmer, D.; Benkovic, S.J.; Kohen, A. Effects of the donor-acceptor distance and dynamics on hydride tunneling in the dihydrofolate reductase catalyzed reaction. J. Am. Chem. Soc. 2012, 134, 1738–1745. [Google Scholar] [CrossRef] [PubMed]
- Hammes-Schiffer, S.; Watney, J.B. Hydride transfer catalysed by Escherichia coli and Bacillus subtilis dihydrofolate reductase: Coupled motions and distal mutations. Philos. Trans. R. Soc. B 2006, 361, 1365–1373. [Google Scholar] [CrossRef]
- Rajagopalan, P.T.R.; Stefan, L.; Benkovic, S.J. Coupling Interactions of Distal Residues Enhance Dihydrofolate Reductase Catalysis: Mutaional Effects on Hydride Transfer Rates. Biochemistry 2002, 41, 12618–12628. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.G.; Murkin, A.S.; Schramm, V.L. Femtosecond dynamics coupled to chemical barrier crossing in a Born-Oppenheimer enzyme. Proc. Natl. Acad. Sci. USA 2011, 108, 18661–18665. [Google Scholar] [CrossRef] [PubMed]
- Kipp, D.R.; Silva, R.G.; Schramm, V.L. Mass-Dependent Bond Vibrational Dynamics Influence Catalysis by HIV-1 Protease. J. Am. Chem. Soc. 2011, 133, 19358–19361. [Google Scholar] [CrossRef] [PubMed]
- Toney, M.D.; Castro, J.N.; Addington, T.A. Heavy-Enzyme Kinetic Isotope Effects on Proton Transfer in Alanine Racemase. J. Am. Chem. Soc. 2013, 135, 2509–2511. [Google Scholar] [CrossRef] [PubMed]
- Pudney, C.R.; Guerriero, A.; Baxter, N.J.; Johannissen, L.O.; Waltho, J.P.; Hay, S.; Scrutton, N.S. Fast Protein Motions Are Coupled to Enzyme H-Transfer Reactions. J. Am. Chem. Soc. 2013, 135, 2512–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luk, L.Y.P.; Javier Ruiz-Pernía, J.; Dawson, W.M.; Roca, M.; Loveridge, E.J.; Glowacki, D.R.; Harvey, J.N.; Mulholland, A.J.; Tuñón, I.; Moliner, V.; et al. Unraveling the role of protein dynamics in dihydrofolate reductase catalysis. Proc. Natl. Acad. Sci. USA 2013, 110, 16344–16349. [Google Scholar] [CrossRef]
- Świderek, K.; Javier Ruiz-Pernía, J.; Moliner, V.; Tuñón, I. Heavy enzymes- experimental and computational insights in enzyme dynamics. Curr. Opin. Chem. Biol. 2014, 21, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Turowski, M.; Yamakawa, N.; Meller, J.; Kimata, K.; Ikegami, T.; Hosoya, K.; Tanaka, N.; Thornton, E.R. Deuterium Isotope Effects on Hydrophobic Interactions: The Importance of Dispersion Interactions in the Hydrophobic Phase. J. Am. Chem. Soc. 2003, 125, 13836–13849. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, A.K.; Bowie, J.U. Evaluation of C–H⋯O Hydrogen Bonds in Native and Misfolded Proteins. J. Mol. Biol. 2002, 322, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Weak H-bonds. Comparisons of CHO to NHO in proteins and PHN to direct PN interactions. Phys. Chem. Chem. Phys. 2011, 13, 13860–13872. [Google Scholar]
- Anand, S.; Anbarasu, A.; Sethumadhavan, R. Influence of C-H…π Hydrogen Bonds in Interleukins. In Silico Biol. 2008, 8, 261–273. [Google Scholar] [PubMed]
- Carreras, C.W.; Santi, D.V. The Catalytic Mechanism and Structure of Thymidylate Synthase. Annu. Rev. Biochem. 1995, 64, 721–762. [Google Scholar] [CrossRef] [PubMed]
- Finer-Moore, J.S.; Santi, D.V.; Stroud, R.M. Lessons and Conclusions from Dissecting the Mechanism of a Bisubstrate Enzyme: Thymidylate Synthase Mutagenesis, Function, and Structure. Biochemistry 2002, 42, 248–256. [Google Scholar] [CrossRef]
- Stroud, R.M.; Finer-Moore, J.S. Conformational Dynamics along an Enzymatic Reaction Pathway: Thymidylate Synthase, “the Movie”. Biochemistry 2003, 42, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Spencer, H.T.; Villafranca, J.E.; Appleman, J.R. Kinetic scheme for thymidylate synthase from Escherichia coli: Determination from measurements of ligand binding, primary and secondary isotope effects, and pre-steady-state catalysis. Biochemistry 1997, 36, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Hong, B.; Mihai, C.; Kohen, A. Vibrationally Enhanced Hydrogen Tunneling in the Escherichia coli Thymidylate Synthase Catalyzed Reaction. Biochemistry 2004, 43, 1998–2006. [Google Scholar] [CrossRef] [PubMed]
- Kanaan, N.; Ferrer, S.; Martí, S.; Garcia-Viloca, M.; Kohen, A.; Moliner, V. Temperature Dependence of the Kinetic Isotope Effects in Thymidylate Synthase. A Theoretical Study. J. Am. Chem. Soc. 2011, 133, 6692–6702. [Google Scholar] [CrossRef]
- Kanaan, N.; Marti, S.; Moliner, V.; Kohen, A. QM/MM Study of Thymidylate Synthase: Enzymatic Motions and the Temperature Dependence of the Rate Limiting Step. J. Phys. Chem. A 2009, 113, 2176–2182. [Google Scholar] [CrossRef] [PubMed]
- Kanaan, N.; Marti, S.; Moliner, V.; Kohen, A. A Quantum Mechanics/Molecular Mechanics Study of the Catalytic Mechanism of the Thymidylate Synthase. Biochemistry 2007, 46, 3704–3713. [Google Scholar] [CrossRef] [PubMed]
- Kanaan, N.; Roca, M.; Tunon, I.; Marti, S.; Moliner, V. Application of Grote-Hynes Theory to the Reaction Catalyzed by Thymidylate Synthase. J. Phys. Chem. B 2010, 114, 13593–13600. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ferrer, S.; Moliner, V.; Kohen, A. QM/MM Calculations Suggest a Novel Intermediate Following the Proton Abstraction Catalyzed by Thymidylate Synthase. Biochemistry 2013, 52, 2348–2358. [Google Scholar] [CrossRef] [PubMed]
- Newby, Z.; Lee, T.T.; Morse, R.J.; Liu, Y.; Liu, L.; Venkatraman, P.; Santi, D.V.; Finer-Moore, J.S.; Stroud, R.M. The Role of Protein Dynamics in Thymidylate Synthase Catalysis: Variants of Conserved 2'-Deoxyuridine 5'-Monophosphate (dUMP)-Binding Tyr-261. Biochemistry 2006, 45, 7415–7428. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.A. H and Other Transfers in Enzymes and in Solution: Theory and Computations, a Unified View. 2. Applications to Experiment and Computations. J. Phys. Chem. B 2007, 111, 6643–6654. [Google Scholar]
- Marcus, R.A. Enzymatic catalysis and transfers in solution. I. Theory and computations, a unified view. J. Chem. Phys. 2006, 125, 194504. [Google Scholar]
- Hay, S.; Scrutton, N.S. Good vibrations in enzyme-catalysed reactions. Nat. Chem. 2012, 4, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Borgis, D.C.; Lee, S.; Hynes, J.T. A dynamical theory of nonadiabatic proton and hydrogen atom transfer reaction rates in solution. Chem. Phys. Lett. 1989, 162, 19–26. [Google Scholar] [CrossRef]
- Maley, G.F.; Maley, F.; Baugh, C.M. Differential inhibition of host and viral thymidylate synthetases by folylpolyglutamates. J. Biol. Chem. 1979, 254, 7485–7487. [Google Scholar] [PubMed]
- Rode, W.; Jastreboff, M. Effects of Mg2+ and adenine nucleotides on thymidylate synthetase from different mouse tumors. Mol. Cell. Biochem. 1984, 60, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sapienza, P.J.; Abeysinghe, T.; Luzum, C.; Lee, A.L.; Finer-Moore, J.S.; Stroud, R.M.; Kohen, A. Mg2+ Binds to the Surface of Thymidylate Synthase and Affects Hydride Transfer at the Interior Active Site. J. Am. Chem. Soc. 2013, 135, 7583–7592. [Google Scholar] [CrossRef] [PubMed]
- Kohen, A.; Cannio, R.; Bartolucci, S.; Klinman, J.P. Enzyme dynamics and hydrogen tunneling in a thermophilic alcohol dehydrogenase. Nature 1999, 399, 496–499. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, P.; Abeysinghe, T.; Kohen, A. Linking Protein Motion to Enzyme Catalysis. Molecules 2015, 20, 1192-1209. https://doi.org/10.3390/molecules20011192
Singh P, Abeysinghe T, Kohen A. Linking Protein Motion to Enzyme Catalysis. Molecules. 2015; 20(1):1192-1209. https://doi.org/10.3390/molecules20011192
Chicago/Turabian StyleSingh, Priyanka, Thelma Abeysinghe, and Amnon Kohen. 2015. "Linking Protein Motion to Enzyme Catalysis" Molecules 20, no. 1: 1192-1209. https://doi.org/10.3390/molecules20011192
APA StyleSingh, P., Abeysinghe, T., & Kohen, A. (2015). Linking Protein Motion to Enzyme Catalysis. Molecules, 20(1), 1192-1209. https://doi.org/10.3390/molecules20011192