
Selective C–C Coupling Reaction of Dimethylphenol to Tetramethyldiphenoquinone Using Molecular Oxygen Catalyzed by Cu Complexes Immobilized in Nanospaces of Structurally-Ordered Materials

Abstract
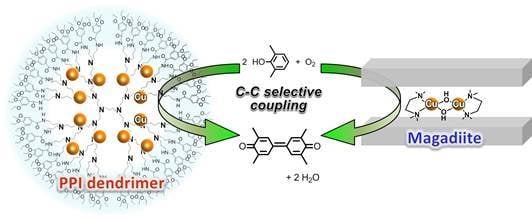
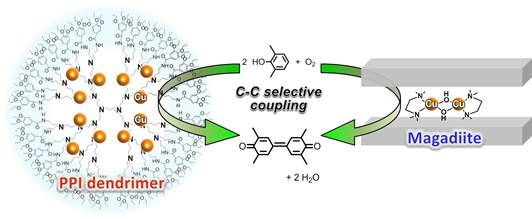
:
1. Introduction
2. Results
2.1. Preparation of Immobilized Cu Catalysts
2.1.1. PPI Dendrimer-Encapsulated Cu Catalyst
2.1.2. Magadiite-Immobilized Cu Catalyst
2.2. Characterization of Immobilized Cu Catalysts
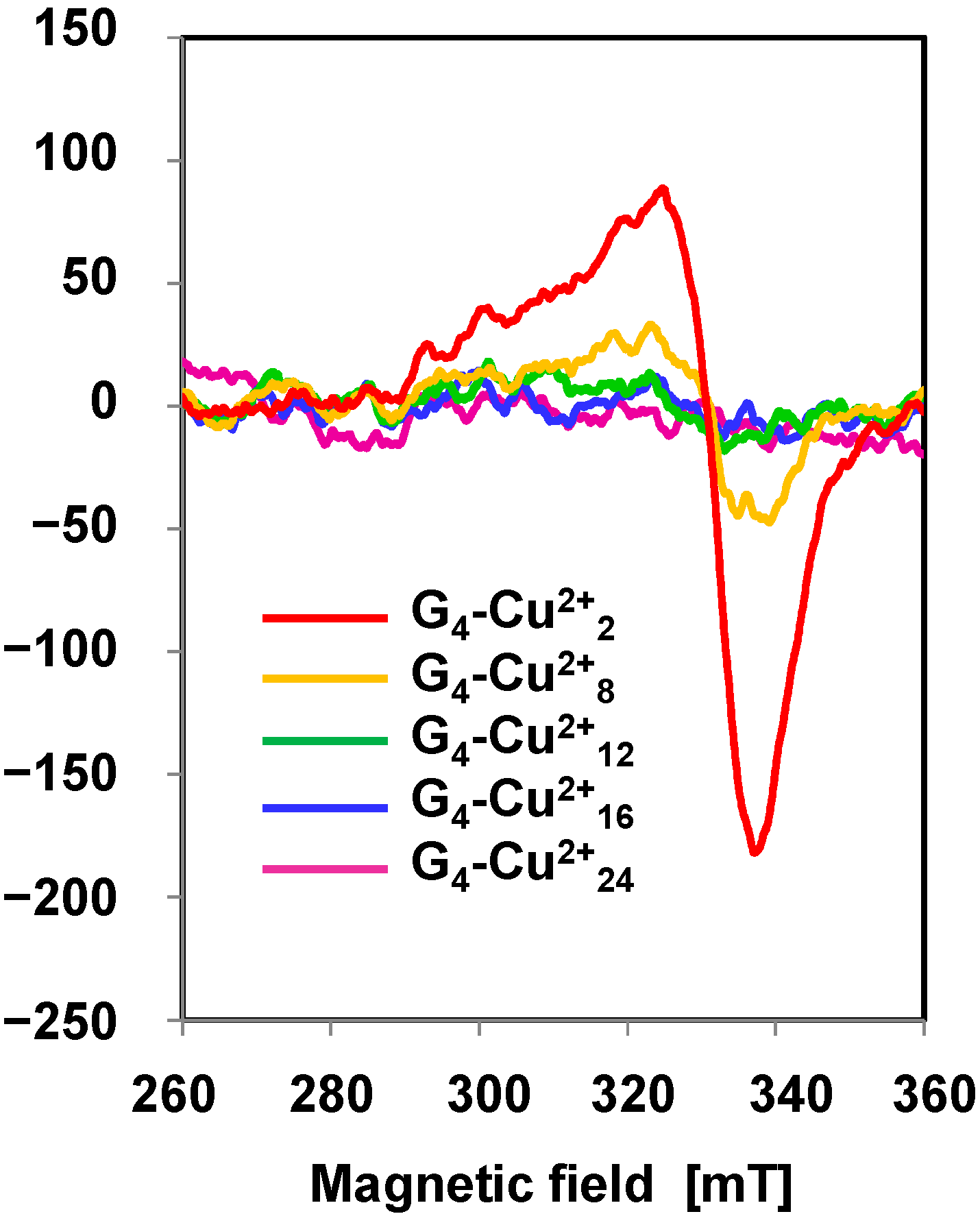
2.2.1. PPI Dendrimer-Encapsulated Cu Catalyst


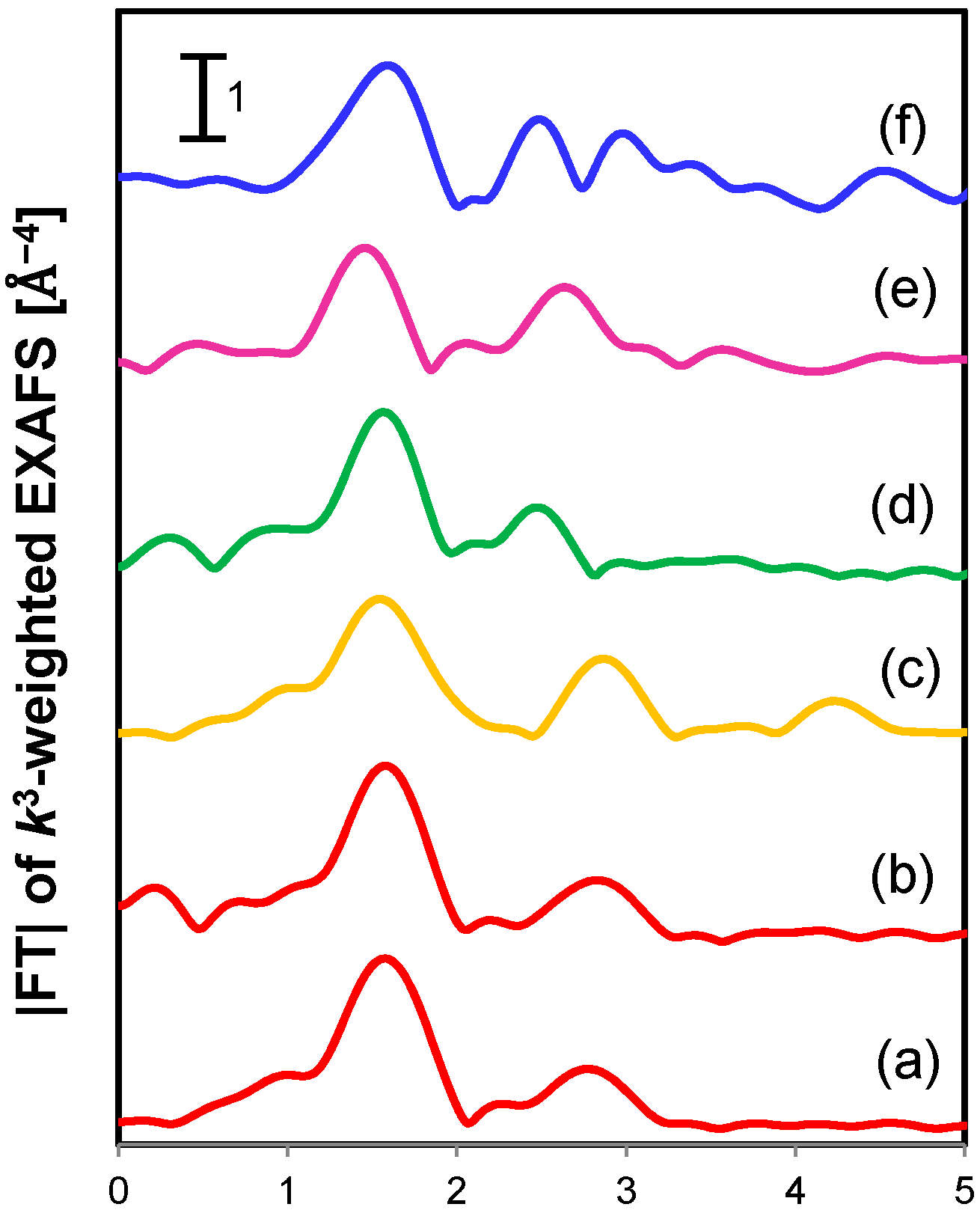
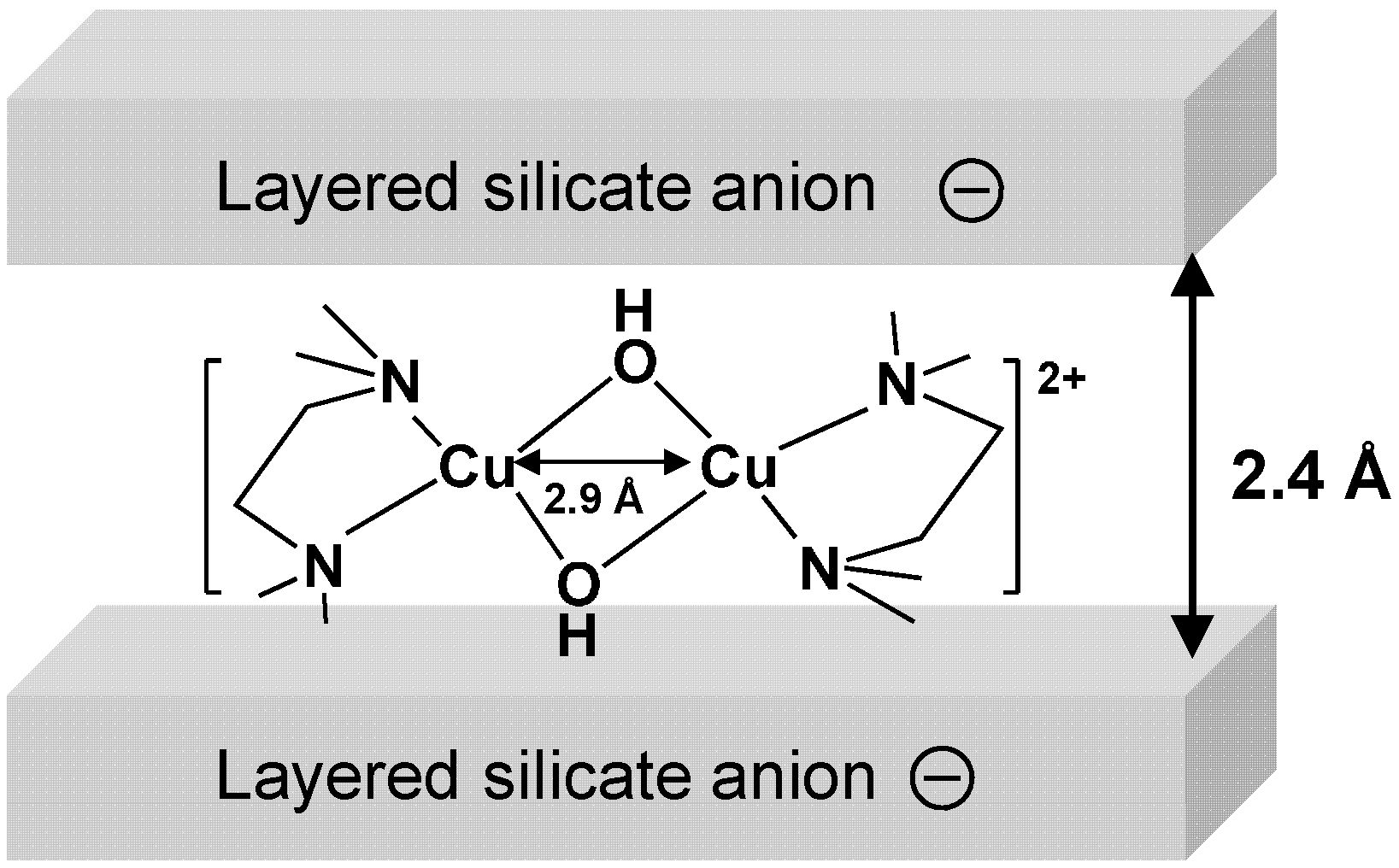
2.2.2. Magadiite-Immobilized Cu Catalyst


| Sample | Shell | CN b | R c [Å] | σ2 d [Å2] |
|---|---|---|---|---|
| [Cu(OH)TMEDA]2Cl2 | Cu-O/N | 4.0 | 2.02 | - |
| Cu-Cu | 1.0 | 2.99 | - | |
| Cu2+-magadiite (fresh) | Cu-O/N | 4.4 | 1.99 | 0.0016 |
| Cu-Cu | 0.9 | 2.97 | 0.0042 | |
| Cu2+-magadiite (used) | Cu-O/N | 4.5 | 1.98 | 0.0060 |
| Cu-Cu | 1.1 | 2.93 | 0.0022 |


2.3. Oxidative Coupling of DMP Using Immobilized Cu Complex Catalysts
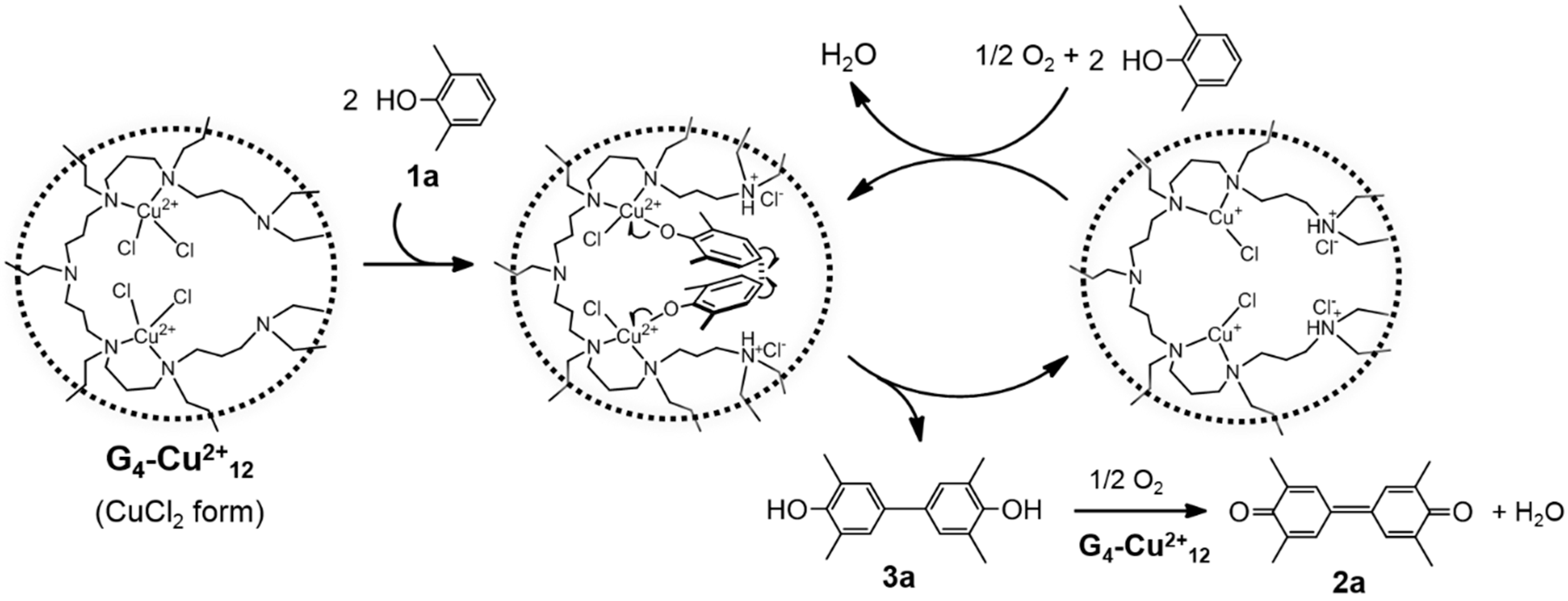
2.3.1. G4-Cu2+n-Catalyzed Oxidative Coupling of DMP

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Solvent | Time [h] | Conv. a [%] | Sel. to | Yield a [%] | ||
|---|---|---|---|---|---|---|---|---|
| C-C b [%] | 2a | 3a | 4a | |||||
| 1 | G4-Cu2+2 | CHCl3 | 6 | 9 | 44 | 4 | 0 | 4 |
| 2 | G4-Cu2+8 | CHCl3 | 6 | 25 | 68 | 8 | 9 | 7 |
| 3 | G4-Cu2+12 | CHCl3 | 6 | 67 | 97 | 55 | 10 | 2 |
| 4 | G4-Cu2+16 | CHCl3 | 6 | 34 | 88 | 15 | 15 | 4 |
| 5 | G4-Cu2+24 | CHCl3 | 6 | 16 | 87 | 7 | 7 | 2 |
| 6 | G4-Cu2+12 | CHCl3 | 18 | >99 | 97 | 97 | trace | 2 |
| 7 | G4-Cu2+12c | CHCl3 | 6 | 29 | 96 | 22 | 6 | 1 |
| 8 | CuCl2-TEA | CHCl3 | 6 | 9 | 44 | 1 | 3 | 5 |
| 9 | CuCl2-TMPDA | CHCl3 | 6 | 98 | 46 | 41 | 4 | 52 |
| 10 | PEI-Cu2+ | CHCl3 | 6 | 11 | 63 | 3 | 4 | 1 |
| 11 | G4-Cu2+12 | MeOH | 6 | 97 | 46 | 32 | 12 | 47 |
| 12 | G4-Cu2+12 | CH3CN | 6 | 9 | 33 | 0 | 3 | 0 |
| 13 | G4-Cu2+12 | TFT | 18 | >99 | 96 | 96 | trace | 2 |
| 14 d | G4-Cu2+12 | CHCl3 | 24 | >99 | 97 | 97 e | trace | 2 |

2.3.2. Heterogeneous Oxidative Coupling Reaction of DMP Using Cu2+-Magadiite
| Entry | Catalyst | Time [h] | Conv. b [%] | Yield b [%] | |
|---|---|---|---|---|---|
| 2a | 4a | ||||
| 1 | Cu2+-magadiite | 12 | 75 | 67 | 3 |
| 2 | Cu2+-magadiite | 18 | >99 | 95 | 4 |
| 3 c | Cu2+-magadiite | 18 | >99 | 94 | 4 |
| 4 d | Cu2+-magadiite | 18 | >99 | 94 | 4 |
| 5 e | Cu2+-magadiite | 48 | >99 | 95 | 3 |
| 6 | Cu2+-SiO2 | 12 | >99 | 60 | 3 |
| 7 | Cu2+-mordenite | 12 | 51 | 27 | 2 |
| 8 | Cu2+(mono)-magadiite | 12 | 15 | 2 | 2 |

3. Discussion

4. Experimental Section
4.1. Preparation of G4-Cu2+n
4.2. Oxidative Coupling of DMP Using G4-Cu2+n
4.3. Preparation of Cu2+-Magadiite
4.4. Oxidative Coupling of DMP Using Cu2+-Magadiite
4.5. Cu2+-Magadiite-Catalyzed Oxidative Coupling of DMP Using Continuous Flow Reactor
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Nishihashi, S.; Hirahashi, T. Enzymatic Preparation of Biphenol Derivatives and Diphenoquinone Derivatives. JP2007129952A, 31 May 2007. [Google Scholar]
- Borsenberger, P.M.; Gruenbaum, W.T.; Oregan, M.B.; Rossi, L.J. Electron transport in acceptor doped polymers: The role of group dipole moments. J. Polym. Sci. Part B: Polym. Phys. 1995, 33, 2143–2149. [Google Scholar] [CrossRef]
- Van Aert, H.A.M.; van Gendersen, M.H.P.; van Steenpaal, G.J.M.L.; Nelissen, L.; Meijer, E.W. Modified poly(2,6-dimethyl-1,4-phenylene ether)s prepared by redistribution. Macromolecules 1997, 30, 6056–6066. [Google Scholar] [CrossRef]
- Wariishi, H.; Kondo, A.; Nishihashi, S. Preparation of Phenol Dimer with High Selectivity by Dimerization of Phenols with Manganese(III). JP2008081449A, 10 April 2008. [Google Scholar]
- Villemin, D.; Sauvaget, F. Dry Synthesis under microwave irradiation: A rapid and efficient coupling of naphthols. Synlett 1994, 8, 435–436. [Google Scholar] [CrossRef]
- Hirano, M.; Ishii, T.; Morimoto, T. Oxidation by cobalt(III) acetate. Part 13. Oxidation of substituted phenols with cobalt(III) acetate in acetic acid. Bull. Chem. Soc. Jpn. 1991, 64, 1434–1436. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Finet, J.-P.; Giannotti, C.; Halley, F. The chemistry of pentavalent organobismuth reagents: Part XI. Reactions with sterically hindered phenols. Tetrahedron 1988, 44, 4483–4494. [Google Scholar] [CrossRef]
- Nishio, H.; Itoh, N.; Nagashima, M.; Kurosawa, K. Choice of manganese(III) complexes for the synthesis of 4,4'-biphenyldiols and 4,4'-diphenoquiones. Bull. Chem. Soc. Jpn. 1992, 65, 620–621. [Google Scholar] [CrossRef]
- Fujisawa, K.; Iwata, Y.; Kitajima, N.; Higashimura, H.; Kubota, M.; Miyashita, Y.; Yamada, Y.; Okamoto, K.; Moro-oka, Y. Synthesis, structure and reactivity of phenoxo copper(II) complexes, Cu(Oar)(HB(3,5-Pri2pz)3) (Ar = C6H4–4-F, 2,6-Me2C6H3, 2,6-But2C6H3). Chem. Lett. 1999, 28, 739–740. [Google Scholar] [CrossRef]
- Hong, Z.; Zhi-Quan, P.; Qin-Hui, L.; Guang-Quan, M.; De-Liang, L.; Jiu-Tong, C. Study on the crystal structure of a macrocycle and tyrosinase activity of its dinuclear copper complexes. Chin. J. Chem. 2005, 23, 835–842. [Google Scholar] [CrossRef]
- Matsushita, M.; Kamata, K.; Yamaguchi, K.; Mizuno, N. Heterogeneously catalyzed aerobic oxidative biaryl coupling of 2-naphthols and substituted phenols in water. J. Am. Chem. Soc. 2005, 127, 6632–6640. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-H.; Menga, X.-G.; Panga, Q.-H.; Liub, S.-D.; Li, J.-M.; Dua, J.; Hu, C.-W. Metal complexes catalyzed oxidative coupling of 2,6-dimethylphenol in micellar media. J. Mol. Catal. A Chem. 2011, 328, 88–92. [Google Scholar] [CrossRef]
- It is known that the oxidative coupling of 2,6-tert-butylphenol gives only the C–C coupling products using copper catalysts. See Refs. [14,15]
- Fujiyama, H.; Kohara, I.; Iwai, K.; Nishiyama, S.; Tsuruya, S.; Masai, M. Liquid-phase oxidation of 2,6-di-tert-butylphenol with Cu-impregnated MCM-41 catalysts in the presence of alkali metals. J. Catal. 1999, 188, 417–425. [Google Scholar] [CrossRef]
- Kohara, I.; Fujiyama, H.; Iwai, K.; Nishiyama, S.; Tsuruya, S. Catalytic activity of Cu ion-exchanged Na·MCM-41 in the liquid-phase oxidation of 2,6-di-tert-butylphenol. J. Mol. Catal. A. Chem. 2000, 153, 93–101. [Google Scholar] [CrossRef]
- Pandey, G.; Muralikrishna, C.; Bhalerao, U.T. Mushroom tyrosinase catalyzed coupling of hindered phenols: A novel approach for the synthesis of diphenoquinones and bisphenols. Tetrahedron Lett. 1990, 26, 3771–3774. [Google Scholar] [CrossRef]
- Gupta, R.; Mukherjee, R. Catalytic oxidation of hindered phenols by a copper(I) complex and dioxygen. Tetrahedron Lett. 2000, 41, 7763–7767. [Google Scholar] [CrossRef]
- Liao, B.-S.; Liu, Y.-H.; Peng, S.-M.; Liu, S.-T. Efficient oxidative coupling of 2,6-disubstituted phenol catalyzed by a dicopper(II) complex. Dalton Trans. 2012, 41, 1158–1164. [Google Scholar] [CrossRef] [PubMed]
- Lissel, M.; in de Wal, H.J.; Neumann, R. Oxidation of activated phenols by dioxygen catalyzed by the H5PV2Mo10O40 heteropolyanion. Tetrahedron Lett. 1992, 33, 1795–1798. [Google Scholar] [CrossRef]
- Jansen, J.F. G.A.; de Brabander-van den Berg, E.M.M.; Meijer, E.W. Encapsulation of guest molecules into dendritic box. Science 1994, 266, 1226–1229. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Takanashi, K. Synthesis and functionality of dendrimer with finely controlled metal assembly. Polymer 2008, 49, 4033–4041. [Google Scholar] [CrossRef]
- Boisselier, E.; Diallo, A.K.; Salmon, L.; Ornelas, C.; Ruiz, J.; Astruc, D. Encapsulation and stabilization of gold nanoparticles with “Click” polyethyleneglycol dendrimers. J. Am. Chem. Soc. 2010, 132, 2729–2742. [Google Scholar] [CrossRef] [PubMed]
- Lemon, B.I.; Crooks, R.M. Preparation and characterization of dendrimer-encapsulated CdS semiconductor quantum dots. J. Am. Chem. Soc. 2000, 122, 12886–12887. [Google Scholar] [CrossRef]
- Quali, A.; Caminade, A.-M. Dendrimers: Towards Catalytic, Material and Biomedical Uses, 1st ed.; John Wiley & Sons: Chichester, UK, 2011; pp. 183–195. [Google Scholar]
- Ooe, M.; Murata, M.; Mizugaki, T.; Ebitani, K.; Kaneda, K. Supramolecular catalysts by encapsulating palladium complexes within dendrimers. J. Am. Chem. Soc. 2004, 126, 1604–1605. [Google Scholar] [CrossRef] [PubMed]
- Ooe, M.; Murata, M.; Mizugaki, T.; Ebitani, K.; Kaneda, K. Dendritic nanoreactors encapsulating Pd particles for substrate-specific hydrogenation of olefins. Nano Lett. 2002, 2, 999–1002. [Google Scholar] [CrossRef]
- Maeno, Z.; Mitsudome, T.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Selective synthesis of Rh5 carbonyl clusters within a polyamine dendrimer for chemoselective reduction of nitro aromatics. Chem. Commun. 2014, 50, 6526–6529. [Google Scholar] [CrossRef]
- Kibata, T.; Mitsudome, T.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Investigation of size-dependent properties of sub-nanometer palladium clusters encapsulated within a polyamine dendrimer. Chem. Commun. 2013, 49, 167–169. [Google Scholar] [CrossRef]
- Maeno, Z.; Okao, M.; Mitsudome, T.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Regioselective oxidative coupling of 2,6-dimethylphenol to tetramethyldiphenoquinone using polyamine dendrimer-encapsulated Cu catalysts. RSC Adv. 2013, 3, 9662–9665. [Google Scholar] [CrossRef]
- Niu, Y.; Crooks, R.M. Preparation of dendrimer-encapsulated metal nanoparticles using organic solvents. Chem. Mater. 2003, 15, 3463–3467. [Google Scholar] [CrossRef]
- Koper, G.J.M.; van Genderen, M.H.P.; Elissen-Román, C.; Baars, M.W.P.L.; Meijer, E.W.; Borkovec, M. Protonation mechanism of poly(propylene imine) dendrimers and some associated oligo amines. J. Am. Chem. Soc. 1997, 119, 6512–6521. [Google Scholar] [CrossRef]
- For details, see our previous paper Ref. [29]
- Kosuge, K.; Yamazaki, A.; Tsunashima, A.; Otsuka, R. Hydrothermal synthesis of magadiite and kenyaite. J. Ceram. Soc. Jpn. 1992, 100, 326–331. [Google Scholar] [CrossRef]
- Schwieger, W.; Selvam, T.; Gravenhorst, O.; Pfänder, N.; Schlögl, R.; Mabande, G.T.P. Intercalation of [Pt(NH3)4]2+ ions into layered sodium silicate magadiite: A useful method to enhance their stabilisation in a highly dispersed state. J. Phys. Chem. Solids 2004, 65, 413–420. [Google Scholar] [CrossRef]
- Zhang, Z.; Seangkerdsub, S.; Dai, S. Intersurface ion-imprinting synthesis on layered magadiite hosts. Chem. Mater. 2003, 15, 2921–2925. [Google Scholar] [CrossRef]
- Ogawa, M. Photoisomerization of azobenzene in the interlayer space of magadiite. J. Mater. Chem. 2002, 12, 3304–3307. [Google Scholar] [CrossRef]
- Kala, U.L.; Suma, S.; Kurup, M.R. P.; Krishnan, S.; John, R.P. Synthesis, spectral characterization and crystal structure of copper(II) complexes of 2-hydroxyacetophenone-N(4)-phenyl semicarbazone. Polyhedron 2007, 26, 1427–1435. [Google Scholar] [CrossRef]
- Pande, S.; Weir, M.G.; Zaccheo, B.A.; Crooks, R.M. Synthesis, characterization, and electrocatalysis using Pt and Pd dendrimer-encapsulated nanoparticles prepared by galvanic exchange. New J. Chem. 2011, 35, 2054–2060. [Google Scholar] [CrossRef]
- Pate, J.E.; Ross, P.K.; Thamann, T.J.; Reed, C.A.; Karlin, K.D.; Sorell, T.N.; Solomon, E.I. Spectroscopic studies of the charge transfer and vibrational features of binuclear copper(II) azide complexes: Comparison to the coupled binuclear copper active site in met azide hemocyanin and tyrosinase. J. Am. Chem. Soc. 1989, 111, 5198–5209. [Google Scholar] [CrossRef]
- Tromp, M.; van Strijdonck, G.P.F.; van Berkel, S.S.; van den Hoogenband, A.; Feiters, M.C.; de Bruin, B.; Fiddy, S.G.; van der Eerden, A.M.J.; van Bokhoven, J.A.; van Leeuwen, P.W.N.M.; et al. Multitechnique approach to reveal the mechanism of copper(II)-catalyzed arylation reactions. Organometallics 2010, 29, 3085–3097. [Google Scholar] [CrossRef]
- Choy, J.-H.; Yoon, J.-B.; Jung, H. Polarization-dependent X-ray absorption spectroscopic study of [Cu(cyclam)]2+-intercalated saponite. J. Phys. Chem. B 2002, 106, 11120–11126. [Google Scholar] [CrossRef]
- Wells, A.F. 333. The crystal structure of anhydrous cupric chloride, and the stereochemistry of the cupric atom. J. Chem. Soc. 1947, 1670–1675. [Google Scholar] [CrossRef]
- Fransson, G.; Lundberg, B.K.S. Metal complexes with mixed ligands. 4. The crystal structure of tetrakisimidazole Cu(II) sulphate, Cu(C3H4N2)4SO4. Acta Chem. Scand. 1972, 26, 3969–3976. [Google Scholar] [CrossRef]
- Mitamura, Y.; Komori, Y.; Hayashi, S.; Sugahara, Y.; Kuroda, K. Interlamellar esterification of H-magadiite with aliphatic alcohols. Chem. Mater. 2001, 13, 3747–3753. [Google Scholar] [CrossRef]
- Moura, A.O.; Prado, A.G. S. Effect of thermal dehydration and rehydration on Na-magadiite structure. J. Colloid Interface Sci. 2009, 330, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, D.; Kuroda, K. Design of silicate nanostructures by interlayer alkoxysilylation of layered silicates (magadiite and kenyaite) and subsequent hydrolysis of alkoxy groups. New J. Chem. 2006, 30, 277–284. [Google Scholar] [CrossRef]
- See Supplementary Materials
- Zhang, K.; Lam, K.-F.; Albela, B.; Xue, T.; Khrouz, L.; Hou, Q.-W.; Yuan, E.-H.; He, M.-Y.; Bonneviot, L. Mononuclear-dinuclear equilibrium of grafted copper complexes confined in the nanochannels of MCM-41-silica. Chem. Eur. J. 2011, 17, 14258–14266. [Google Scholar] [CrossRef] [PubMed]
- Cu2+(mono)-magadiite was prepared from Cu(ethylenediamine)2(ClO4)2 and magadiite in the same manner as reported in Ref. [50]
- Choy, J.-H.; Kim, D.-K.; Park, J.-C.; Choi, S.-N.; Kim, Y.-J. Intracrystalline and electronic structure of copper(II) complexes stabilized in two-dimensional aliminosilicate. Inorg. Chem. 1997, 36, 189–195. [Google Scholar] [CrossRef]
- Peisach, J.; Blumberg, W.E. Structural implications derived from the analysis of electron paramagnetic resonance spectra of natural and artificial copper proteins. Arch. Biochem. Biophys. 1974, 165, 691–708. [Google Scholar] [CrossRef] [PubMed]
- In each of these catalytic systems, the ratio of tertiary amino groups to Cu2+ was adjusted to equal that of G4-Cu2+12
- Takahashi, Y.; Mitsudome, T.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Highly atom-efficient and chemoselective reduction of ketones in the presence of aldehydes using heterogeneous catalysts. Green Chem. 2013, 15, 2695–2698. [Google Scholar] [CrossRef]
- Mitsudome, T.; Takahashi, Y.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Hydrogenation of sulfoxides to sulfides under mild conditions using ruthenium nanoparticle catalysts. Angew. Chem. Int. Ed. 2014, 53, 8348–8351. [Google Scholar] [CrossRef]
- Kresta, J.; Tkáč, A.; Přikryl, R.; Malik, L. Ion-radical mechanism of the polymerization of 2,6-dimethylphenol by oxidative coupling catalysed by CuCl2-amine complex. Makromol. Chem. 1975, 176, 157–175. [Google Scholar] [CrossRef]
- The molecular sizes of DMP and DPQ were estimated by Chem 3D calculations
- De Brabander-van den Berg, E.M.M.; Meijer, E.W. Poly(propylene imine) dendrimers: Large-scale synthesis by hetereogeneously catalyzed hydrogenations. Angew. Chem. Int. Ed. 1993, 32, 1308–1311. [Google Scholar] [CrossRef]
- Sample Availability: Samples are not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maeno, Z.; Mitsudome, T.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Selective C–C Coupling Reaction of Dimethylphenol to Tetramethyldiphenoquinone Using Molecular Oxygen Catalyzed by Cu Complexes Immobilized in Nanospaces of Structurally-Ordered Materials. Molecules 2015, 20, 3089-3106. https://doi.org/10.3390/molecules20023089
Maeno Z, Mitsudome T, Mizugaki T, Jitsukawa K, Kaneda K. Selective C–C Coupling Reaction of Dimethylphenol to Tetramethyldiphenoquinone Using Molecular Oxygen Catalyzed by Cu Complexes Immobilized in Nanospaces of Structurally-Ordered Materials. Molecules. 2015; 20(2):3089-3106. https://doi.org/10.3390/molecules20023089
Chicago/Turabian StyleMaeno, Zen, Takato Mitsudome, Tomoo Mizugaki, Koichiro Jitsukawa, and Kiyotomi Kaneda. 2015. "Selective C–C Coupling Reaction of Dimethylphenol to Tetramethyldiphenoquinone Using Molecular Oxygen Catalyzed by Cu Complexes Immobilized in Nanospaces of Structurally-Ordered Materials" Molecules 20, no. 2: 3089-3106. https://doi.org/10.3390/molecules20023089
APA StyleMaeno, Z., Mitsudome, T., Mizugaki, T., Jitsukawa, K., & Kaneda, K. (2015). Selective C–C Coupling Reaction of Dimethylphenol to Tetramethyldiphenoquinone Using Molecular Oxygen Catalyzed by Cu Complexes Immobilized in Nanospaces of Structurally-Ordered Materials. Molecules, 20(2), 3089-3106. https://doi.org/10.3390/molecules20023089