
Neferine Attenuates the Protein Level and Toxicity of Mutant Huntingtin in PC-12 Cells via Induction of Autophagy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
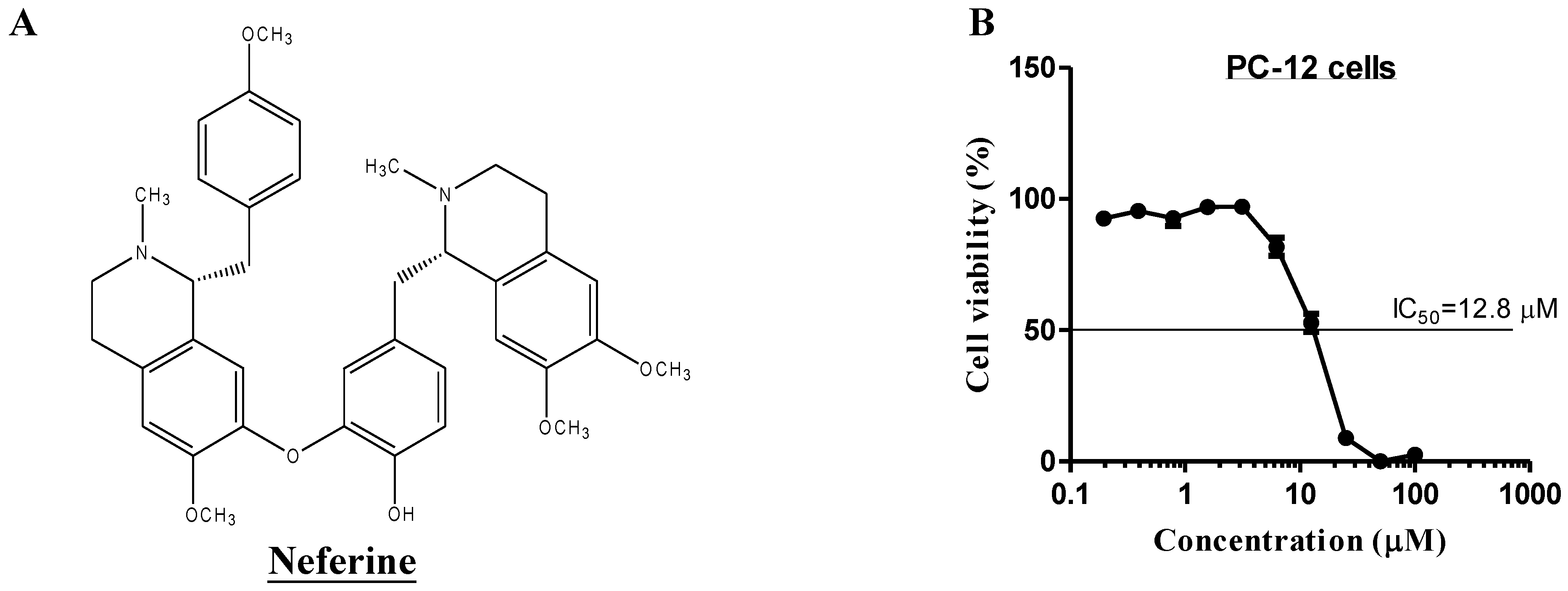
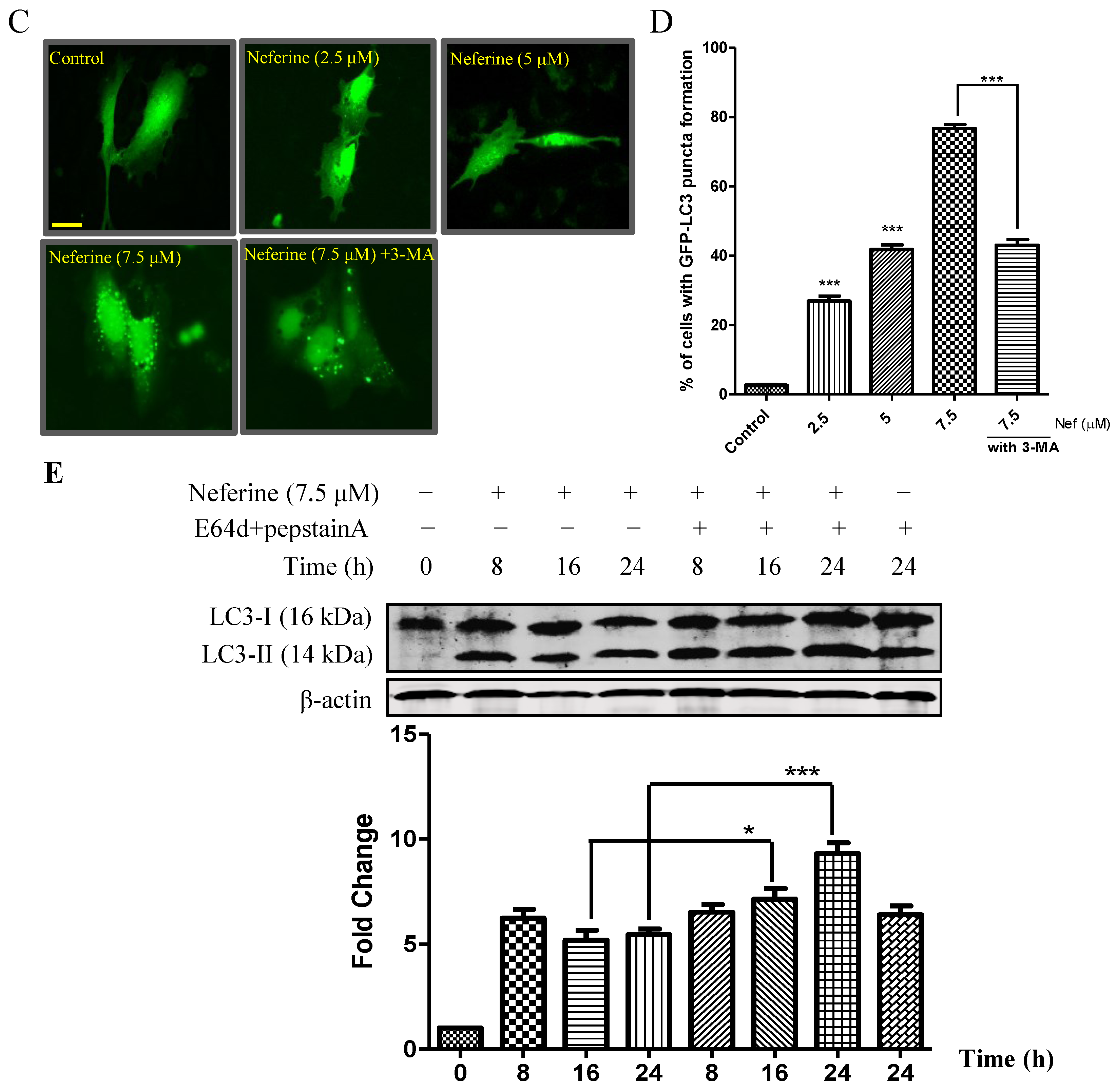
2.1. Neferine Induces Autophagy in PC-12 Cells


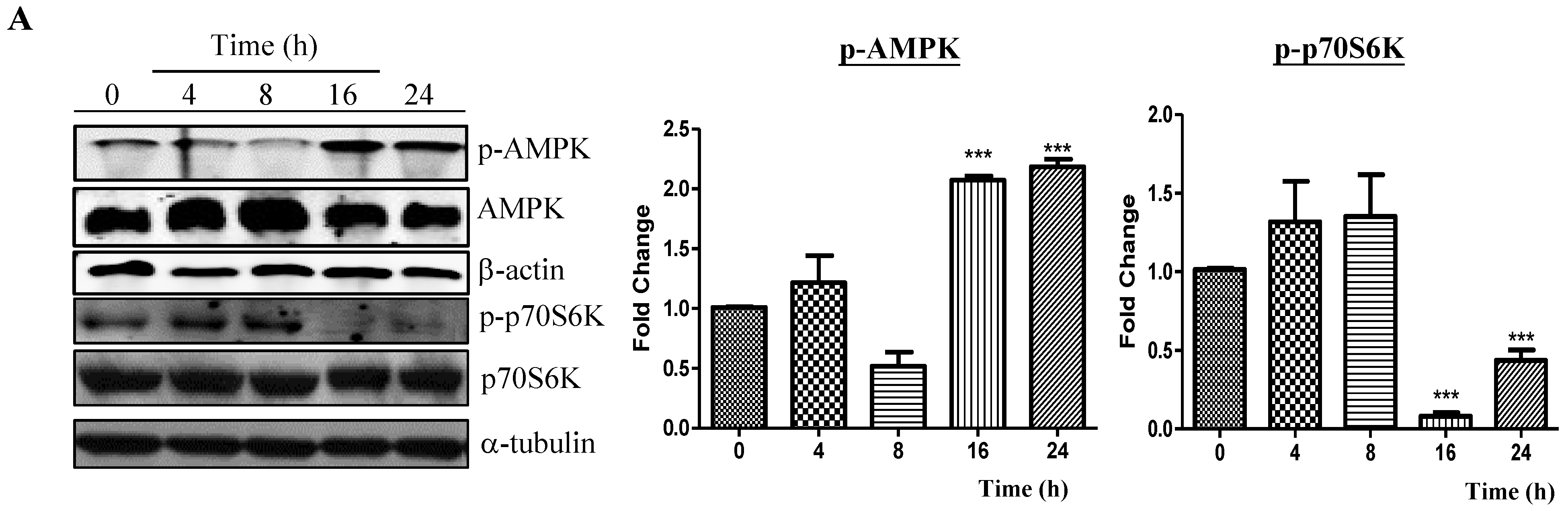
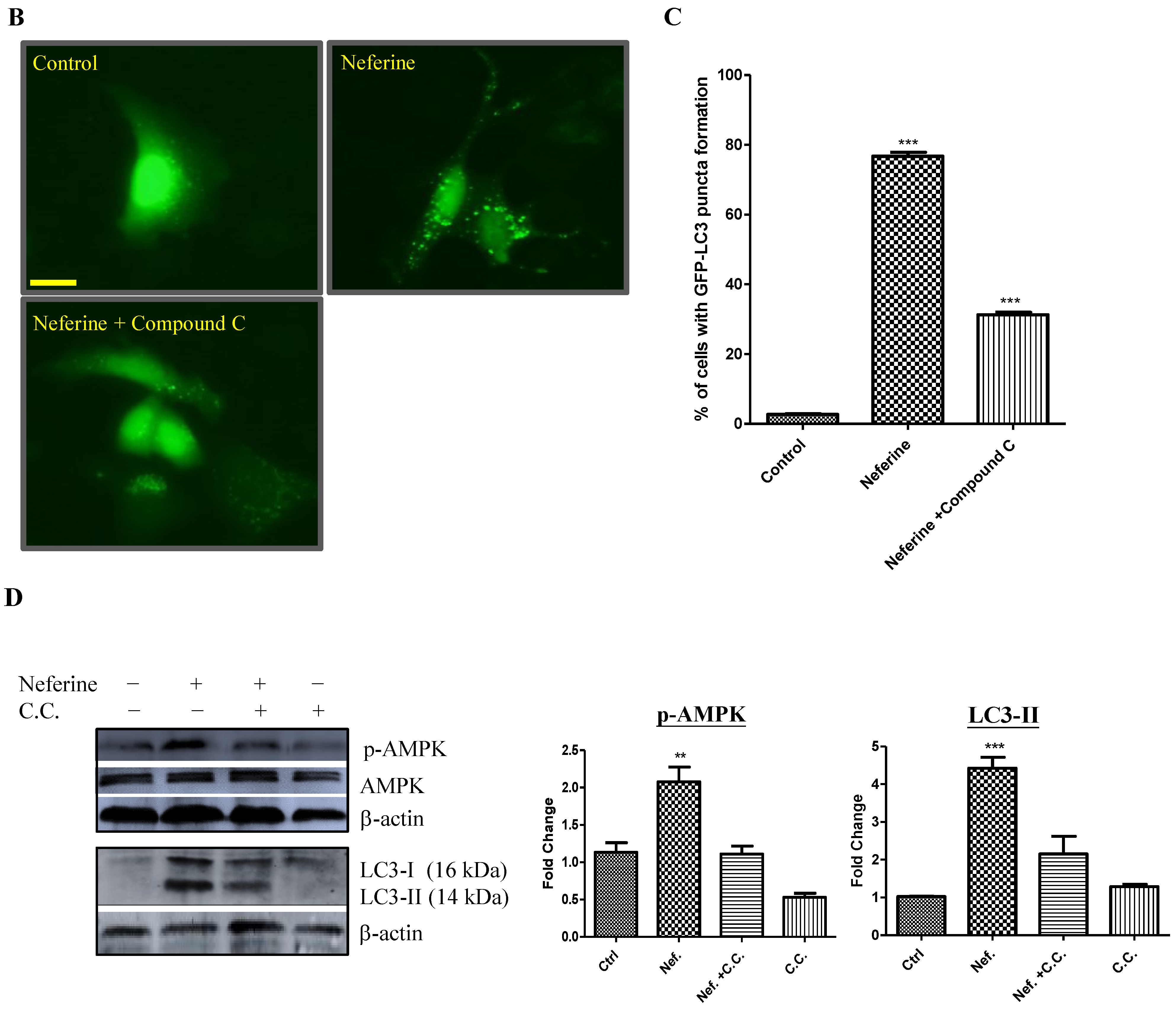
2.2. Neferine Induces Autophagy through the mTOR-AMPK-Dependent Pathway


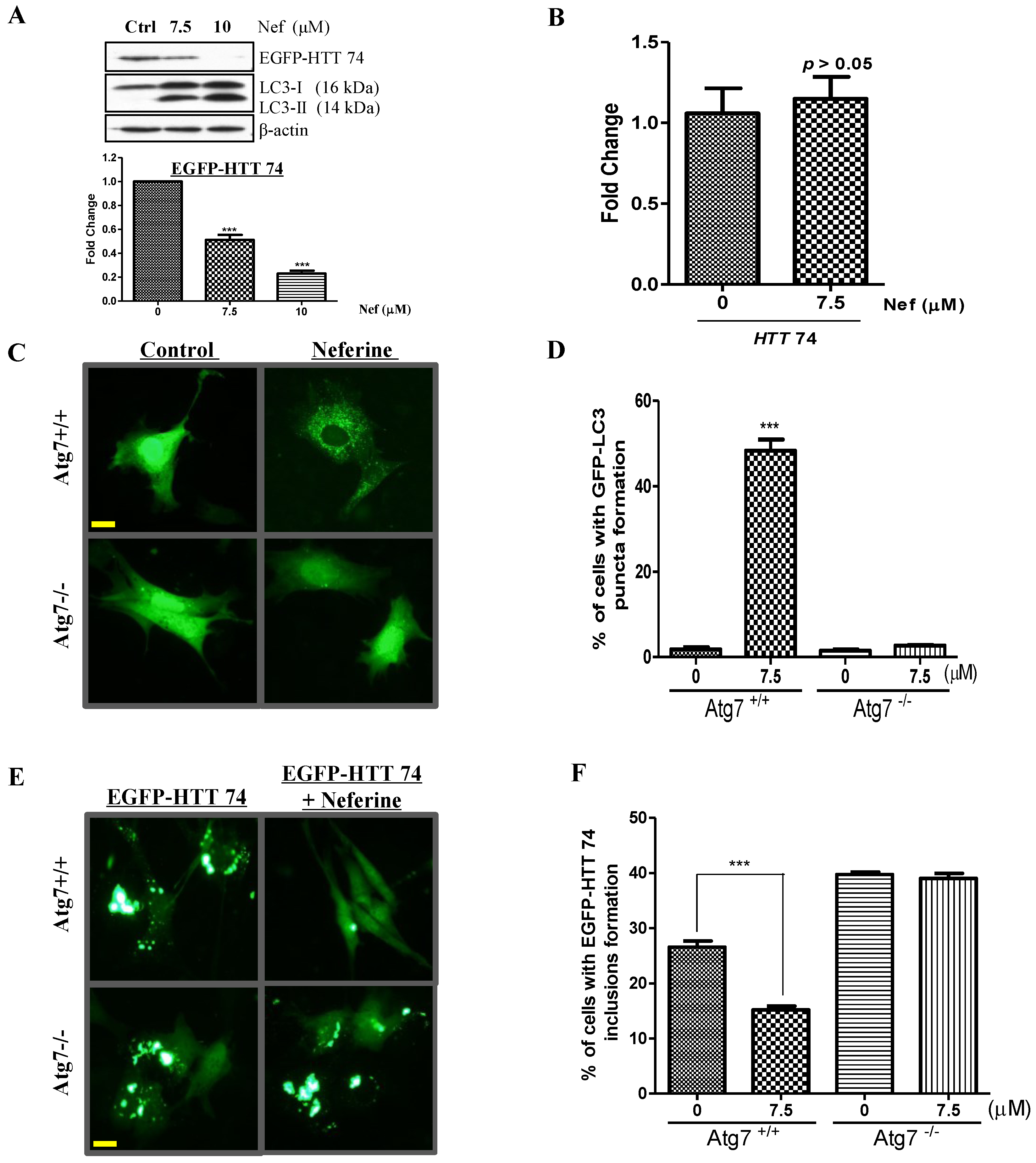
2.3. Neferine Enhances the Clearance of Huntingtin Protein and Inclusion in an Atg7-Dependent Manner


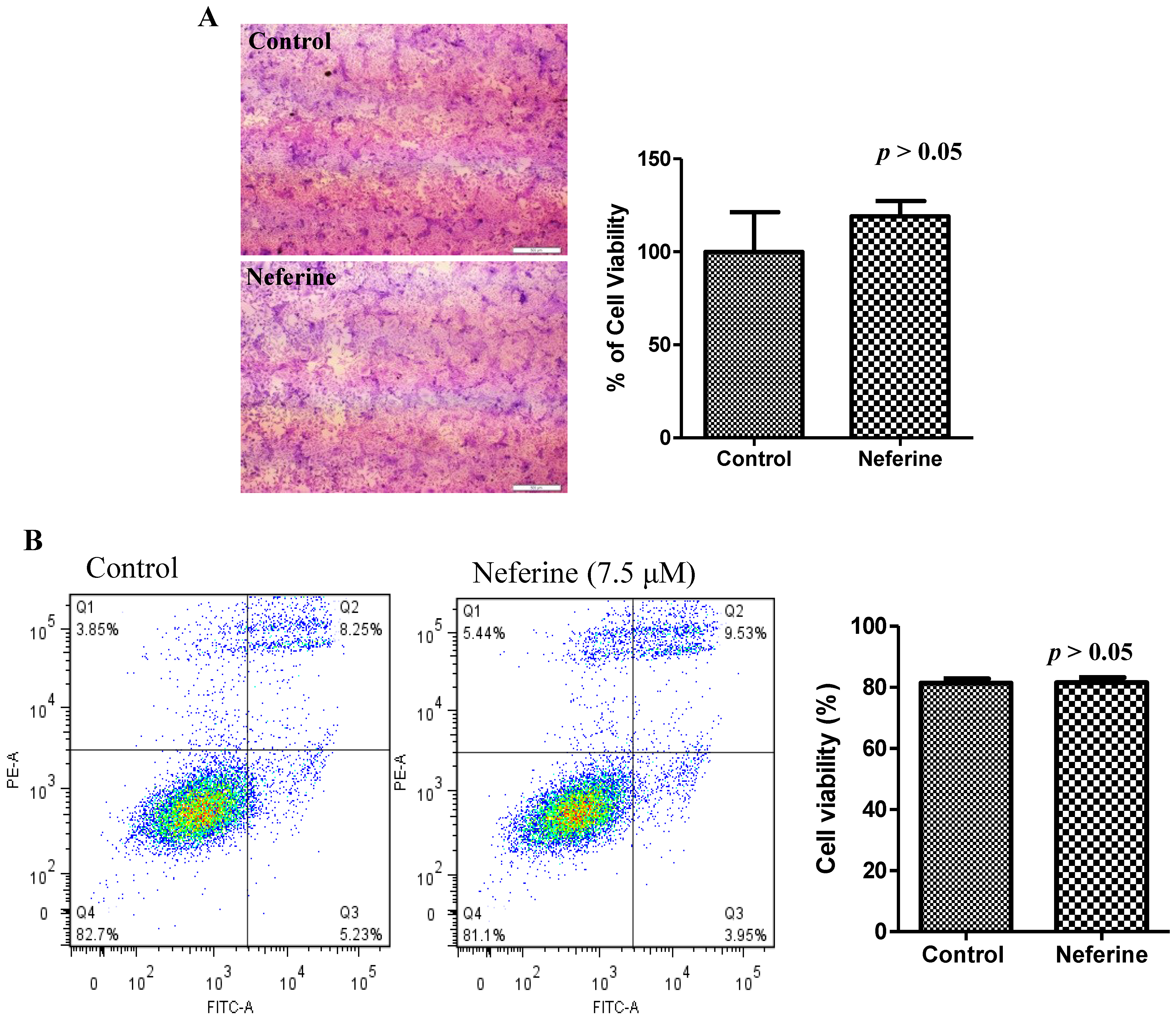
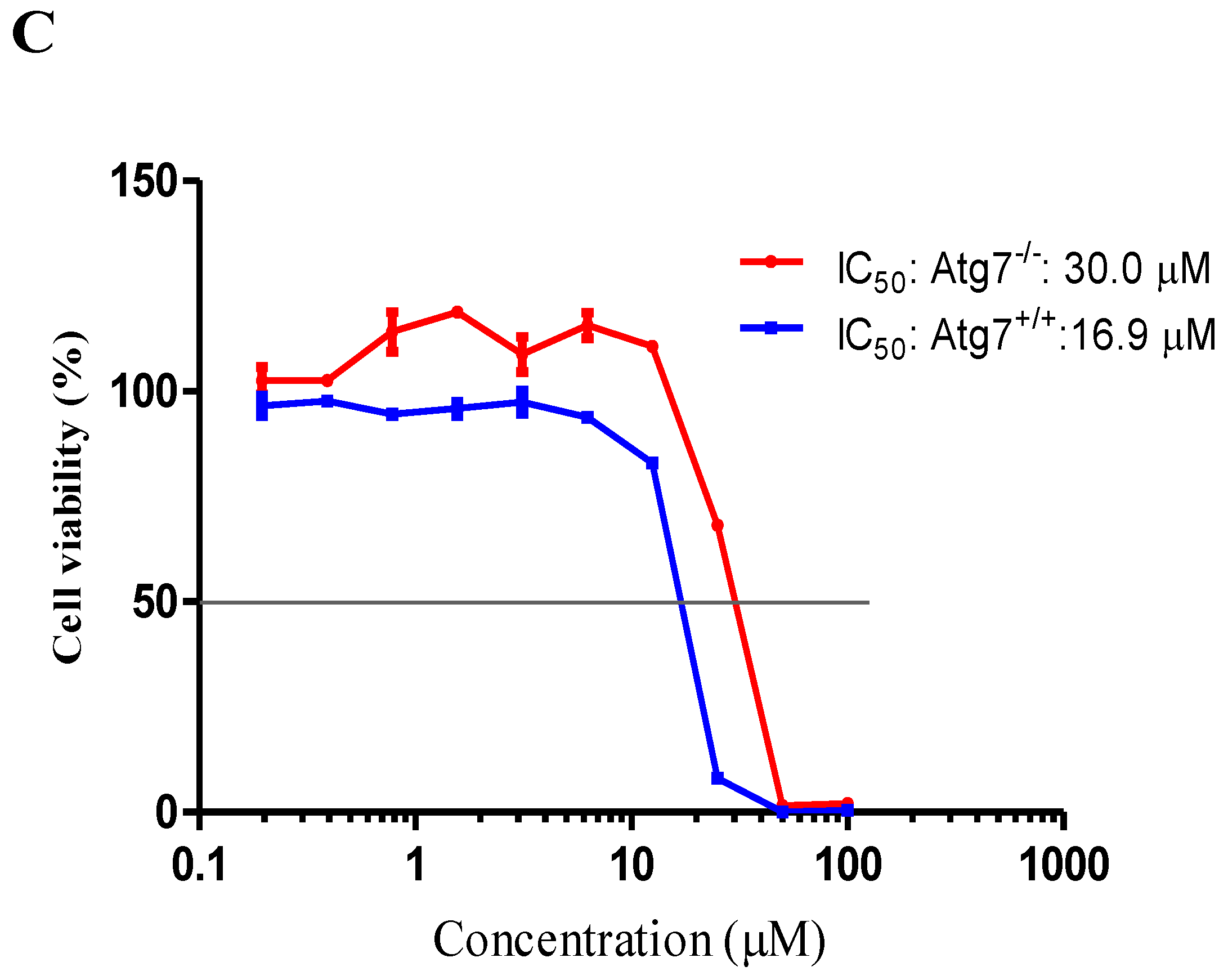
2.4. Neferine Confers No Obvious Cytotoxicity at Its Effective Working Concentration


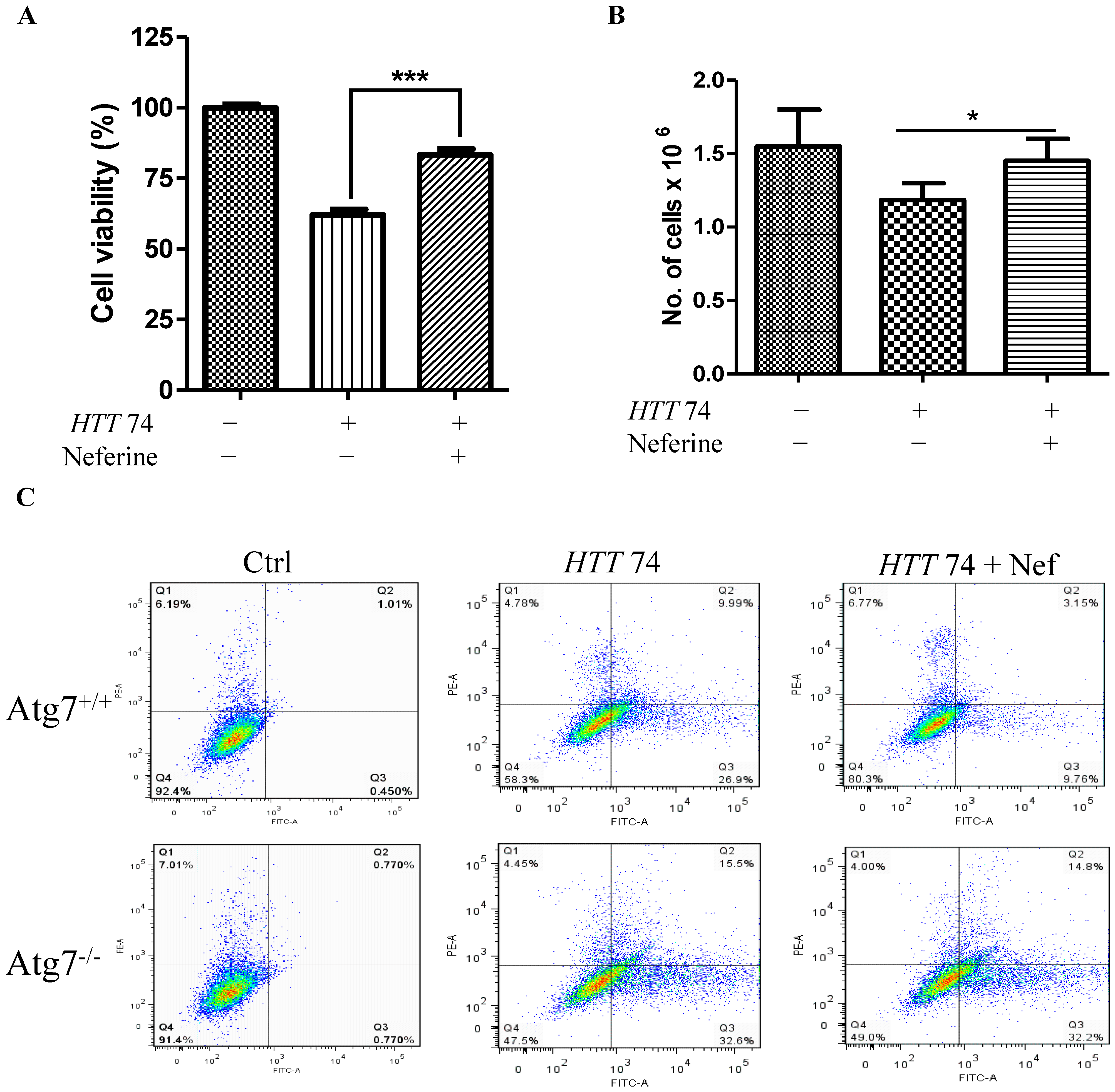
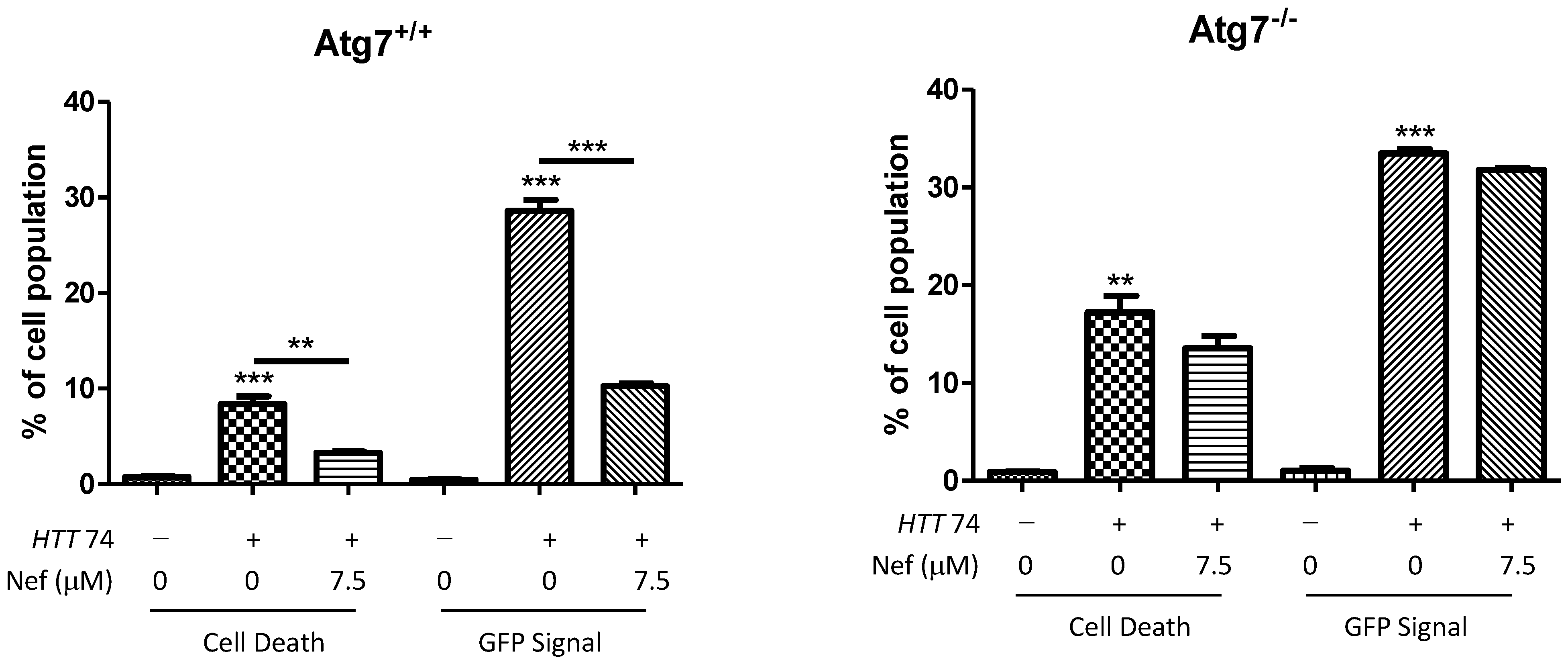
2.5. Neferine Reduces the Toxicity of Mutant Huntingtin in Cells


3. Experimental Section
3.1. Reagents, Chemicals, Antibodies and Plasmids
3.2. Cell Culture
3.3. Quantification of GFP-LC3 Puncta Formation
3.4. Cytotoxicity Assays and Flow Cytometry
3.5. Western Blot Analysis
3.6. Detection of Mutant Huntingtin Protein and Inclusions
3.7. Real-Time PCR Analysis
3.8. Statistical Analysis
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Fecto, F.; Yan, J.; Vemula, S.P.; Liu, E.; Yang, Y.; Chen, W.; Zheng, J.G.; Shi, Y.; Siddique, N.; Arrat, H.; et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2011, 68, 1440–1446. [Google Scholar]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. alpha-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar]
- Shibata, M.; Lu, T.; Furuya, T.; Degterev, A.; Mizushima, N.; Yoshimori, T.; MacDonald, M.; Yankner, B.; Yuan, J. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J. Biol. Chem. 2006, 281, 14474–14485. [Google Scholar] [CrossRef]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.; Menzies, F.M.; Renna, M.; Acevedo-Arozena, A.; Corrochano, S.; Sadiq, O.; Brown, S.D.; Rubinsztein, D.C. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2010, 19, 2144–2153. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Perlstein, E.O.; Imarisio, S.; Pineau, S.; Cordenier, A.; Maglathlin, R.L.; Webster, J.A.; Lewis, T.A.; O’kane, C.J.; Schreiber, S.L.; et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat. Chem. Biol. 2007, 3, 331–338. [Google Scholar]
- Rubinsztein, D.C. Lessons from animal models of Huntington’s disease. Trends Genet. 2002, 18, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.; Afaq, F.; Khan, N.; Johnson, J.J.; Khusro, F.H.; Mukhtar, H. Fisetin induces autophagic cell death through suppression of mTOR signaling pathway in prostate cancer cells. Carcinogenesis 2010, 31, 1424–1433. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Gestwicki, J.E.; Murphy, L.O.; Klionsky, D.J. Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 2007, 6, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Pallet, N.; Legendre, C. Adverse events associated with mTOR inhibitors. Expert Opin. Drug Saf. 2012, 12, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.Y.; Zhang, S.D.; Liu, S.; He, B.M. Protective effect of neferine on endothelial cell nitric oxide production induced by lysophosphatidylcholine: The role of the DDAH-ADMA pathway. Can. J. Physiol. Pharmacol. 2011, 89, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Li, Y.; Cao, P.; Xie, Z.; Qin, Z. Synergistic effect of hyperthermia and neferine on reverse multidrug resistance in adriamycin-resistant SGC7901/ADM gastric cancer cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2011, 31, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Poornima, P.; Weng, C.F.; Padma, V.V. Neferine from Nelumbo nucifera induces autophagy through the inhibition of PI3K/Akt/mTOR pathway and ROS hyper generation in A549 cells. Food Chem. 2013, 141, 3598–3605. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.A.; Jin, S.E.; Choi, R.J.; Kim, D.H.; Kim, Y.S.; Ryu, J.H.; Kim, D.W.; Son, Y.K.; Park, J.J.; Choi, J.S. Anti-amnesic activity of neferine with antioxidant and anti-inflammatory capacities, as well as inhibition of ChEs and BACE1. Life Sci. 2010, 87, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar]
- Wong, V.K.; Li, T.; Law, B.Y.; Ma, E.D.; Yip, N.C.; Michelangeli, F.; Law, C.K.; Zhang, M.M.; Lam, K.Y.; Chan, P.L.; et al. Saikosaponin-d, a novel SERCA inhibitor, induces autophagic cell death in apoptosis-defective cells. Cell Death Dis. 2013, 4, e720. [Google Scholar]
- Wu, Y.T.; Tan, H.L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005, 1, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Law, B.Y.; Wang, M.; Ma, D.L.; Al-Mousa, F.; Michelangeli, F.; Cheng, S.H.; Ng, M.H.; To, K.F.; Mok, A.Y.; Ko, R.Y.; et al. Alisol B, a novel inhibitor of the sarcoplasmic/endoplasmic reticulum Ca(2+) ATPase pump, induces autophagy, endoplasmic reticulum stress, and apoptosis. Mol. Cancer Ther. 2010, 9, 718–730. [Google Scholar]
- Law, B.Y.; Chan, W.K.; Xu, S.W.; Wang, J.R.; Bai, L.P.; Liu, L.; Wong, V.K. Natural small-molecule enhancers of autophagy induce autophagic cell death in apoptosis-defective cells. Sci. Rep. 2014, 4, 5510. [Google Scholar] [CrossRef] [PubMed]
- Bjorkoy, G.; Lamark, T.; Pankiv, S.; Overvatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Klionsky, D.J. Protein turnover via autophagy: Implications for metabolism. Annu. Rev. Nutr. 2007, 27, 19–40. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, H.; Nieminen, A.; Saarela, M.; Kallioniemi, A.; Klefstrom, J.; Hautaniemi, S.; Monni, O. Deciphering downstream gene targets of PI3K/mTOR/p70S6K pathway in breast cancer. BMC Genomics 2008, 9, 348. [Google Scholar] [CrossRef] [PubMed]
- Hindle, J.V. Ageing, neurodegeneration and Parkinson’s disease. Age Ageing 2010, 39, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- McEwan, D.G.; Dikic, I. The Three Musketeers of Autophagy: Phosphorylation, ubiquitylation and acetylation. Trends Cell Biol. 2011, 21, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.G.; Wong, V.K.; Xu, S.W.; Chan, W.K.; Ng, C.I.; Liu, L.; Law, B.Y. Onjisaponin B Derived from Radix Polygalae Enhances Autophagy and Accelerates the Degradation of Mutant alpha-Synuclein and Huntingtin in PC-12 Cells. Int. J. Mol. Sci. 2013, 14, 22618–22641. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [PubMed]
- Narain, Y.; Wyttenbach, A.; Rankin, J.; Furlong, R.A.; Rubinsztein, D.C. A molecular investigation of true dominance in Huntington’s disease. J. Med. Genet. 1999, 36, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; DiFiglia, M.; Heintz, N.; Nixon, R.A.; Qin, Z.H.; Ravikumar, B.; Stefanis, L.; Tolkovsky, A. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 2005, 1, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Renna, M.; Jimenez-Sanchez, M.; Sarkar, S.; Rubinsztein, D.C. Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases. J. Biol. Chem. 2010, 285, 11061–11067. [Google Scholar] [CrossRef] [PubMed]
- Nagata, E.; Saiardi, A.; Tsukamoto, H.; Okada, Y.; Itoh, Y.; Satoh, T.; Itoh, J.; Margolis, R.L.; Takizawa, S.; Sawa, A.; et al. Inositol hexakisphosphate kinases induce cell death in Huntington disease. J. Biol. Chem. 2011, 286, 26680–26686. [Google Scholar]
- Nagata, E.; Saiardi, A.; Tsukamoto, H.; Satoh, T.; Itoh, Y.; Itoh, J.; Shibata, M.; Takizawa, S.; Takagi, S. Inositol hexakisphosphate kinases promote autophagy. Int. J. Biochem. Cell Biol. 2010, 42, 2065–2071. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.D.; Ranum, L.P. Repeat-associated non-ATG (RAN) translation in neurological disease. Hum. Mol. Genet. 2013, 22, R45–R51. [Google Scholar] [CrossRef] [PubMed]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.K.; Yang, H.Q.; Chen, S.D. Traditional Chinese medicine: A promising candidate for the treatment of Alzheimer’s disease. Transl. Neurodegener. 2013, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, H.; Zhao, X.; Lin, X.; Tan, C.; Cao, G.; Wang, Z. Neuroprotective effects of salidroside against beta-amyloid-induced oxidative stress in SH-SY5Y human neuroblastoma cells. Neurochem. Int. 2010, 57, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar]
- Ved, H.S.; Koenig, M.L.; Dave, J.R.; Doctor, B.P. Huperzine A, a potential therapeutic agent for dementia, reduces neuronal cell death caused by glutamate. Neuroreport 1997, 8, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, V.K.W.; Wu, A.G.; Wang, J.R.; Liu, L.; Law, B.Y.-K. Neferine Attenuates the Protein Level and Toxicity of Mutant Huntingtin in PC-12 Cells via Induction of Autophagy. Molecules 2015, 20, 3496-3514. https://doi.org/10.3390/molecules20033496
Wong VKW, Wu AG, Wang JR, Liu L, Law BY-K. Neferine Attenuates the Protein Level and Toxicity of Mutant Huntingtin in PC-12 Cells via Induction of Autophagy. Molecules. 2015; 20(3):3496-3514. https://doi.org/10.3390/molecules20033496
Chicago/Turabian StyleWong, Vincent Kam Wai, An Guo Wu, Jing Rong Wang, Liang Liu, and Betty Yuen-Kwan Law. 2015. "Neferine Attenuates the Protein Level and Toxicity of Mutant Huntingtin in PC-12 Cells via Induction of Autophagy" Molecules 20, no. 3: 3496-3514. https://doi.org/10.3390/molecules20033496
APA StyleWong, V. K. W., Wu, A. G., Wang, J. R., Liu, L., & Law, B. Y. -K. (2015). Neferine Attenuates the Protein Level and Toxicity of Mutant Huntingtin in PC-12 Cells via Induction of Autophagy. Molecules, 20(3), 3496-3514. https://doi.org/10.3390/molecules20033496