
Anisotropy in Bone Demineralization Revealed by Polarized Far-IR Spectroscopy

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Design of a Sample Chamber for Hydrated Biological Samples
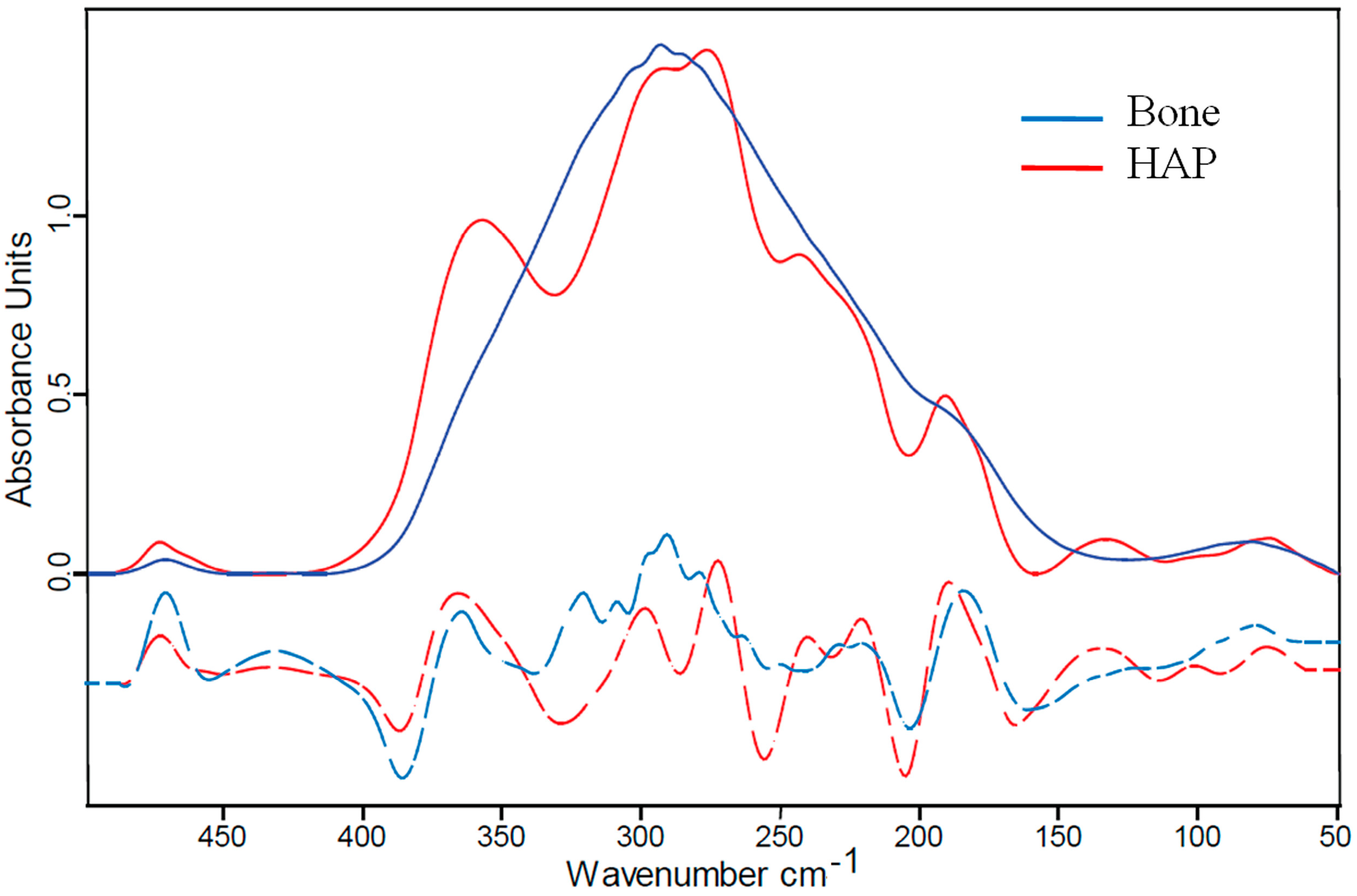
2.2. Far IR Spectra of Bone and Its Components (Hydroxyapaptite and Collagen)
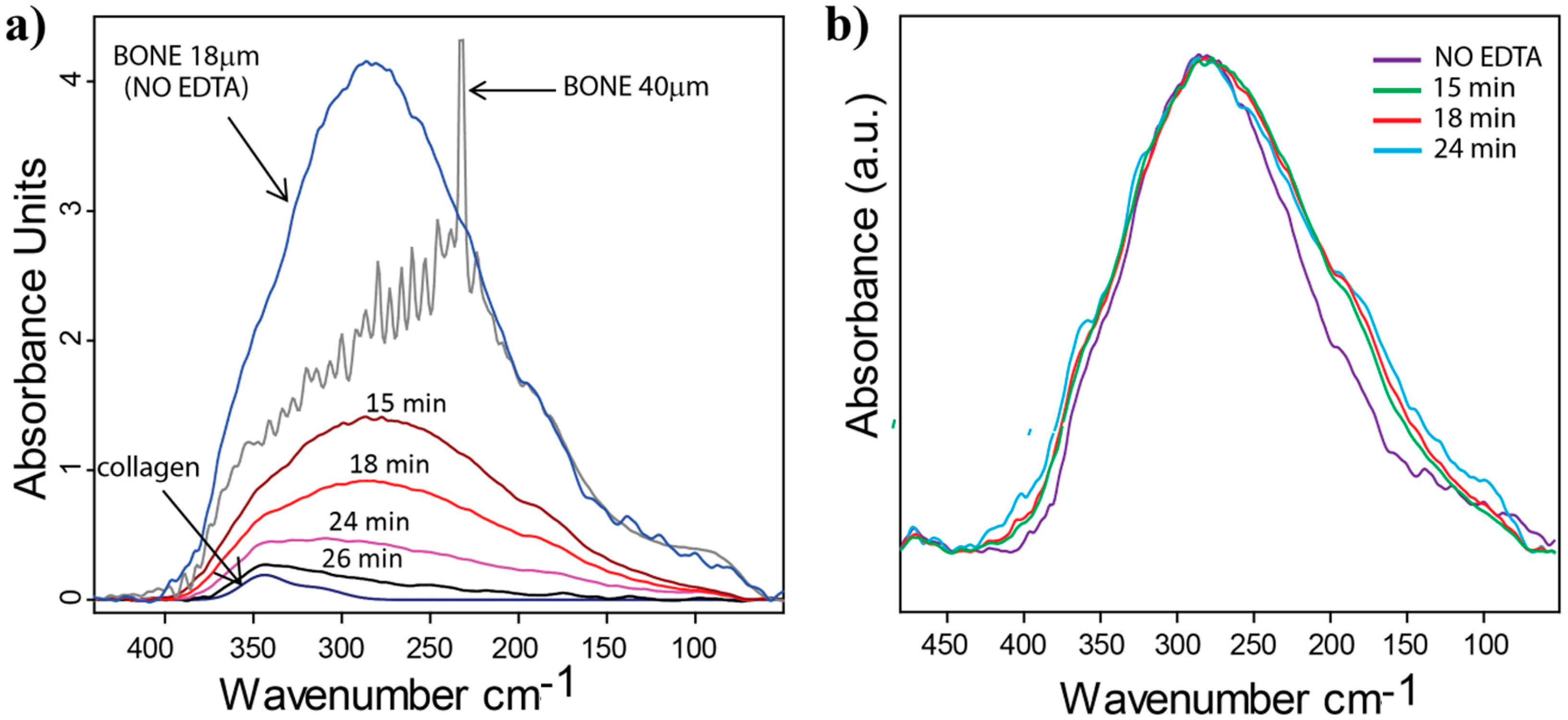
2.3. Far-IR Spectroscopy of Bone Demineralization



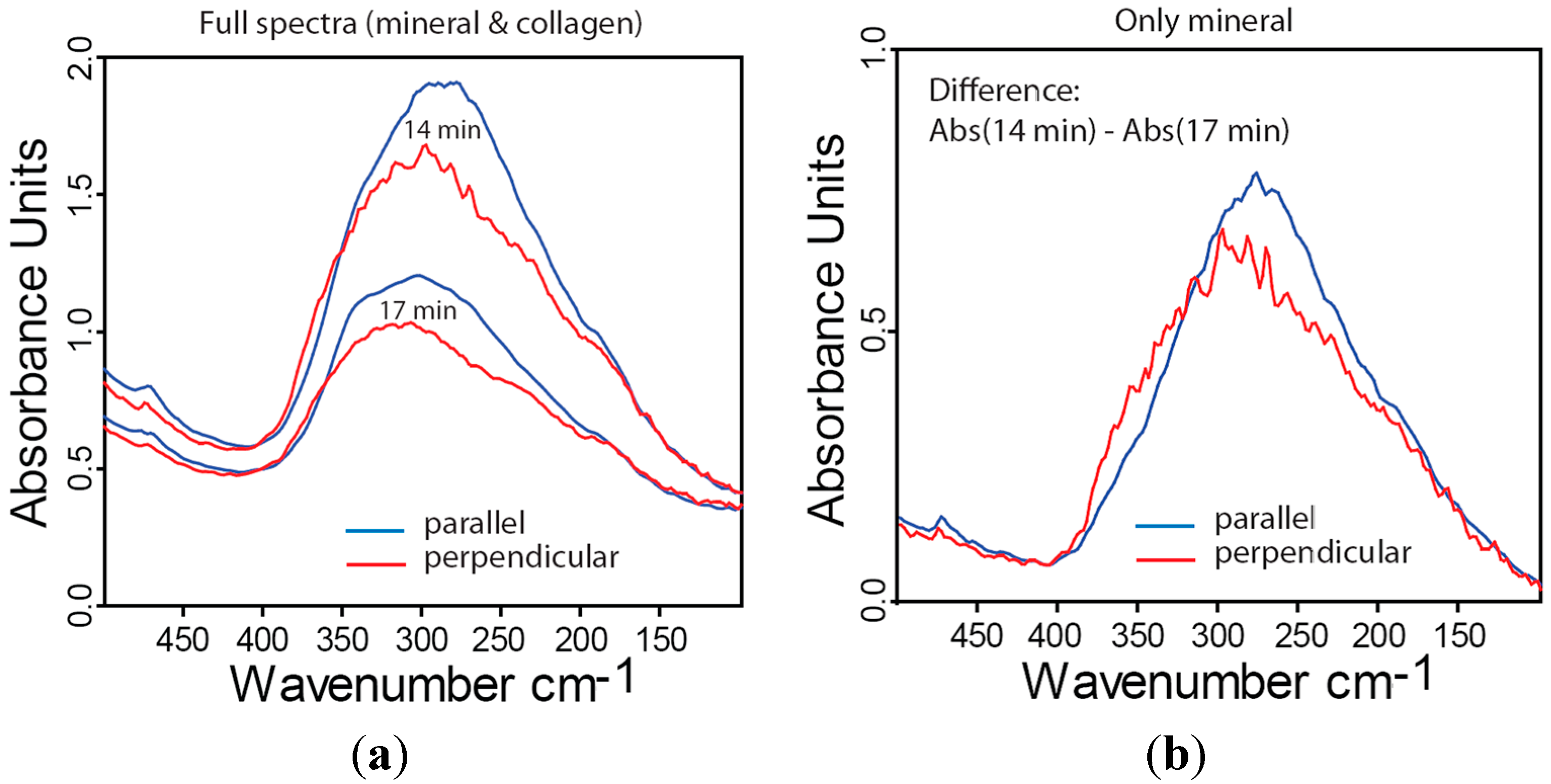
2.4. Far-IR Linear Dichroism of Bone and Bone Mineral

2.5. Far-IR Linear Dichroism of Collagen from Fully Demineralized Bone

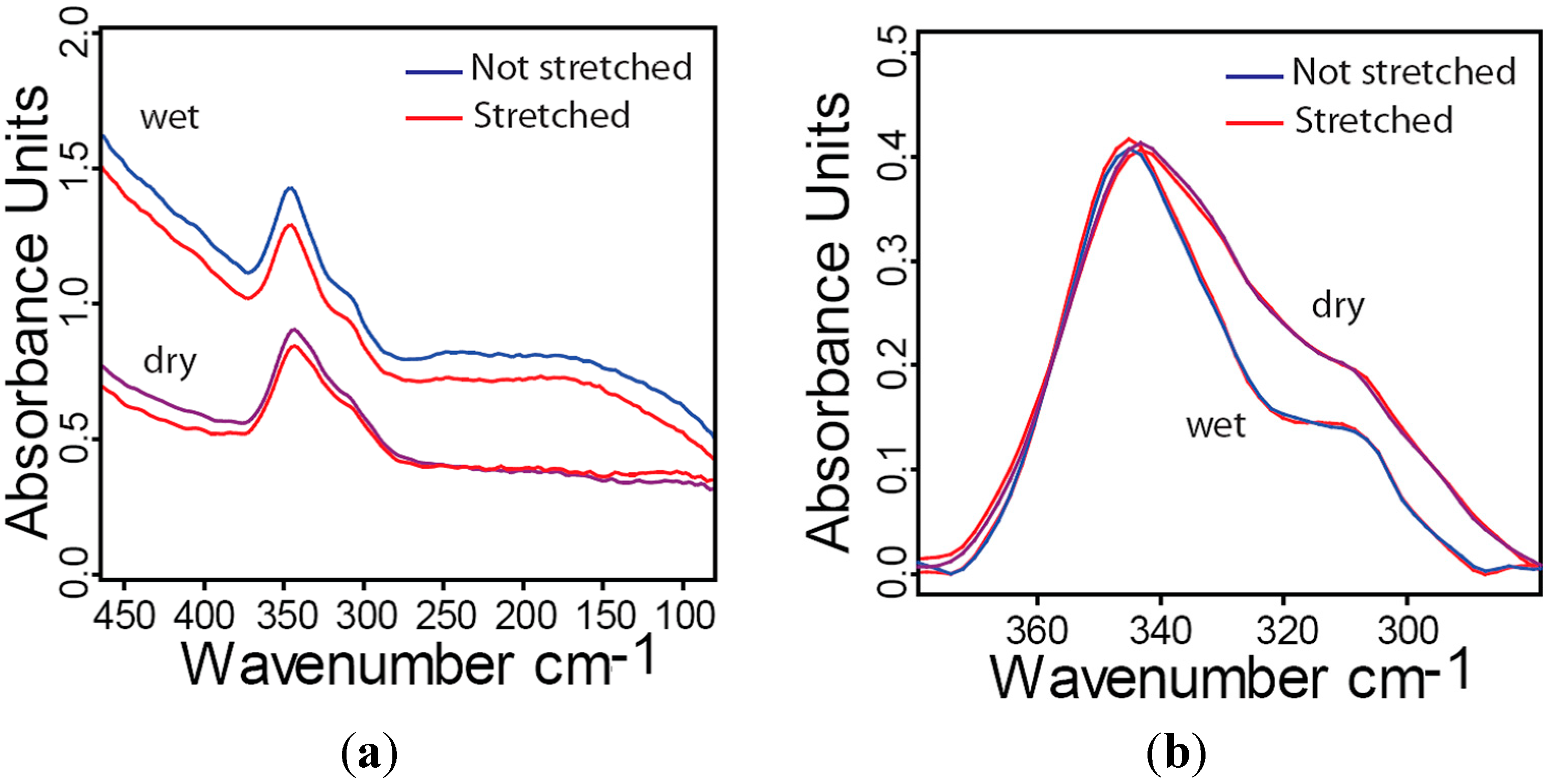
2.6. Influence of Hydration and Stress on Far-IR Spectra of Collagen

3. Discussion
4. Experimental Section
4.1. Far-IR at BESSY
4.2. Controlled Environment Sample Chamber
4.3. Sample Preparation and Measurement Procedure
4.4. Data Evaluation
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Weiner, S.; Wagner, H.D. The material bone: Structure mechanical function relations. Ann. Rev. Mater. Sci. 1998, 28, 271–298. [Google Scholar] [CrossRef]
- Fratzl, P.; Weinkamer, R. Nature’s hierarchical materials. Prog. Mater. Sci. 2007, 52, 1263–1334. [Google Scholar] [CrossRef]
- Roschger, P.; Paschalis, E.P.; Fratzl, P.; Klaushofer, K. Bone mineralization density distribution in health and disease. Bone 2008, 42, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Currey, J.D. Bones: Structure and Mechanics; Princeton University Press: Princeton, NJ, USA, 2002. [Google Scholar]
- Ruffoni, D.; Fratzl, P.; Roschger, P.; Klaushofer, K.; Weinkamer, R. The bone mineralization density distribution as a fingerprint of the mineralization process. Bone 2007, 40, 1308–1319. [Google Scholar] [CrossRef] [PubMed]
- Nudelman, F.; Pieterse, K.; George, A.; Bomans, P.H.H.; Friedrich, H.; Brylka, L.J.; Hilbers, P.A.J.; de With, G.; Sommerdijk, N.A.J.M.; Nudelman, F.; et al. The role of collagen in bone apatite formation in the presence of hydroxyapatite nucleation inhibitors. Nat. Mater. 2010, 9, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Mahamid, J.; Sharir, A.; Gur, D.; Zelzer, E.; Addadi, L.; Weiner, S. Bone mineralization proceeds through intracellular calcium phosphate loaded vesicles: A cryo-electron microscopy study. J. Struct. Biol. 2011, 174, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Bennet, M.; Akiva, A.; Faivre, D.; Malkinson, G.; Yaniv, K.; Abdelilah-Seyfried, S.; Fratzl, P.; Masic, A. Simultaneous Raman Microspectroscopy and Fluorescence Imaging of Bone Mineralization in Living Zebrafish Larvae. Biophys. J. 2014, 106, L17–L19. [Google Scholar] [CrossRef] [PubMed]
- Crane, N.J.; Popescu, V.; Morris, M.D.; Steenhuis, P.; Ignelzi, M.A., Jr. Raman spectroscopic evidence for octacalcium phosphate and other transient mineral species deposited during intramembranous mineralization. Bone 2006, 39, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.Y.; Chauret, N.; Cromlish, W.; Desmarais, S.; Duong le, T.; Falgueyret, J.P.; Kimmel, D.B.; Lamontagne, S.; Leger, S.; LeRiche, T.; et al. The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg. Med. Chem. Lett. 2008, 18, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.G.; Rogers, M.J. Bisphosphonates: From the laboratory to the clinic and back again. Bone 1999, 25, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Teitelbaum, S.L. Bone Resorption by Osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Ruffoni, D.; Lambers, F.M.; Schulte, F.A.; Kuhn, G.; Kollmannsberger, P.; Weinkamer, R.; Muller, R. Mineralization kinetics in murine trabecular bone quantified by time-lapsed in vivo micro-computed tomography. Bone 2013, 56, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Callis, G.; Sterchi, D. Decalcification of Bone: Literature Review and Practical Study of Various Decalcifying Agents. Methods, and Their Effects on Bone Histology. J. Histotechnol. 1998, 21, 49–58. [Google Scholar] [CrossRef]
- Bowman, S.M.; Zeind, J.; Gibson, L.J.; Hayes, W.C.; McMahon, T.A. The tensile behavior of demineralized bovine cortical bone. J. Biomech. 1996, 29, 1497–501. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.M.; Goh, J.C.; Karunanithy, R.; Low, S.L.; Das De, S.; Bose, K. Effect of decalcification on bone mineral content and bending strength of feline femur. Calcif. Tissue Int. 1995, 56, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Unal, M.; Yang, S.; Akkus, O. Molecular spectroscopic identification of the water compartments in bone. Bone 2014, 67, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Hassenkam, T.; Fantner, G.E.; Cutroni, J.A.; Weaver, J.C.; Morse, D.E.; Hansma, P.K. High-resolution AFM imaging of intact and fractured trabecular bone. Bone 2004, 35, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Heymann, J.A. W.; Hayles, M.; Gestmann, I.; Giannuzzi, L.A.; Lich, B.; Subramaniam, S. Site-specific 3D imaging of cells and tissues with a dual beam microscope. J. Struct. Biol. 2006, 155, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Reznikov, N.; Shahar, R.; Weiner, S. Three-dimensional structure of human lamellar bone: The presence of two different materials and new insights into the hierarchical organization. Bone 2014, 59, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Reznikov, N.; Almany-Magal, R.; Shahar, R.; Weiner, S. Three-dimensional imaging of collagen fibril organization in rat circumferential lamellar bone using a dual beam electron microscope reveals ordered and disordered sub-lamellar structures. Bone 2013, 52, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Fratzl, P.; Groschner, M.; Vogl, G.; Plenk, H., Jr.; Eschberger, J.; Fratzl-Zelman, N.; Koller, K.; Klaushofer, K. Mineral crystals in calcified tissues: A comparative study by SAXS. J. Bone Miner. Res. 1992, 7, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Pabisch, S.; Wagermaier, W.; Zander, T.; Li, C.; Fratzl, P. Imaging the nanostructure of bone and dentin through small- and wide-angle X-ray scattering. Methods Enzymol. 2013, 532, 391–413. [Google Scholar] [PubMed]
- Wagermaier, W.; Gupta, H.S.; Gourrier, A.; Burghammer, M.; Roschger, P.; Fratzl, P. Spiral twisting of fiber orientation inside bone lamellae. Biointerphases 2006, 1, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Li, C.; Manjubala, I.; Wagermaier, W.; Kuhnisch, J.; Kolanczyk, M.; Mundlos, S.; Knaus, P.; Fratzl, P. Fetal and postnatal mouse bone tissue contains more calcium than is present in hydroxyapatite. J. Struct. Biol. 2011, 176, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Gupta, H.S.; Seto, J.; Wagermaier, W.; Zaslansky, P.; Boesecke, P.; Fratzl, P. Cooperative deformation of mineral and collagen in bone at the nanoscale. Proc. Natl. Acad. Sci. USA 2006, 103, 17741–17746. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.M.; Vairavamurthy, V.; Chance, M.R.; Mendelsohn, R.; Paschalis, E.P.; Betts, F.; Boskey, A.L. In situ analysis of mineral content and crystallinity in bone using infrared micro-spectroscopy of the ν4 PO43- vibration. Biochim. Biophys. Acta Gen. Subj. 2001, 1527, 11–19. [Google Scholar] [CrossRef]
- Paschalis, E.P.; Betts, F.; DiCarlo, E.; Mendelsohn, R.; Boskey, A.L. FTIR microspectroscopic analysis of normal human cortical and trabecular bone. Calcif. Tissue Int. 1997, 61, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Boskey, A.; Mendelsohn, R. Infrared analysis of bone in health and disease. J. Biomed. Opt. 2005, 10, 031102–0311029. [Google Scholar] [CrossRef] [PubMed]
- Carden, A.; Morris, M.D. Application of vibrational spectroscopy to the study of mineralized tissues (Review). J. Biomed. Opt. 2000, 5, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Duboeuf, F.; Burt-Pichat, B.; Farlay, D.; Suy, P.; Truy, E.; Boivin, G. Bone quality and biomechanical function: A lesson from human ossicles. Bone 2015, 73, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Takata, S.; Shibata, A.; Yonezu, H.; Yamada, T.; Takahashi, M.; Abbaspour, A.; Yasui, N. Biophysic evaluation of bone quality-application of Fourier transform infrared spectroscopy and phosphorus-31 solid-state nuclear magnetic resonance spectroscopy. J. Med. Investig. 2004, 51, 133–138. [Google Scholar] [CrossRef]
- Gamsjaeger, S.; Mendelsohn, R.; Boskey, A.L.; Gourion-Arsiquaud, S.; Klaushofer, K.; Paschalis, E.P. Vibrational Spectroscopic Imaging for the Evaluation of Matrix and Mineral Chemistry. Curr. Osteoporos. Rep. 2014, 12, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Makowski, A.J.; Patil, C.A.; Mahadevan-Jansen, A.; Nyman, J.S. Polarization control of Raman spectroscopy optimizes the assessment of bone tissue. J. Biomed. Opt. 2013, 18, 055005. [Google Scholar] [CrossRef]
- Janko, M.; Davydovskaya, P.; Bauer, M.; Zink, A.; Stark, R.W. Anisotropic Raman scattering in collagen bundles. Opt. Lett. 2010, 35, 2765–2767. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, S.J.; Landis, W.J.; Boskey, A.L.; Mendelsohn, R. Polarized FT-IR microscopy of calcified turkey leg tendon. Connect. Tissue Res. 1996, 34, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Kazanci, M.; Roschger, P.; Paschalis, E.P.; Klaushofer, K.; Fratzl, P. Bone osteonal tissues by Raman spectral mapping: Orientation-composition. J. Struct. Biol. 2006, 156, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Kazanci, M.; Wagner, H.D.; Manjubala, N.I.; Gupta, H.S.; Paschalis, E.; Roschger, P.; Fratzl, P. Raman imaging of two orthogonal planes within cortical bone. Bone 2007, 41, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Carnelli, D.; Vena, P.; Dao, M.; Ortiz, C.; Contro, R. Orientation and size-dependent mechanical modulation within individual secondary osteons in cortical bone tissue. J. R. Soc. Interface 2013, 10, 20120953. [Google Scholar] [CrossRef] [PubMed]
- Falgayrac, G.; Facq, S.; Leroy, G.; Cortet, B.; Penel, G. New method for Raman investigation of the orientation of collagen fibrils and crystallites in the Haversian system of bone. Appl. Spectrosc. 2010, 64, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, M.; Sahar, N.D.; Wilson, R.H.; Mycek, M.A.; Pleshko, N.; Kohn, D.H.; Morris, M.D. Quantitative polarized Raman spectroscopy in highly turbid bone tissue. J. Biomed. Opt. 2010, 15, 037001. [Google Scholar] [CrossRef] [PubMed]
- Galvis, L.; Dunlop, J.W.C.; Duda, G.; Fratzl, P.; Masic, A. Polarized Raman Anisotropic Response of Collagen in Tendon: Towards 3D Orientation Mapping of Collagen in Tissues. PLoS One 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Gamsjaeger, S.; Masic, A.; Roschger, P.; Kazanci, M.; Dunlop, J.W.C.; Klaushofer, K.; Paschalis, E.P.; Fratzl, P. Cortical bone composition and orientation as a function of animal and tissue age in mice by Raman spectroscopy. Bone 2010, 47, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Masic, A.; Bertinetti, L.; Schuetz, R.; Galvis, L.; Timofeeva, N.; Dunlop, J.W.C.; Seto, J.; Hartmann, M.A.; Fratzl, P. Observations of Multiscale, Stress-Induced Changes of Collagen Orientation in Tendon by Polarized Raman Spectroscopy. Biomacromolecules 2011, 12, 3989–3996. [Google Scholar] [CrossRef] [PubMed]
- Schrof, S.; Varga, P.; Galvis, L.; Raum, K.; Masic, A. 3D Raman mapping of the collagen fibril orientation in human osteonal lamellae. J. Struct. Biol. 2014, 187, 266–275. [Google Scholar] [CrossRef]
- Mantsch, H.H.; Naumann, D. Terahertz spectroscopy: The renaissance of far infrared spectroscopy. J. Mol. Struct. 2010, 964, 1–4. [Google Scholar] [CrossRef]
- Gordon, P.L.; Huang, C.; Lord, R.C.; Yannas, I.V. Far-infrared spectrum of collagen. Macromolecules 1974, 7, 954. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, M.; Saito, S.; Fukunaga, K.; Hosako, I. Absorption Properties of Oriented Collagen in Terahertz Band. In Proceedings of the 2011 36th International Conference on Infrared, Millimeter, and Terahertz Waves (Irmmw-Thz), Houston, TX, USA, 2–7 October 2011.
- Mizuno, M.; Yamada, A.; Fukunaga, K.; Kojima, H. Terahertz observation of salt penetration in collagen fibers. In Proceedings of the 2014 39th International Conference on Infrared, Millimeter, and Terahertz waves (IRMMW-THz), Tucson, AZ, USA, 14–19 September 2014; pp. 1–2.
- Knyazev, B.A.; Gerasimov, V.V.; Gonchar, A.M.; Kolosova, N.G. Using of terahertz radiation for monitoring of senile osteoporosis development. In Proceedings of the 2007 Joint 32nd International Conference on Infrared and Millimeter Waves and 15th International Conference on Terahertz Electronics, Cardiff, UK, 2–9 September 2007; pp. 555–556.
- Stringer, M.R.; Lund, D.N.; Foulds, A.P.; Uddin, A.; Berry, E.; Miles, R.E.; Davies, A.G. The analysis of human cortical bone by terahertz time-domain spectroscopy. Phys. Med. Biol. 2005, 50, 3211. [Google Scholar] [CrossRef] [PubMed]
- Brusentsova, T.N.; Peale, R.E.; Maukonen, D.; Harlow, G.E.; Boesenberg, J.S.; Ebel, D. Far infrared spectroscopy of carbonate minerals. Am. Miner. 2010, 95, 1515–1522. [Google Scholar] [CrossRef]
- Angino, E.E. Far Infrared (500–30 Cm-1) Spectra of Some Carbonate Minerals. Am. Miner. 1967, 52, 137–148. [Google Scholar]
- Peatman, W.B.; Schade, U. A brilliant infrared light source at BESSY. Rev. Sci. Instrum. 2001, 72, 1620–1624. [Google Scholar] [CrossRef]
- Almany Magal, R.; Reznikov, N.; Shahar, R.; Weiner, S. Three-dimensional structure of minipig fibrolamellar bone: Adaptation to axial loading. J. Struct. Biol. 2014, 186, 253–264. [Google Scholar] [CrossRef]
- Locke, M. Structure of long bones in mammals. J. Morphol. 2004, 262, 546–565. [Google Scholar] [CrossRef] [PubMed]
- Masic, A.; Bertinetti, L.; Schuetz, R.; Chang, S.W.; Metzger, H.; Buehler, M.J.; Fratzl, P. Osmotic pressure induced tensile forces in tendon collagen. Nat. Commun. 2015, 6, 5942. [Google Scholar] [CrossRef] [PubMed]
- Balan, E.; Delattre, S.; Roche, D.; Segalen, L.; Morin, G.; Guillaumet, M.; Blanchard, M.; Lazzeri, M.; Brouder, C.; Salje, E.H. Line-broadening effects in the powder infrared spectrum of apatite. Phys. Chem. Miner. 2011, 38, 111–122. [Google Scholar] [CrossRef]
- Awonusi, A.; Morris, M.; Tecklenburg, M. Carbonate Assignment and Calibration in the Raman Spectrum of Apatite. Calcif. Tissue Int. 2007, 81, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Gupta, H.S.; Wagermaier, W.; Zickler, G.A.; Raz-Ben Aroush, D.; Funari, S.S.; Roschger, P.; Wagner, H.D.; Fratzl, P. Nanoscale Deformation Mechanisms in Bone. Nano Lett. 2005, 5, 2108–2111. [Google Scholar] [CrossRef] [PubMed]
- Puxkandl, R.; Zizak, I.; Paris, O.; Keckes, J.; Tesch, W.; Bernstorff, S.; Purslow, P.; Fratzl, P. Viscoelastic properties of collagen: Synchrotron radiation investigations and structural model. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2002, 357, 191–197. [Google Scholar] [CrossRef]
- Faist, J.; Capasso, F.; Sivco, D.L.; Sirtori, C.; Hutchinson, A.L.; Cho, A.Y. Quantum cascade laser. Science 1994, 264, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Lendl, B.; Frank, J.; Schindler, R.; Müller, A.; Beck, M.; Faist, J. Mid-infrared quantum cascade lasers for flow injection analysis. Anal. Chem. 2000, 72, 1645–1648. [Google Scholar] [CrossRef] [PubMed]
- Schade, U.; Roseler, A.; Korte, E.H.; Scheer, M.; Peatman, W.B. Measured characteristics of infrared edge radiation from BESSY II. Nucl. Instrum. Methods Phys. Res. Sect. A 2000, 455, 476–486. [Google Scholar] [CrossRef]
- Sample Availability: Samples are not available from authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schuetz, R.; Fix, D.; Schade, U.; Aziz, E.F.; Timofeeva, N.; Weinkamer, R.; Masic, A. Anisotropy in Bone Demineralization Revealed by Polarized Far-IR Spectroscopy. Molecules 2015, 20, 5835-5850. https://doi.org/10.3390/molecules20045835
Schuetz R, Fix D, Schade U, Aziz EF, Timofeeva N, Weinkamer R, Masic A. Anisotropy in Bone Demineralization Revealed by Polarized Far-IR Spectroscopy. Molecules. 2015; 20(4):5835-5850. https://doi.org/10.3390/molecules20045835
Chicago/Turabian StyleSchuetz, Roman, Dmitri Fix, Ulrich Schade, Emad F. Aziz, Nadya Timofeeva, Richard Weinkamer, and Admir Masic. 2015. "Anisotropy in Bone Demineralization Revealed by Polarized Far-IR Spectroscopy" Molecules 20, no. 4: 5835-5850. https://doi.org/10.3390/molecules20045835
APA StyleSchuetz, R., Fix, D., Schade, U., Aziz, E. F., Timofeeva, N., Weinkamer, R., & Masic, A. (2015). Anisotropy in Bone Demineralization Revealed by Polarized Far-IR Spectroscopy. Molecules, 20(4), 5835-5850. https://doi.org/10.3390/molecules20045835