
Pi-pi Stacking Mediated Cooperative Mechanism for Human Cytochrome P450 3A4

Abstract
:1. Introduction
2. Results and Discussion
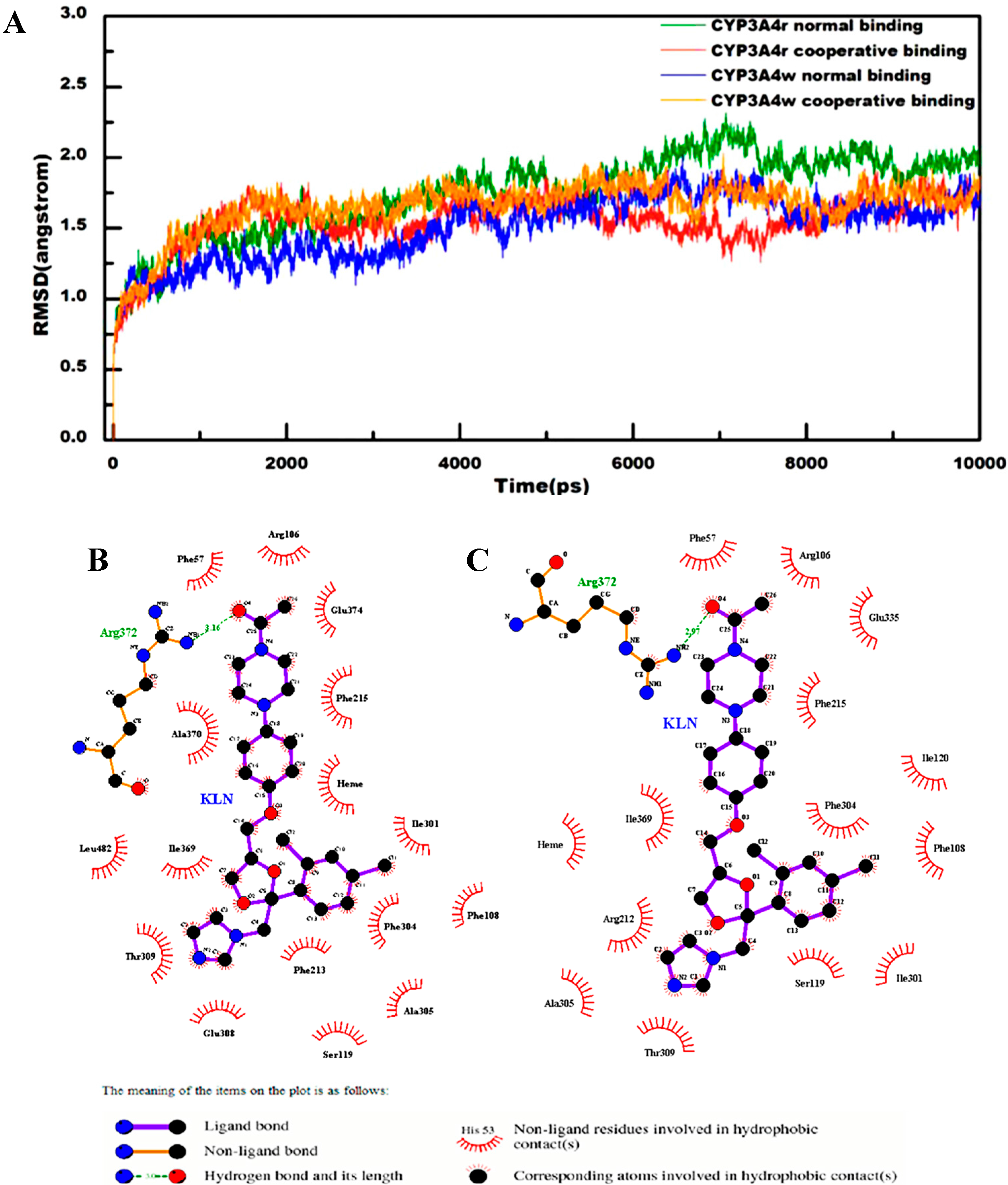
2.1. General Analysis of Molecular Dynamics Trajectories
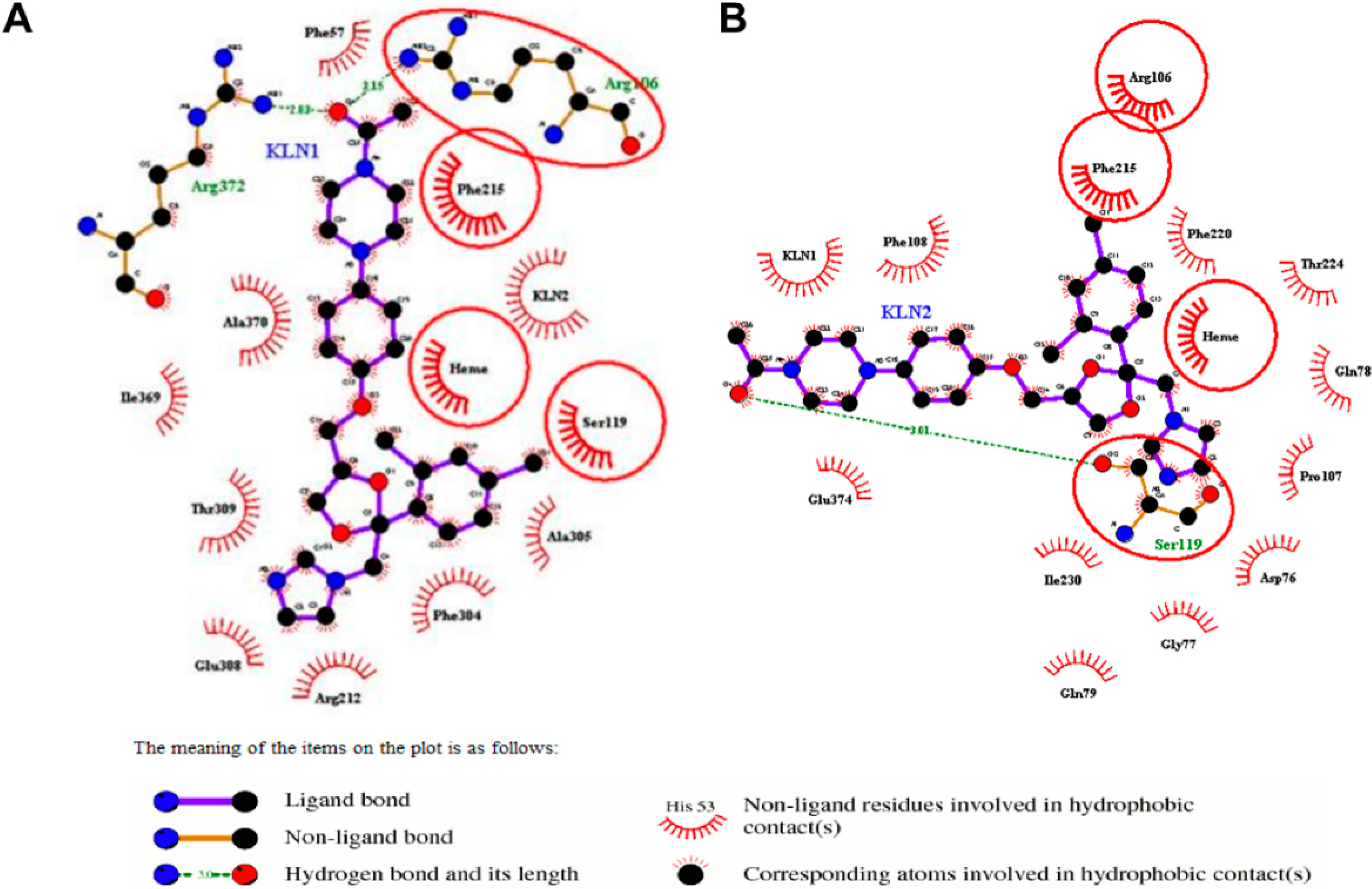
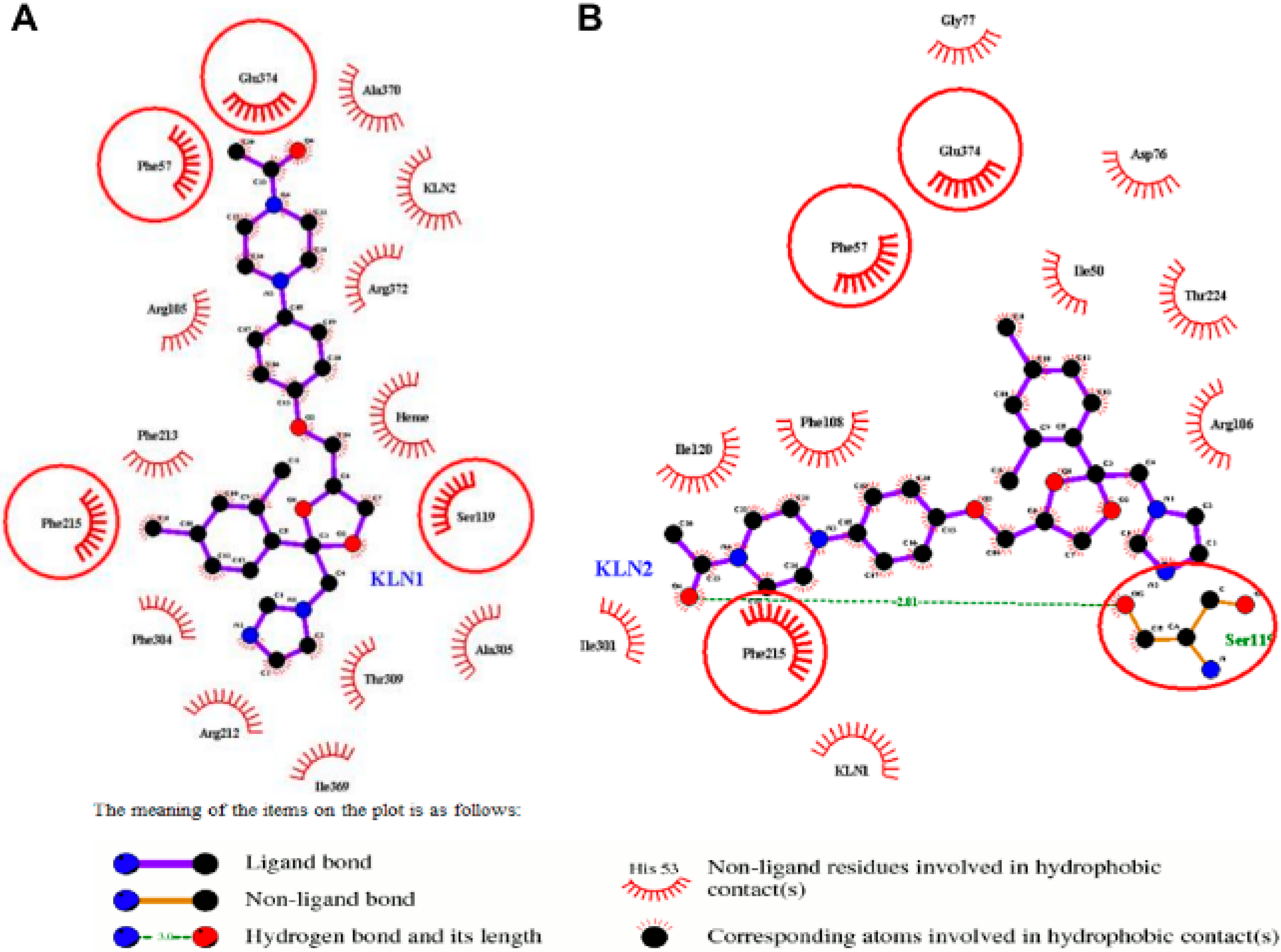
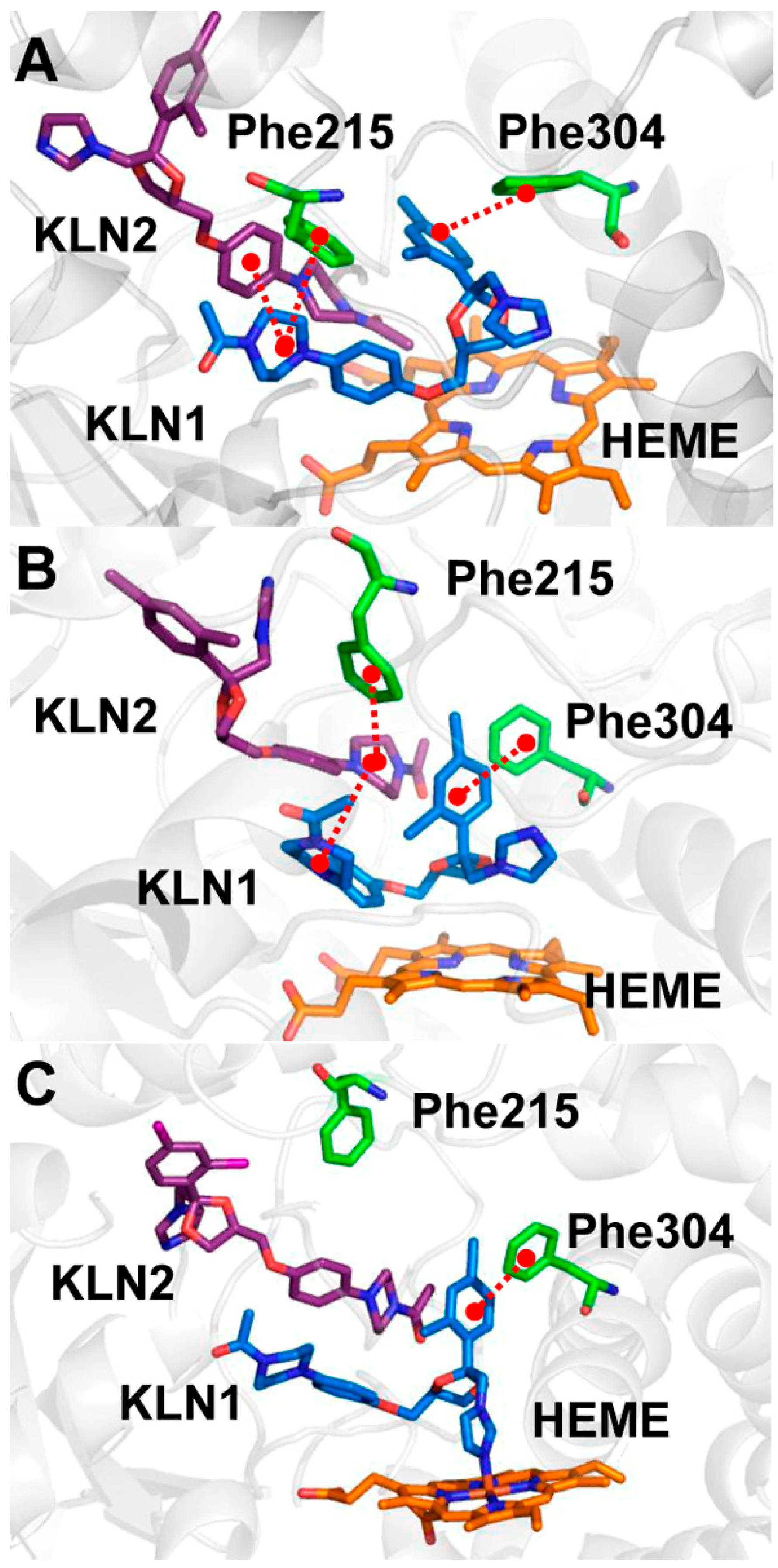
2.2. Interactions for Normal and Cooperative Binding



2.3. Binding Free Energy Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energies (kcal/mol) | CYP3A4t | CYP3A4w | ||
|---|---|---|---|---|
| Normal | Cooperative | Normal | Cooperative | |
| ΔEele | −20.80 ± 2.71 | −25.68 ± 3.08 | −31.63 ± 3.45 | −25.29 ± 3.57 |
| ΔEvdw | −65.33 ± 2.46 | −74.99 ± 2.57 | −57.44 ± 2.37 | −75.63 ± 3.42 |
| ΔEgas | −86.13 ± 3.34 | −100.67 ± 3.68 | −89.06 ± 3.17 | −100.92 ± 4.35 |
| ΔGnonpolar/PB | −8.37 ± 0.21 | −8.44 ± 0.14 | −7.79 ± 0.06 | −7.84 ± 0.15 |
| ΔGsol/PB | 47.27 ± 3.84 | 53.11 ± 3.15 | 59.24 ± 2.46 | 56.43 ± 2.67 |
| ΔGele/PB | 34.84 ± 3.64 | 35.87 ± 4.17 | 35.40 ± 4.15 | 38.97 ± 3.86 |
| ΔGbind/PB | −38.86 ± 4.00 | −47.56 ± 4.36 | −29.83 ± 4.26 | −44.50 ± 4.45 |
| ΔGnonpolar/GB | −8.37 ± 0.21 | −8.44 ± 0.14 | −7.79 ± 0.16 | −7.84 ± 0.15 |
| ΔGsol/GB | 31.72 ± 2.50 | 33.72 ± 2.21 | 38.10 ± 2.85 | 34.11 ± 2.04 |
| ΔGele/GB | 19.29 ± 2.23 | 16.48 ± 2.44 | 14.26 ± 2.44 | 16.65 ± 2.82 |
| ∆ Gbind/GB | −54.41 ± 2.64 | −66.95 ± 3.12 | −50.97 ± 3.22 | −66.81 ± 3.74 |
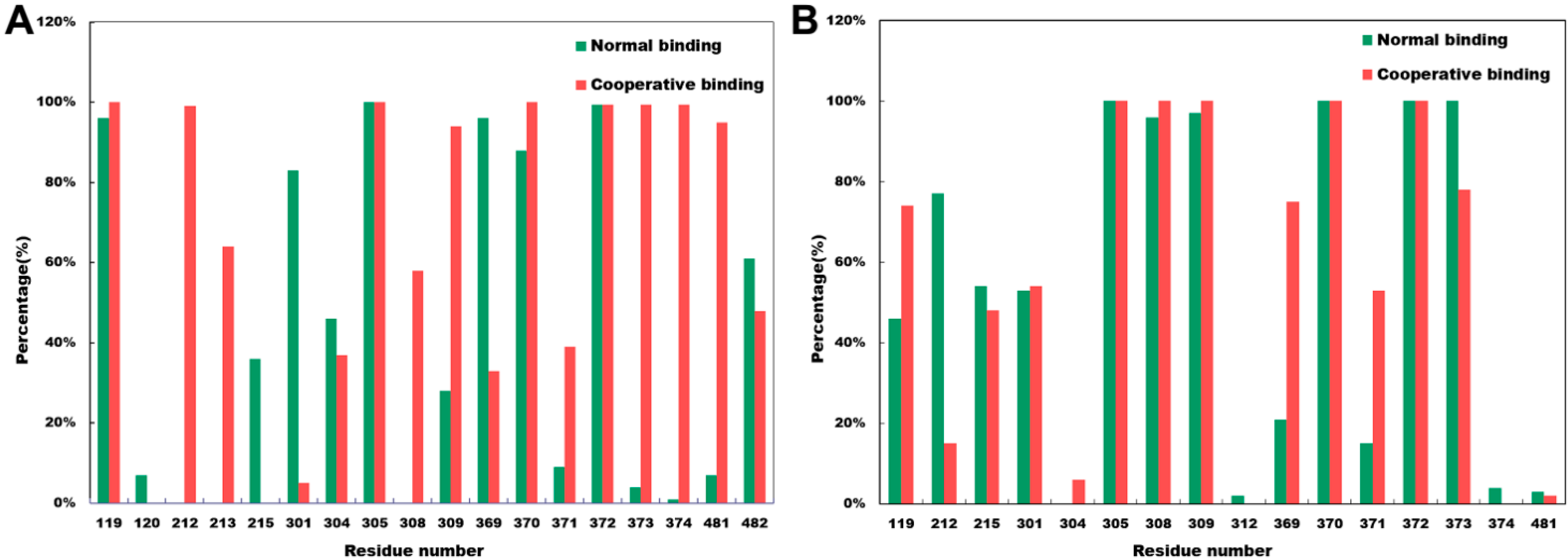
2.4. Close Contact Analysis

| Res. No. | Res. Name | ΔG Contribution | SRS Reg. | |
|---|---|---|---|---|
| Normal | Cooperative | |||
| 57 | Phenylalanine | −0.79 ± 0.38 | −0.96 ± 0.33 | 1 |
| 106 | Arginine | −0.89 ± 0.33 | −2.79 ± 0.76 | 1 |
| 108 | Phenylalanine | −0.86 ± 0.27 | 0.04 ± 0.05 | 1 |
| 212 | Arginine | −1.75 ± 0.37 | −1.71 ± 0.29 | 2 and 3 |
| 215 | Phenylalanine | −2.03 ± 0.35 | −1.76 ± 0.30 | 2 and 3 |
| 301 | Isoleucine | −0.80 ± 0.20 | −0.71 ± 0.18 | 4 |
| 304 | Phenylalanine | −1.56 ± 0.34 | −2.14 ± 0.31 | 4 |
| 305 | Alanine | −0.94 ± 0.25 | −1.30 ± 0.26 | 4 |
| 308 | Glutamic acid | −0.92 ± 0.37 | −0.62 ± 0.34 | 4 |
| 309 | Threonine | −1.34 ± 0.29 | −1.19 ± 0.47 | 4 |
| 369 | Isoleucine | −1.39 ± 0.32 | −1.63 ± 0.33 | 5 |
| 370 | Alanine | −1.59 ± 0.37 | −1.04 ± 0.41 | 5 |
| 371 | Methionine | −0.26 ± 0.07 | −1.24 ± 0.36 | 5 |
| 372 | Arginine | −4.31 ± 0.69 | −4.25 ± 0.79 | 5 |
| 373 | Leucine | −0.83 ± 0.17 | −0.38 ± 0.11 | 5 |
| 374 | Glutamic acid | −1.09 ± 0.70 | −1.35 ± 0.45 | 5 |
| 482 | Leucine | −1.03 ± 0.22 | −0.60 ± 0.15 | 6 |
| Res. No. | Res. Name | ΔG Contribution | SRS Reg. | |
|---|---|---|---|---|
| Normal | Cooperative | |||
| 57 | Phenylalanine | −0.83 ± 0.25 | −0.24 ± 0.22 | 1 |
| 105 | Arginine | −0.19 ± 0.13 | −2.41 ± 0.58 | 1 |
| 106 | Arginine | −0.80 ± 0.33 | −0.70 ± 0.20 | 1 |
| 108 | Phenylalanine | −0.73 ± 0.24 | 0.06 ± 0.03 | 1 |
| 119 | Serine | −0.42 ± 0.28 | −0.78 ± 0.46 | 1 |
| 212 | Arginine | −3.10 ± 0.66 | −3.83 ± 1.00 | 2 and 3 |
| 215 | Phenylalanine | −1.89 ± 0.39 | −0.43 ± 0.22 | 2 and 3 |
| 301 | Isoleucine | −0.64 ± 0.26 | −0.46 ± 0.14 | 4 |
| 304 | Phenylalanine | −1.56 ± 0.42 | −1.07 ± 0.33 | 4 |
| 305 | Alanine | −1.42 ± 0.17 | −1.30 ± 0.26 | 4 |
| 309 | Threonine | −0.77 ± 0.21 | −0.37 ± 0.36 | 4 |
| 369 | Isoleucine | −1.12 ± 0.43 | −0.95 ± 0.28 | 5 |
| 370 | Alanine | −0.73 ± 0.17 | −1.19 ± 0.46 | 5 |
| 371 | Methionine | −1.75 ± 0.29 | −0.30 ± 0.07 | 5 |
| 372 | Arginine | −4.49 ± 0.58 | −1.86 ± 0.59 | 5 |
| 373 | Leucine | −0.78 ± 0.17 | −1.56 ± 0.41 | 5 |
| 374 | Glutamic acid | 2.48 ± 0.79 | 0.44 ± 0.56 | 5 |
| 482 | Leucine | −1.24 ± 0.49 | −2.26 ± 0.36 | 6 |
2.5. Pi-Pi Stacking in the Cooperative Binding Models

3. Materials and Computational Methods
3.1. Constructing Computational Models
3.2. Molecular Dynamics Simulation
3.3. Binding Free Energy Estimation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chen, Q.; Zhang, T.; Wang, J.F.; Wei, D.Q. Advances in human cytochrome p450 and personalized medicine. Curr. Drug Metab. 2011, 12, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.R. Cytochrome P450 and the individuality of species. Arch. Biochem. Biophys. 1999, 369, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.F.; Zhang, C.C.; Chou, K.C.; Wei, D.Q. Structure of cytochrome P450s and personalized drug. Curr. Med. Chem. 2009, 16, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Cytochrome p450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.R.; Field, J.; Goodwin, B.; Bierach, S.; Tran, M.; Lehnert, A.; Liddle, C. Transgenic mouse models of human CYP3A4 gene regulation. Mol. Pharmacol. 2003, 64, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.M.; Auchus, R.J. Two surfaces of cytochrome b5 with major and minor contributions to CYP3A4-catalyzed steroid and nifedipine oxygenation chemistries. Arch. Biochem. Biophys. 2014, 541, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Shahrokh, K.; Cheatham, T.E., 3rd; Yost, G.S. Conformational dynamics of CYP3A4 demonstrate the important role of Arg212 coupled with the opening of ingress, egress and solvent channels to dehydrogenation of 4-hydroxy-tamoxifen. Biochim. Biophys. Acta 2012, 1820, 1605–1617. [Google Scholar]
- Keshava, C.; McCanlies, E.C.; Weston, A. CYP3A4 polymorphisms–potential risk factors for breast and prostate cancer: A HuGE review. Am. J. Epidemiol. 2004, 160, 825–841. [Google Scholar] [CrossRef] [PubMed]
- Atkins, W.M. Non-Michaelis-Menten kinetics in cytochrome P450-catalyzed reactions. Ann. Rev. Pharmacol. Toxicol. 2005, 45, 291–310. [Google Scholar] [CrossRef]
- Harlow, G.R.; Halpert, J.R. Analysis of human cytochrome P450 3A4 cooperativity: Construction and characterization of a site-directed mutant that displays hyperbolic steroid hydroxylation kinetics. Proc. Natl. Acad. Sci. USA 1998, 95, 6636–6641. [Google Scholar] [CrossRef] [PubMed]
- Davydov, D.R.; Halpert, J.R.; Renaud, J.P.; Hui Bon Hoa, G. Conformational heterogeneity of cytochrome P450 3A4 revealed by high pressure spectroscopy. Biochem. Biophys. Res. Commun. 2003, 312, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Kenworthy, K.E.; Clarke, S.E.; Andrews, J.; Houston, J.B. Multisite kinetic models for CYP3A4: Simultaneous activation and inhibition of diazepam and testosterone metabolism. Drug Metab. Dispos. 2001, 29, 1644–1651. [Google Scholar] [PubMed]
- Bren, U.; Oostenbrink, C. Cytochrome P450 3A4 inhibition by ketoconazole: Tackling the problem of ligand cooperativity using molecular dynamics simulations and free-energy calculations. J. Chem. Inf. Model. 2012, 52, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Fishelovitch, D.; Hazan, C.; Hirao, H.; Wolfson, H.J.; Nussinov, R.; Shaik, S. QM/MM study of the active species of the human cytochrome P450 3A4, and the influence thereof of the multiple substrate binding. J. Phys. Chem. B 2007, 111, 13822–13832. [Google Scholar] [CrossRef] [PubMed]
- Fishelovitch, D.; Hazan, C.; Shaik, S.; Wolfson, H.J.; Nussinov, R. Structural dynamics of the cooperative binding of organic molecules in the human cytochrome P450 3A4. J. Am. Chem. Soc. 2007, 129, 1602–1611. [Google Scholar] [CrossRef] [PubMed]
- Isin, E.M.; Guengerich, F.P. Kinetics and thermodynamics of ligand binding by cytochrome P450 3A4. J. Biol. Chem. 2006, 281, 9127–9136. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wei, D.Q.; Wang, J.F.; Li, Y.X. A negative cooperativity mechanism of human CYP2E1 inferred from molecular dynamics simulations and free energy calculations. J. Chem. Inf. Model. 2011, 51, 3217–3225. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, B.Y.; Hao, P.; Li, X.; Li, Y.X.; Wang, J.F. Pi-pi Stacking mediated drug-drug interactions in human CYP2E1. Proteins 2013, 81, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Ping, J.; Wang, Y.J.; Wang, J.F.; Li, X.; Li, Y.X.; Hao, P. Negatively cooperative binding properties of human cytochrome P450 2E1 with monocyclic substrates. Curr. Drug Metab. 2012, 13, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, V.H.; Ribeiro, V.; Martel, P.J. Analysis of binding modes of ligands to multiple conformations of CYP3A4. Biochim. Biophys. Acta 2010, 1804, 2036–2045. [Google Scholar] [CrossRef] [PubMed]
- Yuki, H.; Honma, T.; Hata, M.; Hoshino, T. Prediction of sites of metabolism in a substrate molecule, instanced by carbamazepine oxidation by CYP3A4. Bioorg. Med. Chem. 2012, 20, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Graf, M.M.H.; Bren, U.; Haltrich, D.; Oostenbrink, C. Molecular dynamics simulations give insight into D-glucose dioxidation at C2 and C3 by Agaricus meleagris pyranose dehydrogenase. J. Comput. Aided Mol. Des. 2013, 27, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Petsko, G.A. Aromatic-aromatic interaction: A mechanism of protein structure stabilization. Science 1985, 229, 23–28. [Google Scholar] [CrossRef] [PubMed]
- McGaughey, G.B.; Gagne, M.; Rappe, A.K. pi-Stacking interactions. Alive and well in proteins. J. Biol. Chem. 1998, 273, 15458–15463. [Google Scholar]
- Bren, U.; Fuchs, J.E.; Oostenbrink, C. Cooperative binding of aflatoxin B1 by cytochrome P450 3A4: A computational study. Chem. Res. Toxicol. 2014, 27, 2136–2147. [Google Scholar] [CrossRef] [PubMed]
- Tauer, T.P.; Sherrill, C.D. Beyond the benzene dimer: An investigation of the additivity of pi-pi interactions. J. Phys. Chem. A 2005, 109, 10475–10478. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.K.; Wester, M.R.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-A resolution. J. Biol. Chem. 2004, 279, 38091–38094. [Google Scholar]
- Williams, P.A.; Cosme, J.; Vinkovic, D.M.; Ward, A.; Angove, H.C.; Day, P.J.; Vonrhein, C.; Tickle, I.J.; Jhoti, H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 2004, 305, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, R.E.; Alexov, E.G.; Gunner, M.R. Combining conformational flexibility and continuum electrostatics for calculating pK(a)s in proteins. Biophys. J. 2002, 83, 1731–1748. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar]
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated docking of flexible ligands: Applications of AutoDock. J. Mol. Recognit. 1996, 9, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Oda, A.; Yamaotsu, N.; Hirono, S. New AMBER force field parameters of heme iron for cytochrome P450s determined by quantum chemical calculations of simplified models. J. Comput. Chem. 2005, 26, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Bren, M.; Florian, J.; Mavri, J.; Bren, U. Do all pieces make a whole?Thiele cumulants and the free energy decompositions. Theor. Chem. Acc. 2007, 117, 535–540. [Google Scholar] [CrossRef]
- Bren, U.; Vaclav, M.; Florian, J. Decomposition of the salvation free energies of deoxyribonucleoside triphosphates using the free energy perturbation method. J. Phys. Chem. B 2006, 110, 12782–12788. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples are not availability from authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fa, B.; Cong, S.; Wang, J. Pi-pi Stacking Mediated Cooperative Mechanism for Human Cytochrome P450 3A4. Molecules 2015, 20, 7558-7573. https://doi.org/10.3390/molecules20057558
Fa B, Cong S, Wang J. Pi-pi Stacking Mediated Cooperative Mechanism for Human Cytochrome P450 3A4. Molecules. 2015; 20(5):7558-7573. https://doi.org/10.3390/molecules20057558
Chicago/Turabian StyleFa, Botao, Shan Cong, and Jingfang Wang. 2015. "Pi-pi Stacking Mediated Cooperative Mechanism for Human Cytochrome P450 3A4" Molecules 20, no. 5: 7558-7573. https://doi.org/10.3390/molecules20057558
APA StyleFa, B., Cong, S., & Wang, J. (2015). Pi-pi Stacking Mediated Cooperative Mechanism for Human Cytochrome P450 3A4. Molecules, 20(5), 7558-7573. https://doi.org/10.3390/molecules20057558