2. Results and Discussion
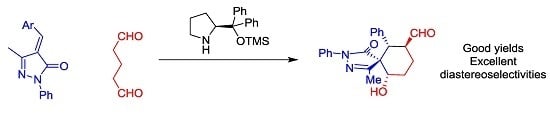
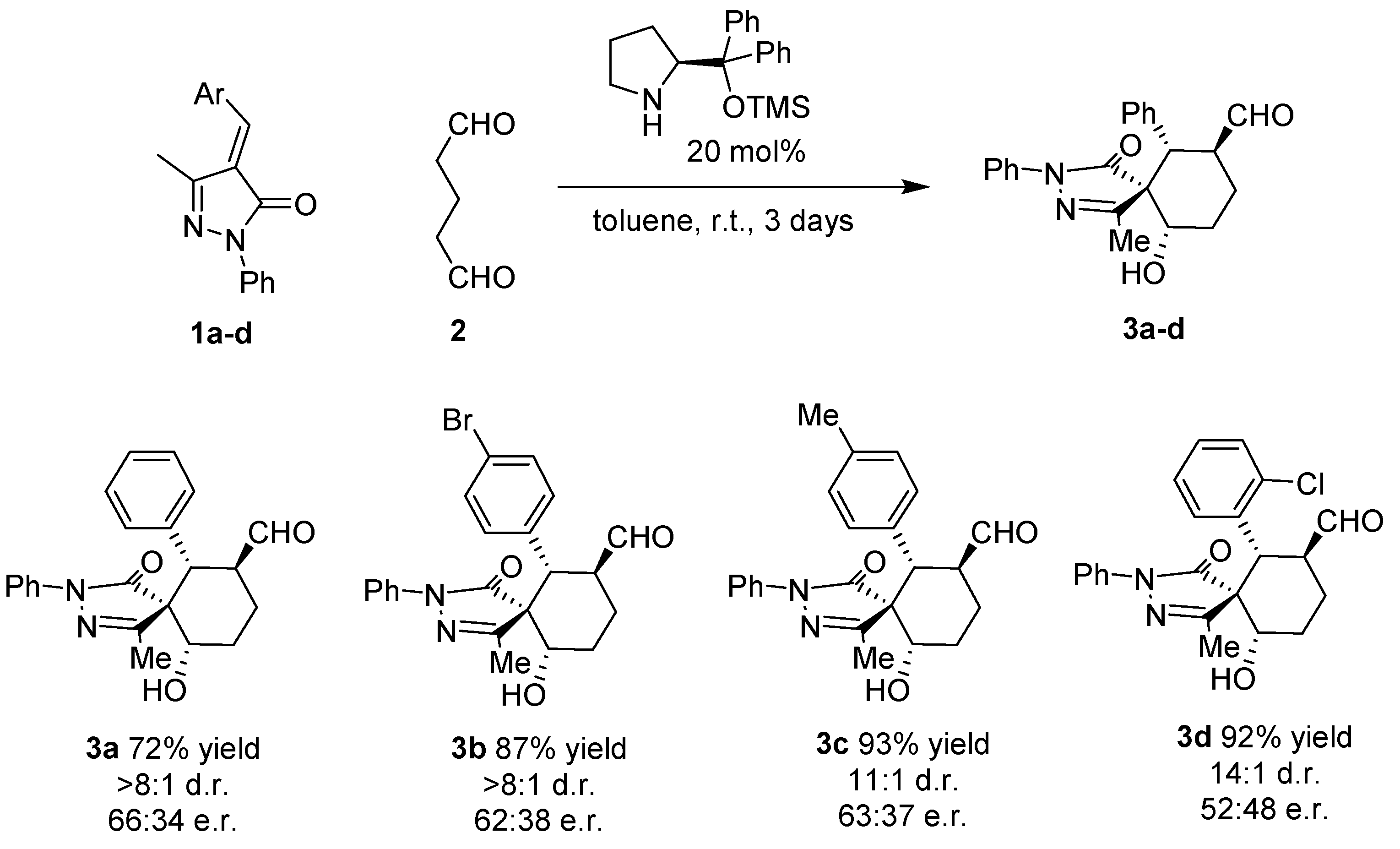
As a model reaction we choose methylenepyrazolone
1a and the dialdehyde
2 that will get access to six-membered ring fused spiropyrazolones. We tested several solvents, catalysts and temperatures as is summarized in
Table 1.
Table 1.
Conditions screening.
a ![Molecules 20 08574 i001]()
Table 1.
Conditions screening. a ![Molecules 20 08574 i001]()
| Entry | Solvent | Catalyst | Conversion b | d.r. c | e.r. d |
|---|
| 1 | CH2Cl2 | I | 100% | 7:1 | 66:34 |
| 2 | Toluene | I | 100% | 10:1 | 66:34 |
| 3 | p-Xylene | I | 100% | 10:1 | 53:47 |
| 4 | AcOEt | I | 100% | 10:1 | 63:37 |
| 5 | CHCl3 | I | 100% | 5:1 | 53:47 |
| 6 | THF | I | 100% | 5:1 | 52:48 |
| 7 e | Toluene | I | 100% | 10:1 | 59:41 |
| 8 | Toluene | II | n.r. | - | - |
| 9 | Toluene | III | n.r. | - | - |
| 10 f | Toluene | I | 100% | 2:1 | 66:34 |
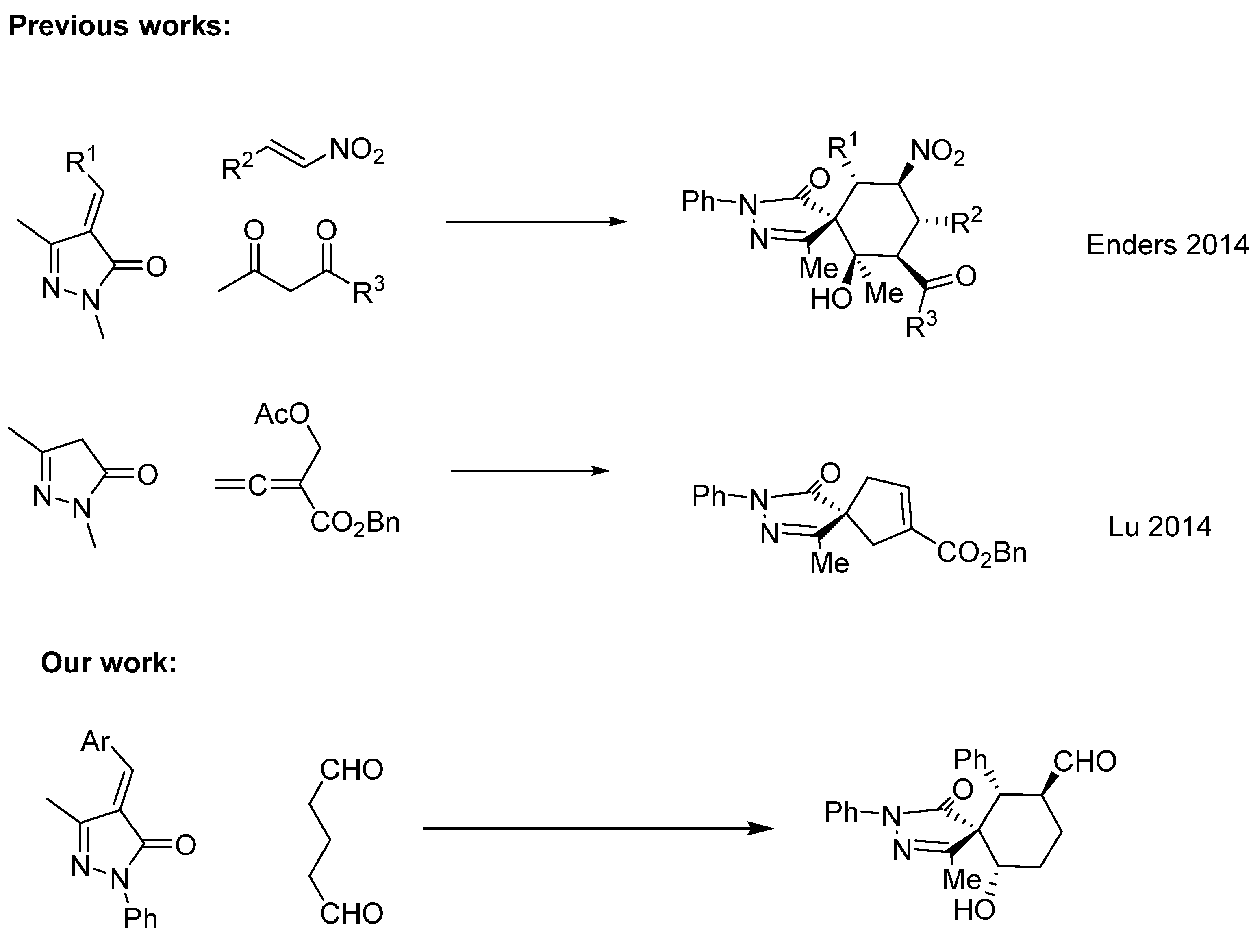
Despite the issues associated with the present reaction like autoaldolization of the dialdehyde, to our delight the reaction catalyzed by Jørgensen-Hayashi catalyst
I rendered the final product in good conversion, good to excellent diastereoselectivities but low enantioselectivities in all the solvents tested. Better diastereoselectivities were obtained with aromatic solvents (toluene and
p-xylene entries 2, 3 and 7;
Table 1) achieving 10:1 d.r. but with low enantioselectivities (66:34). The use of other solvents such as CH
2Cl
2, CHCl
3 or THF (entries 1, 5 and 6;
Table 1) renders the final compounds with slightly worst diastereo- and enantioselectivities. The use of an additive such as benzoic acid have little effect on the outcome of the reaction only giving marginally worse enantioselectivities. Remarkably, the use of catalyst
II did not render any cyclization product; when Proline (
III) was used as catalyst the only product obtained was the aldol product of the dialdehyde (entries 8, and 9;
Table 1). Decreasing the reaction temperature to −20 °C results in reduces the diastereoselectivity of the reaction without increasing the enantioselectivity. With the best conditions in hand we studied the scope of the reaction with different unsaturated pyrazolones. As it is shown in
Figure 2, the reaction affords the final spiro compounds with excellent yields and diastereoselectivities but very low enantioselectivities.
As shown in
Figure 2, the reaction works with several substituents in the para position Br (
3b), Me (
3c) rendering the final compound almost diastereopure in excellent yields. When a halogen was placed in the ortho position of the aromatic ring the diastereoselectivity was increased to 14:1 but we got an almost racemic compound (
3d).
Figure 2.
Reaction scope.
Figure 2.
Reaction scope.
Next we tried the reaction using butandial as a dialdehyde source in order to have access to the cyclopentane derivatives. Unfortunately in all the reactions tested we only got decomposition of the starting material and no signals of any new product.
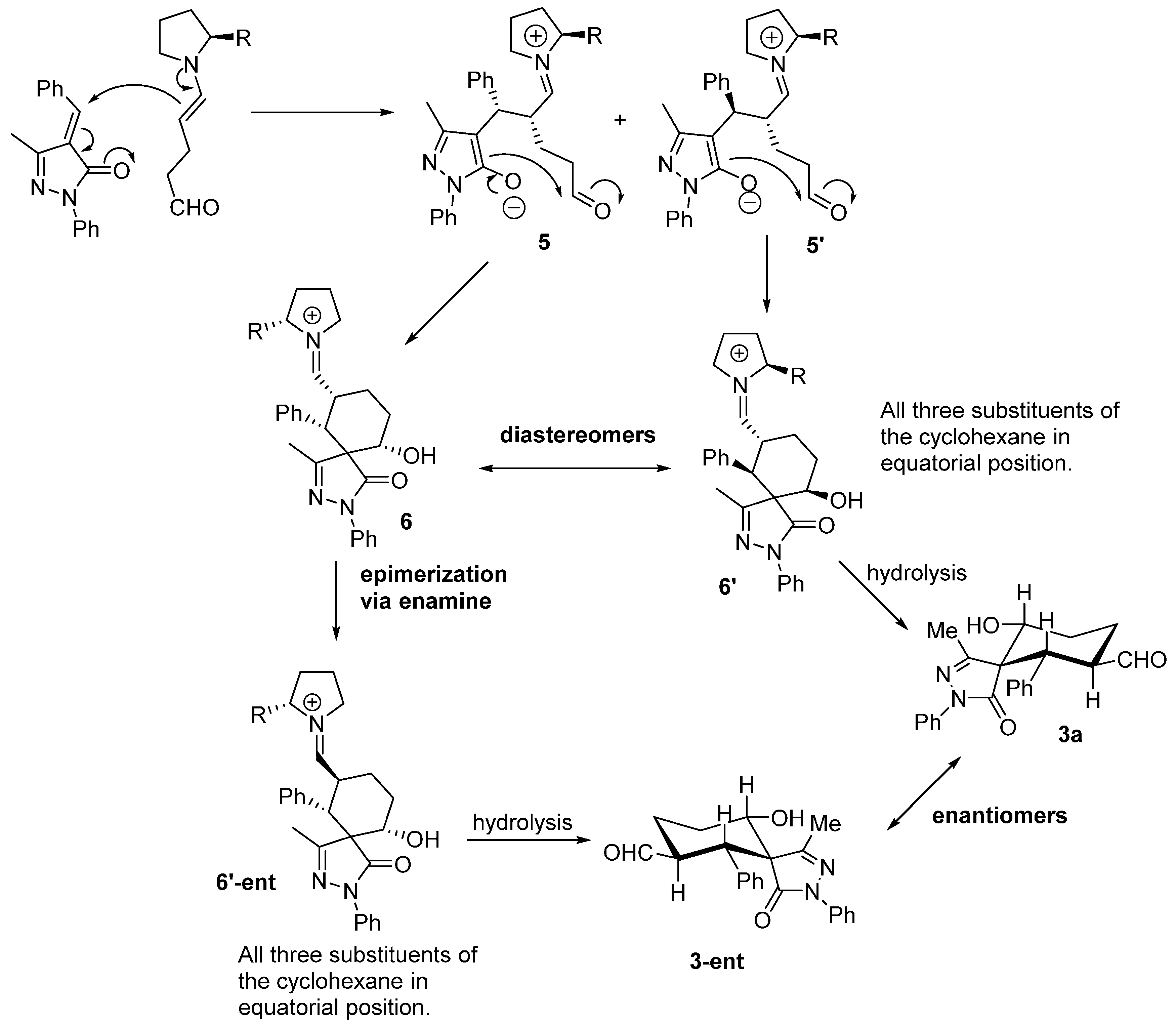
Surprisingly all the compounds in
Figure 2 were obtained in extremely good diastereoselectivities but low enantioselectivities. This suggests an epimerization mechanism that renders the most stable diastereomer as product. The proposed mechanism for the reaction is shown in
Figure 3.
Figure 3.
Proposed mechanism.
Figure 3.
Proposed mechanism.
The reaction starts with the enamine formation between dialdehyde
2 and the secondary amine catalyst (
I). Next, the enamine
4 reacts with the Michael acceptor
1 to form adduct
5. The catalyst efficiently shields one of the faces of the enamine, for this reason the attack takes place on the other side of the enamine. The formed enolate in the pyrazole ring undergoes an intramolecular aldol reaction with the other aldehyde group to form the intermediate
6, which on hydrolysis renders the final product
3a and regenerate the catalyst ready for a new catalytic cycle. Based on this mechanism what is expected is to have high enantioselectivities and poor diastereoselectivities. One possible explanation for the results obtained (poor ee and extremely good d.r.) could be the epimerization in the α-carbaldehyde position via an enamine intermediate. As shown in
Figure 4 the first enamine addition should be highly stereospecific in the alpha position of the enamine because it is directly controlled by the catalyst, but probably we will get a mixture of diastereomers in the benzylic position (
5,
5'). The following aldol reaction will be termodynamically controlled affording the most stable compound in which the benzylic and the hydroxyl group present a relative equatorial position in the cyclohexane ring (
6,
6'). Next, an epimerization via enamine takes place to obtain the carbaldehyde substituent in a relative
trans configuration with respect to the Ph in order to get all the substituents in relative equatorial positions of the cyclohexane ring. As is well known, Michael and aldol reactions are reversible reactions that will favour the most thermodynamically stable final compound.
Figure 4.
Plausible explanation for the low enantioselectivity observed.
Figure 4.
Plausible explanation for the low enantioselectivity observed.
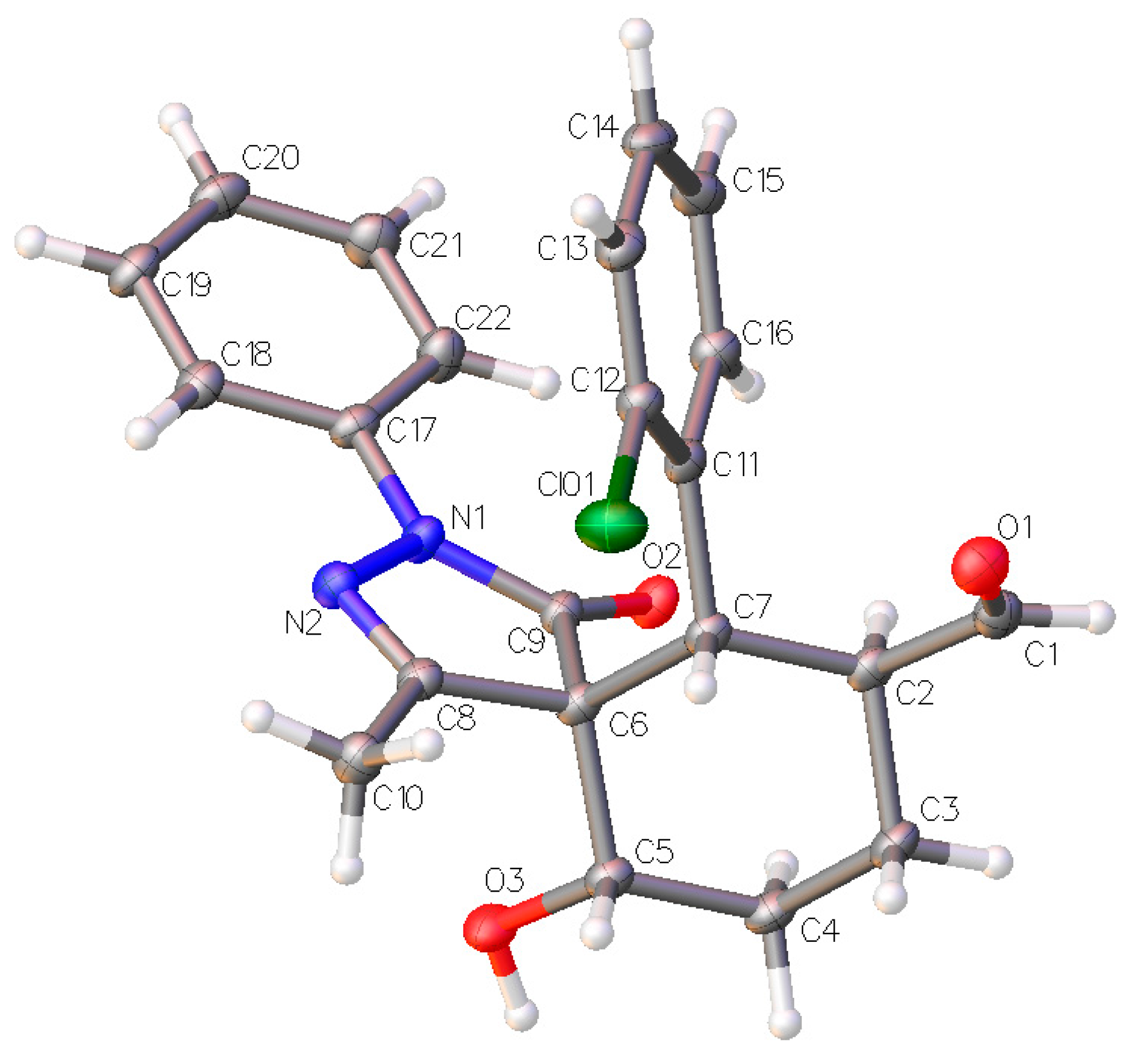
The relative configuration of the major diastereomer was inferred by X-ray crystallography of compound
3d (
Figure 5) and confirms the proposed structure of compound
3a where all the substituents are in equatorial position.
Figure 5.
X-ray structure of compound
3d (major diastereomer). The displacement ellipsoids are drawn at the 50% probability level [
10].
Figure 5.
X-ray structure of compound
3d (major diastereomer). The displacement ellipsoids are drawn at the 50% probability level [
10].
3. Experimental Section
General procedure for the preparation spyropyrazolones 3: To a solution of unsaturated pyrazolone (1) (1 equiv.) in toluene was added glutaraldehyde (3 equiv.) and (S)-2-(diphenyl ((trimethylsilyl) oxy) methyl)pyrroline (0.2 equiv.). The solution was stirred for 3 days and subsequently the solvent was removed by rotary evaporation. The reaction mixture was purified by column chromatography (Hexane/AcOEt). The product was confirmed by 1H-NMR.
3a: Purified by column chromatography (Hexane/AcOEt, 5:1).1H-NMR (CDCl3, 300 MHz): 9.59 ppm (d, 1H, 3J = 2.26 Hz), 7.78 ppm (dd, 2H, J = 1.13 Hz, J = 8.67 Hz), 7.57–7.45 ppm (m, 3H), 7.40–7.27 ppm (m, 5H), 4.12 ppm (ddd, 2H, 3J = 2.26 Hz, 3J = 3.39 Hz, 3J = 12.06 Hz), 3.27 ppm (d, 1H, 3J = 12.06 Hz), 2.83 ppm (ddd, 1H, J = 4.14 Hz, J = 13.19 Hz, J = 26 Hz), 2.34 ppm (s, 3H), 2.20–2.10 ppm (m, 2H), 1.80–163 ppm (m, 2H). 13C-NMR (CDCl3, 75 MHz): 203.0, 172.6, 160.5, 137.4, 135.8, 128.8, 128.7, 127.9, 125.4, 119.7, 71.9, 64.0, 47.5, 46.7, 27.0, 24.5, 13.6. HRMS (ESI) calcd. for C22H23N2O3 (M+H)+ 363.1703, found 363.1710. The enantiomeric excess was determined by HPLC using a Chiralpak ID column (hexane/iPrOH = 80:20, flow rate 1.0 mL/min, λ = 210 nm): tr1 = 13.4, tr2 = 15.6, 32% ee.
3b: Purified by column chromatography (Hexane/AcOEt, 3:2).1H-NMR (CDCl3, 300 MHz): 9.25 ppm (d, 1H, 3J = 1.88 Hz), 7.48 ppm (d, 2H, J = 7.91 Hz), 7.28–7.10 ppm (m, 5H), 6.92 ppm (d, 2H, J = 8.67 Hz), 3.75 ppm (ddd, 2H, 3J = 1.9 Hz, 3J = 3.8 Hz, 3J = 12. Hz), 2.92 ppm (d, 1H, 3J = 11.7 Hz), 2.44 ppm (ddd, 1H, 3J = 4.1 Hz, 3J = 13. Hz, 3J = 26.0 Hz), 1.97 (s, 3H), 1.83–1.73 ppm (m, 2H), 1.3 ppm (d, 2H, J = 3.77 Hz, J = 13.19 Hz, J = 22.23 Hz). 13C-NMR (CDCl3, 75 MHz): 201.3, 171.4, 159.3, 136.2, 134.1, 130.8, 127.7, 124.4, 121.0, 118.5, 70.6, 62.7, 45.7, 45.4, 25.7, 23.3, 12.4. HRMS (ESI) calcd. for C22H22Br79N2O3 (M+H)+ 441.0808, found 441.0812. The enantiomeric excess was determined by HPLC using a Chiralpak ID column (hexane/iPrOH = 80:20, flow rate 1.0 mL/min, λ = 210 nm): tr1 = 11.5, tr2 = 12.7, 24% ee.
3c: 1H-NMR (400 MHz, CDCl3) δ 9.38 (d, J = 2.0 Hz, 1H), 7.66–7.60 (m, 2H), 7.37–7.30 (m, 2H), 7.15 (t, J = 7.4 Hz, 1H), 7.06 (d, J = 8.0 Hz, 2H), 6.96 (d, J = 8.0 Hz, 2H), 3.94 (dd, J = 11.6, 4.6 Hz, 1H), 3.88 (tdd, J = 12.0, 3.6, 2.0 Hz, 1H), 3.02 (d, J = 12.0 Hz, 1H), 2.61 (ddd, J = 25.2, 13.3, 4.1 Hz, 1H), 2.22–2.16 (overlapping signals, 4H), 2.13 (s, 3H), 1.97–1.88 (m, 1H) and 1.49 (ddd, J = 26.1, 13.3, 4.1 Hz, 1H). 13C-NMR (100 MHz, CDCl3) δ 203.2, 172.7, 160.5, 137.8, 137.4, 132.6, 129.5, 128.6, 127.7, 125.3, 119.6, 71.9, 64.1, 47.2, 46.8, 26.9, 24.4, 20.9, 13.5. HRMS (ESI)calcd. for C23H25N2O3 (M+H)+ 377.1860, found 377.1856. The enantiomeric excess was determined by HPLC using a Chiralpak ID column (hexane/iPrOH = 80:20, flow rate 1.0 mL/min, λ = 210 nm): tr1= 12.1, tr2 = 14.3, 26% ee.
3d: 1H-NMR (400 MHz, CDCl3) δ 9.30 (d, J = 2.2 Hz, 1H), 7.71 (dd, J = 7.4, 6.3 Hz, 2H), 7.42–7.30 (m, 4H), 7.18 (t, J = 7.4 Hz, 1H), 7.08 (td, J = 7.6, 1.5 Hz, 1H), 7.02 (td, J = 7.6, 1.5 Hz, 1H), 4.06–3.98 (m, 1H), 3.88 (d, J = 11.9 Hz, 1H), 3.76 (tdd, J = 11.9, 3.4, 2.4 Hz, 1H), 2.65 (ddd, J = 25.4, 13.3, 4.3 Hz, 1H), 2.19 (s, 3H), 2.17–2.13 (m, 1H), 2.11 (d, J = 6.1 Hz, 1H), 1.95 (ddd, J = 9.2, 7.5, 4.1 Hz, 1H) and 1.59 (ddd, J = 25.4, 13.3, 4.1 Hz, 1H).13C-NMR (100 MHz, CDCl3) δ 202.1, 172.8, 161.0, 137.4, 134.0, 133.5, 130.0, 129.2, 128.8, 128.5, 127.6, 125.4, 119.5, 72.2, 63.3, 48.0, 41.9, 26.7, 24.4, 13.7. HRMS (ESI) calcd. for C22H22Cl35N2O3 (M+H)+ 397.1313, found 397.1308. The enantiomeric excess was determined by HPLC using a Chiralpak ID column (hexane/iPrOH = 80:20, flow rate 1.0 mL/min, λ = 210 nm): tr1 = 14.1, tr2 = 16.4, 3% ee.
X-ray: Single clear colourless fragment-shaped crystals of 3d were recrystallised from a mixture of TCM and hexane by slow evaporation. A suitable crystal (0.09 × 0.08 × 0.05 mm3) was selected and mounted on a MITIGEN holder in perfluoroether oil on a Rigaku AFC12 FRE-HF diffractometer (University of Southampton, Southampton, UK). The crystal was kept at T = 100(2) K during data collection. Using Olex, the structure was solved with the ShelXT structure solution program, using the Direct Methods solution method. The model was refined with version of ShelXL using Least Squares. Unit cell parameters were refined against all data. An empirical absorption correction was carried out using CrystalClear software (University of Southampton, Southampton, UK). The crystal structure of 3d was solved by charge flipping methods and refined on Fo2 by full-matrix least-squares refinements using programs of the SHELX-2013 software (University of Southampton, Southampton, UK). All non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were added at calculated positions and refined using a riding model with isotropic displacement parameters based on the equivalent isotropic displacement parameter (Ueq) of the parent atom. Single clear colourless fragment-shaped crystals of were recrystallised from a mixture of TCM and hexane by slow evaporation.
Crystal Data for C
22H
21ClN
2O
3 (
M = 396.86 g/mol): monoclinic, space group P2
1/n (no. 14), a = 9.90175(13) Å, b = 8.02985(9) Å, c = 23.3685(3) Å, β = 96.4448(12),
V = 1846.28(4) Å
3,
Z = 4, T = 100(2) K, μ(CuKα) = 2.056 mm
−1,
Dcalc = 1.428 g/cm
3, 24601 reflections measured (9.36° ≤ 2Θ ≤ 138.082°), 3363 unique (
Rint = 0.0868, R
sigma = 0.0313) which were used in all calculations. The final
R1 was 0.0456 (I > 2σ(I)) and
wR2 was 0.1231 Crystallographic data (excluding structure factors) for the structure
3d have been deposited with the Cambridge Crystallographic Data Centre with CCDC number 1062126. Copies of the data can be obtained, free of charge, on application to Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK, (fax: +44-(0)1223-336033 or e-mail:
[email protected]).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



