3.1. Chemistry Section
3.1.1. General Section
Melting points were determined on a Kofler melting point apparatus and were uncorrected. Thin-layer chromatography (TLC) was accomplished on 0.2-mm precoated plates of silica gel 60 F-254 (Merck, Fontenay-sous-Bois, France). Visualization was made with ultraviolet light (254 and 365 nm) or with a fluorescence indicator. 1H-NMR spectra were recorded on BRUKER AC 300 P (300 MHz) spectrometer, 13C-NMR spectra on a BRUKER AC 300 P (75 MHz) spectrometer. Chemical shifts are expressed in parts per million downfield from tetramethylsilane as an internal standard. Data are given in the following order: δ value, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad), number of protons, coupling constants J is given in Hertz. The mass spectra (HRMS) were taken respectively on a MS/MS ZABSpec Tof Micromass (EBE TOF geometry) at an ionizing potential of 8 eV and on a VARIAN MAT 311 at an ionizing potential of 70 eV in the Centre Régional de Mesures Physiques de l’Ouest (CRMPO, Rennes, France). Reactions under microwave irradiations were realized in the Anton Paar Monowave 300® microwave reactor (Anton-Paar, Courtaboeuf, France) using borosilicate glass vials of 10 mL equipped with snap caps (at the end of the irradiation, cooling reaction was realized by compressed air). The microwave instrument consists of a continuous focused microwave power output from 0 to 800 W for this Monowave 300® apparatus. All the experiments in the microwave reactor were performed using a stirring option. The target temperature was reached with a ramp of 5 min and the chosen microwave power stayed constant to hold the mixture at this temperature. The reaction temperature is monitored using calibrated infrared sensor and the reaction time included the ramp period. The microwave irradiation parameters (power and temperature) were monitored by the Monowave software package of the Monowave 300® microwave reactor. Solvents were evaporated with a BUCHI rotary evaporator. All reagents and solvents were purchased from Acros, Sigma-Aldrich Chimie (Saint-Quentin Fallavier, France), TCI France and Fluka France and were used without further purification.
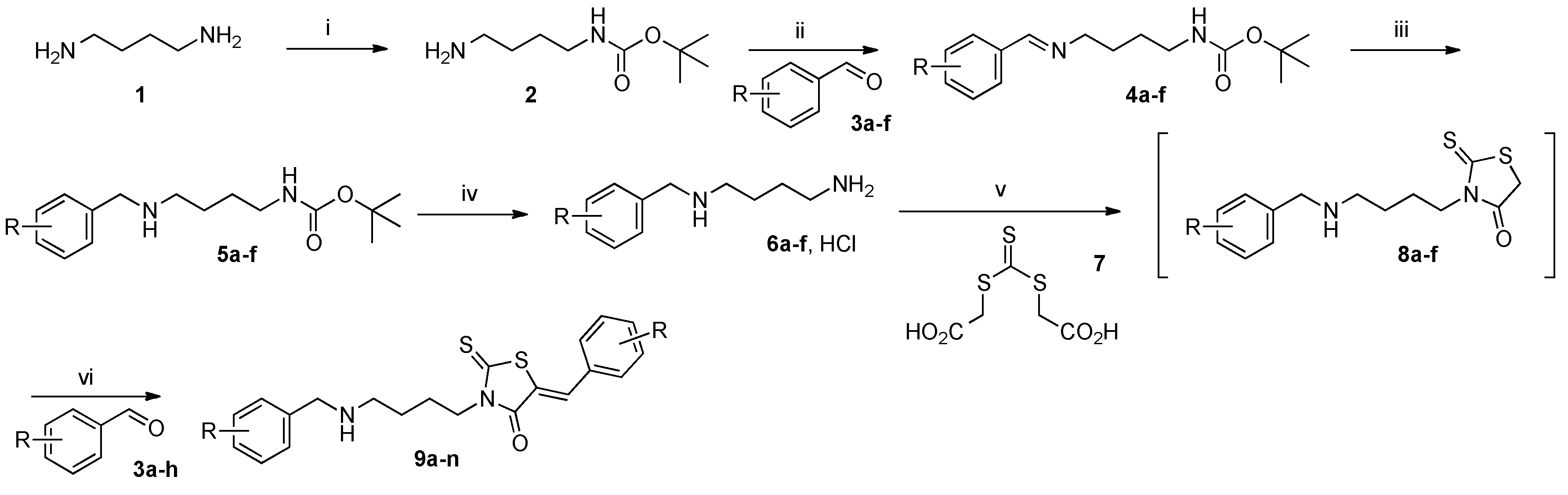
tert-Butyl (4-aminobutyl)carbamate (2). In a 250 mL two-necked round-bottomed flask, provided with magnetic stirrer and condenser, commercial 1,4-diaminobutane 1 (14.3 mL, 12.5 g, 0.14 mol) was solubilized in 69 mL of 1,4-dioxane at room temperature. To this mixture was added dropwise a solution of commercial di-tert-butyldicarbonate (6.5 g, 30 mmol) in 1,4-dioxane (85 mL) over a period of 3 h. After vigorous stirring at 25 °C during a 12 h period, the volatile compounds of the reaction mixture were eliminated in vacuo and into the crude reaction mixture was poured 150 mL of deionized water. The mixture was extracted with methylene chloride (5 × 50 mL), organic phases were collected and dried over magnesium sulfate. The filtrate was concentrated in a rotary evaporator under reduced pressure and was dried under high vacuum (10−2 Torr) at 25 °C for 10 min. The desired carbamate 2 (1.58 g) was obtained as a colourless mobile oil in 84% yield and was further used without purification. 1H-NMR (DMSO-d6) δ: 1.34 (s, 9H, Me3CO); 1.40 (m, 4H, CH2); 2.61 (t, 2H, J = 6.7 Hz, CH2); 3.02 (m, 2H, CH2NH); 4.93 (br s, 2H, NH2); 5.71 (br s, 1H, NH). 13C-NMR (DMSO-d6) δ: 27.2 (CH2); 28.4 (OC(CH3)3); 29.5 (CH2); 40.2 (CH2); 41.1 (CH2); 78.9 (OC(CH3)3); 156.1 (C=O). HRMS, m/z: 189.1600 found (calculated for C9H21N2O2 [M + H]+ requires 189.1603).
3.1.2. Standard Procedure for the Preparation of Aldimines 4a–f from tert-Butyl (4-aminobutyl)Carbamate 2 and Aromatic Aldehyde 3a–f
In a 100 mL two-necked round-bottomed flask, provided with magnetic stirrer and condenser, containing a solution of tert-butyl (4-aminobutyl)carbamate 2 (1.97 g, 10.5 mmol) in anhydrous ether (50 mL) was added dropwise during 30 min, a suspension of commercial aldehyde (10 mmol) in anhydrous ether (30 mL) and molecular sieves (2 g, 3 Å, 8–12 mesh). The crude reaction mixture is stirred vigorously for 16 h at room temperature until the disappearance of aromatic aldehyde 3 controlled by thin-layer chromatography on 0.2-mm precoated plates of silica gel 60 F-254 (Merck). The crude reaction mixture was filtered on filter paper and then concentrated in a rotary evaporator under reduced pressure. The crude residue was dried under high vacuum (10−2 Torr) at 25 °C for 10 min. The desired aldimine 4 was obtained as yellowish viscous oil and was further used without purification.
[4-(Benzylidene-amino)-butyl]-carbamic acid tert-butyl ester (4a). Compound 4a was prepared in 97% yield (2.68 g) from benzaldehyde 3a (1.06 g, 10 mmol) according to the standard procedure as yellowish viscous oil. 1H-NMR (DMSO-d6) δ: 1.37 (s, 9H, Me3C); 1.40–1.48 (m, 2H, CH2, H-2); 1.54–1.62 (m, 2H, CH2, H-3); 2.96 (q, 2H, J = 6.8 Hz, CH2NH, H-1); 3.55 (t, 2H, J = 6.8 Hz, CH2N=, H-4); 6.79–6.83 (br s, 1H, NH); 7.41–7.46 (m, 3H, H-3ʹ, H-4ʹ, H-5ʹ, Ar); 7.71–7.75 (m, 2H, H-2ʹ, H-6ʹ, Ar); 8.32 (s, 1H, N=CH). 13C-NMR (DMSO-d6) δ: 27.3 (C-2); 27.8 (C-3); 28.2 (OC(CH3)3); 39.6 (C-1); 60.1 (C-4); 77.2 (OC(CH3)3); 127.7 (C-2ʹ, C-6ʹ, Ar); 128.5 (C-3ʹ, C-5ʹ, Ar); 130.4 (C-4ʹ, Ar); 136.1 (C-1ʹ, Ar); 155.5 (C=O); 160.5 (N=CH).
{4-[(4-Methoxy-benzylidene)-amino]-butyl}-carbamic acid tert-butyl ester (4b). Compound 4b was prepared in 99% yield (3.03 g) from 4-methoxybenzaldehyde 3b (1.36 g, 10 mmol) according to the standard procedure as yellowish viscous oil. 1H-NMR (DMSO-d6) δ: 1.37 (s, 9H, Me3C); 1.32–1.46 (m, 2H, CH2, H-2); 1.52–1.61 (m, 2H, CH2, H-3); 2.94 (q, 2H, J = 6.9 Hz, CH2NH, H-1); 3.50 (t, 2H, J = 6.8 Hz, CH2N=, H-4); 3.80 (s, 3H, OCH3); 6.77–6.82 (br s, 1H, NH); 6.98 (d, 2H, J = 8.8 Hz, H-2ʹ, H-6ʹ, Ar); 7.66 (d, 2H, J = 8.8 Hz, H-3ʹ, H-5ʹ, Ar); 8.24 (s, 1H, N=CH). 13C-NMR (DMSO-d6) δ: 27.4 (C-2); 28.0 (C-3); 28.2 (OC(CH3)3); 39.6 (C-1); 55.2 (OCH3); 60.1 (C-4); 77.3 (OC(CH3)3); 114.0 (C-2ʹ, C-6ʹ, Ar); 129.0 (C-1ʹ, Ar); 129.3 (C-3ʹ, C-5ʹ, Ar); 155.5 (C=O); 159.7 (N=CH); 161.0 (C-4ʹ, Ar).
{4-[(2-Chloro-benzylidene)-amino]-butyl}-carbamic acid tert-butyl ester (4c). Compound 4c was prepared in 98% yield (3.05 g) from 2-chlorobenzaldehyde 3c (1.41 g, 10 mmol) according to the standard procedure as yellowish viscous oil. 1H-NMR (DMSO-d6) δ: 1.37 (s, 9H, Me3C); 1.34–1.48 (m, 2H, CH2, H-2); 1.56–1.63 (m, 2H, CH2, H-3); 2.96 (q, 2H, J = 6.7 Hz, CH2NH, H-1); 3.61 (t, 2H, J = 6.8 Hz, CH2N=, H-4); 6.78–6.82 (br s, 1H, NH); 7.36–7.52 (m, 3H, H-4ʹ, H-5ʹ, H-6ʹ, Ar); 7.94–7.97 (m, 1H, H-3ʹ, Ar); 8.65 (s, 1H, N=CH). 13C-NMR (DMSO-d6) δ: 27.3 (C-2); 27.7 (C-3); 28.2 (OC(CH3)3); 39.6 (C-1); 60.4 (C-4); 77.2 (OC(CH3)3); 127.4 (C-5ʹ, Ar); 128.0 (C-4ʹ, Ar); 129.8 (C-6ʹ, Ar); 132.0 (C-3ʹ, Ar); 132.7 (C-1ʹ, C-2ʹ, Ar); 133.8 (C-1ʹ, C-2ʹ, Ar); 155.5 (C=O); 156.6 (N=CH).
{4-[(3-Methoxy-benzylidene)-amino]-butyl}-carbamic acid tert-butyl ester (4d). Compound 4d was prepared in 98% yield (3.03 g) from 3-methoxybenzaldehyde 3d (1.36 g, 10 mmol) according to the standard procedure as yellowish viscous oil. 1H-NMR (DMSO-d6) δ: 1.37 (s, 9H, Me3C); 1.36–1.45 (m, 2H, CH2, H-2); 1.54–1.64 (m, 2H, CH2, H-3); 2.96 (q, 2H, J = 6.7 Hz, CH2NH, H-1); 3.54 (t, 2H, J = 6.7 Hz, CH2N=, H-4); 3.78 (s, 3H, OCH3), 6.76–6.82 (br s, 1H, NH); 6.99–7.03 (m, 1H, H-5ʹ, Ar); 7.27–7.38 (m, 3H, H-2ʹ, H-4ʹ, H-6ʹ, Ar); 8.29 (s, 1H, N=CH). 13C-NMR (DMSO-d6) δ: 27.3 (C-2); 27.8 (C-3); 28.2 (OC(CH3)3); 39.6 (C-1); 55.0 (OCH3); 60.1 (C-4); 77.2 (OC(CH3)3); 111.9 (C-5ʹ, Ar); 116.6 (C-6ʹ, Ar); 120.6 (C-4ʹ, Ar); 129.6 (C-2ʹ, Ar); 137.6 (C-1ʹ, Ar); 155.5 (C=O); 159.4 (C-3ʹ, Ar); 160.4 (C=N).
{4-[(Benzo[1,3]dioxol-5-ylmethylene)-amino]-butyl}-carbamic acid tert-butyl ester (4e). Compound 4e was prepared in 98% yield (3.13 g) from 3,4-methylenedioxybenzaldehyde 3e (1.51 g, 10 mmol) according to the standard procedure as yellowish viscous oil. 1H-NMR (DMSO-d6) δ: 1.37 (s, 9H, Me3C); 1.30–1.46 (m, 2H, CH2, H-2); 1.51–1.61 (m, 2H, CH2, H-3); 2.94 (q, 2H, J = 6.9 Hz, CH2NH, H-1); 3.49 (t, 2H, J = 6.7 Hz, CH2N=, H-4); 6.07 (s, 2H, H-2ʹ, Ar), 6.77–6.82 (br s, 1H, NH); 6.97 (d, 1H, J = 8 Hz, H-6ʹ, H-7ʹ, Ar); 7.16–7.20 (m, 1H, H-6ʹ, H-7ʹ, Ar); 7.27–7.28 (m, 1H, H-4ʹ, Ar), 8.20 (s, 1H, N=CH). 13C-NMR (DMSO-d6) δ: 27.3 (C-2); 27.9 (C-3); 28.2 (OC(CH3)3); 39.6 (C-1); 59.8 (C-4); 77.4 (OC(CH3)3); 101.4 (C-2ʹ, Ar); 105.8 (C-6ʹ, Ar); 108.1 (C-7ʹ, Ar); 124.0 (C-4ʹ, Ar); 130.9 (C-5ʹ, Ar); 147.8 (C-3aʹ, C-7aʹ, Ar); 149.3 (C-3aʹ, C-7aʹ, Ar); 155.5 (C=O); 160.0 (C=N).
{4-[(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethylene)-amino]-butyl}-carbamic acid tert-butylester (4f). Compound 4f was prepared in 99% yield (3.31 g) from 2,3-dihydro-benzo[1,4]dioxin-6-carbaldehyde 3f (1.50 g, 10 mmol) according to the standard procedure as yellowish viscous oil. 1H-NMR (DMSO-d6) δ: 1.37 (s, 9H, Me3C); 1.34–1.45 (m, 2H, CH2, H-2); 1.51–1.60 (m, 2H, CH2, H-3); 2.94 (q, 2H, J = 6.8 Hz, CH2NH, H-1); 3.50 (t, 2H, J = 6.7 Hz, CH2N=, H-4); 4.23–4.30 (m, 4H, H-2ʹ, H-3ʹ, Ar), 6.77–6.81 (br s, 1H, NH); 6.88–6.91 (m, 1H, H-7ʹ, Ar); 7.18–7.21 (m, 2H, H-5ʹ, H-8ʹ, Ar); 8.17 (s, 1H, N=CH). 13C-NMR (DMSO-d6) δ: 27.3 (C-2); 27.9 (C-3); 28.2 (OC(CH3)3); 39.6 (C-1); 60.0 (C-4); 63.9 (C-2ʹ, Ar), 64.2 (C-3ʹ, Ar), 77.2 (OC(CH3)3); 116.0 (C-7ʹ, Ar); 117.1 (C-8ʹ, Ar); 121.2 (C-5ʹ, Ar); 129.8 (C-6ʹ, Ar); 143.4 (C-4aʹ, C-8aʹ, Ar); 145.4 (C-4aʹ, C-8aʹ, Ar); 155.5 (C=O); 159.6 (C=N).
3.1.3. Standard Procedure for Reduction of Aldimines 4a–f into N-Boc Monoprotected Diamines 5a–f
In a 50 mL two-necked round-bottomed flask, provided with a magnetic stirrer and reflux condenser, compound 4 (5 mmol) was dissolved in methanol p.a. (30 mL) under vigorous stirring and cooled at 0 °C. To this solution was added by small portions commercial sodium borohydride NaBH4 (0.95 g, 25 mmol) over a period of 20 min. The resulting suspension was stirred at 50 °C for 24 h (monitored by TLC on 0.2-mm precoated plates of silica gel 60 F-254, Merck). After cooling down to room temperature, the volatile compounds were removed in a rotary evaporator under reduced pressure, then deionized water (40 mL) was added in one portion to the crude residue. The mixture was transferred to a separating funnel and was extracted with dichloromethane (3 × 50 mL). The combined organic phases were dried over magnesium sulphate MgSO4, filtered on filter paper and the solvent was eliminated in vacuo. The crude residue was dried under high vacuum (10−2 Torr) at 25 °C for 2 h. The desired compound 5 was obtained as yellowish viscous oil and was further used without purification.
(4-Phenylmethylamino-butyl)-carbamic acid tert-butyl ester (5a). Compound 5a was prepared in 93% yield (1.25 g) from [4-(benzylidene-amino)-butyl]-carbamic acid tert-butyl ester 4a (1.38 g, 5 mmol) according to the standard procedure that gave 5a as yellowish viscous oil. 1H-NMR (DMSO-d6) δ: 1.37 (s, 9H, Me3C); 1.34–1.42 (m, 4H, CH2, H-2, H-3); 2.45 (t, 2H, J = 6.2 Hz, CH2NH, H-4); 2.89–2.91 (m, 2H, CH2NH, H-1); 3.66 (s, 2H, ArCH2NH); 6.78–6.82 (br s, 1H, NHCO); 7.18–7.13 (m, 1H, H-4ʹ, Ar); 7.26–7.33 (m, 4H, H-2ʹ, H-3ʹ, H-5ʹ, H-6ʹ, Ar). 13C-NMR (DMSO-d6) δ: 27.4 (C-3); 26.8 (C-2); 39.8 (C-1); 48.4 (C-4); 53.0 (ArCH2NH); 77.2 (OC(CH3)3); 126.3 (C-4ʹ, Ar); 127.8 (C-2ʹ, C-6ʹ, Ar); 128.0 (C-3ʹ, C-5ʹ, Ar); 141,0 (C-1ʹ, Ar); 155,5 (C=O).
[4-(4-Methoxy-phenylmethylamino)-butyl]-carbamic acid tert-butyl ester (5b). Compound 5b was prepared in 99% yield (1.52 g) from {4-[(4-methoxy-benzylidene)-amino]-butyl}-carbamic acid tert-butyl ester 4b (1.53 g, 5 mmol) according to the standard procedure that gave 5b as yellowish viscous oil. 1H-NMR (DMSO-d6) δ: 1.37 (s, 9H, Me3C); 1.33–1.41 (m, 4H, H-2, H-3); 2.45 (t, 2H, J = 6.5 Hz, CH2NH, H-4); 2.88–2.90 (m, 2H, CH2NHCO, H-1); 3.60 (s, 2H, ArCH2NH); 3.72 (s, 3H, OCH3); 6.76–6.80 (br s, 1H, NHCO); 6.85 (d, 2H, J = 8.5 Hz, H-2ʹ, H-6ʹ, Ar); 7.22 (d, 2H, J = 8.6 Hz, H-3ʹ, H-5ʹ, Ar). 13C-NMR (DMSO-d6) δ: 26.6 (C-2); 27.4 (C-3); 28.2 (OC(CH3)3); 39.8 (C-4); 48.2 (C-1); 52.3 (ArCH2NH); 54.9 (OCH3); 77.2 (OC(CH3)3); 113.4 (C-2ʹ, C-6ʹ, Ar); 129.0 (C-3ʹ, C-5ʹ, Ar); 132.6 (C-1ʹ, Ar); 155.5 (C=O); 158.0 (C-4ʹ, Ar).
[4-(2-Chloro-phenylmethylamino)-butyl]-carbamic acid tert-butyl ester (5c). Compound 5c was prepared in 85% yield (1.33 g) from {4-[(2-chloro-benzylidene)-amino]-butyl}-carbamic acid tert-butyl ester 4c (1.55 g, 5 mmol) according to the standard procedure that gave 5c as yellowish viscous oil. 1H-NMR (DMSO-d6) δ: 1.37 (s, 9H, Me3C); 1.34–1.43 (m, 4H, CH2, H-2, H-3, 4H); 2.47–2.52 (m, 2H, ArCH2NH, H-4); 2.88–2.92 (m, 2H, CH2NHCO, H-1); 3.75 (s, 2H, ArCH2NH); 6.77–6.80 (br s, 1H, NHCO); 7.21–7.33 (m, 2H, H-4ʹ, H-5ʹ, Ar); 7.37–7.4 (m, 1H, H-6ʹ, Ar); 7.5–7.53 (m, 1H, H-3ʹ, Ar). 13C-NMR (DMSO-d6) δ: 26.7 (C-2); 27.4 (C-3); 28.2 (OC(CH3)3); 39.8 (C-4); 48.5 (C-1); 50.0 (ArCH2NH); 77.2 (OC(CH3)3); 126.9 (C-5ʹ, Ar); 128.0 (C-4ʹ, Ar); 128.9 (C-6ʹ, Ar); 129.6 (C-3ʹ, Ar); 132.5 (C-1ʹ, Ar); 138.2 (C-2ʹ, Ar); 155.5 (C=O).
[4-(3-Methoxy-phenylmethylamino)-butyl]-carbamic acid tert-butyl ester (5d). Compound 5d was prepared in 90% yield (1.39 g) from {4-[(3-methoxy-benzylidene)-amino]-butyl}-carbamic acid tert-butyl ester 4d (1.53 g, 5 mmol) according to the standard procedure that gave 5d as pale pink viscous oil. 1H-NMR (DMSO-d6) δ: 1.37 (s, 9H, Me3C); 1.34–1.46 (m, 4H, H-2, H-3); 2.45 (t, 2H, J = 6.4 Hz, CH2NH, H-4); 2.89–2.91 (m, 2H, CH2NHCO, H-1); 3.64 (s, 2H, ArCH2NH); 3.74 (s, 3H, OCH3); 6.75–6.80 (br s, 1H, NHCO); 6.75–6.91 (m, 3H, H-4ʹ, H-5ʹ, H-6ʹ, Ar); 7.17–7.22 (m, 1H, H-2ʹ, Ar). 13C-NMR (DMSO-d6) δ: 26.8 (C-2); 27.4 (C-3); 28,2 (OC(CH3)3); 39.8 (C-4); 48.4 (C-1); 52.9 (ArCH2NH); 54.8 (OCH3); 77.2 (OC(CH3)3); 111.8 (C-5ʹ, Ar); 113.2 (C-6ʹ, Ar); 120.0 (C-4ʹ, Ar); 128.9 (C-2ʹ, Ar); 142.8 (C-1ʹ, Ar); 155.5 (C=O); 159.2 (C-3ʹ, Ar).
{4-[(Benzo[1,3]dioxol-5-ylmethyl)-amino]-butyl}-carbamic acid tert-butyl ester (5e). Compound 5e was prepared in 97% yield (1.56 g) from {4-[(benzo[1,3]dioxol-5-ylmethylene)-amino]-butyl}-carbamic acid tert-butyl ester 4e (1.60 g, 5 mmol) according to the standard procedure that gave 5e as yellowish viscous oil. 1H-NMR (DMSO-d6) δ: 1.37 (s, 9H, Me3C); 1.23–1.40 (m, 4H, H-2, H-3); 2.43 (t, 2H, J = 6.5 Hz, CH2NH, H-4); 2.89–2.91 (m, 2H, CH2NHCO, H-1); 3.64 (s, 2H, ArCH2NH); 5.96 (s, 2H, H-2ʹ, Ar); 6.73–6.82 (br s, 1H, NHCO); 6.73–6.90 (m, 3H, H-4ʹ, H-6ʹ, H-7ʹ, Ar). 13C-NMR (DMSO-d6) δ: 26.8 (C-2); 27.4 (C-3); 28.2 (OC(CH3)3); 39.8 (C-4); 48.1 (C-1); 52.7 (ArCH2NH); 77.2 (OC(CH3)3); 100.6 (C-2ʹ, Ar); 107.7 (C-6ʹ, Ar); 108.2 (C-7ʹ, Ar); 120.8 (C-4ʹ, Ar); 135.0 (C-5ʹ, Ar); 145.7 (C-3aʹ, C-7aʹ, Ar); 147.1 (C-3aʹ, C-7aʹ, Ar); 155.5 (C=O).
{4-[(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-amino]-butyl}-carbamic acid tert-butyl ester (5f). Compound 5f was prepared in 88% yield (1.48 g) from {4-[(2,3-dihydro-benzo[1,4]dioxin-6-ylmethylene)-amino]-butyl}-carbamic acid tert-butylester 4f (1.67 g, 5 mmol) according to the standard procedure that gave 5f as yellowish viscous oil. 1H-NMR (DMSO-d6) δ: 1.37 (s, 9H, Me3C); 1.33–1.43 (m, 4H, H-2, H-3); 2.42 (t, 2H, J = 6.4 Hz, CH2NH, H-4); 2.88–2.90 (m, 2H, CH2NHCO, H-1); 3.53 (s, 2H, ArCH2NH); 4.20 (s, 4H, H-2ʹ, H-3ʹ, Ar); 6.75–6.80 (br s, 1H, NHCO); 6.75–6.80 (m, 3H, H-5ʹ, H-7ʹ, H-8ʹ, Ar). 13C-NMR (DMSO-d6) δ: 26.8 (C-2); 27.4 (C-3); 28.2 (OC(CH3)3); 39.8 (C-4); 48.2 (C-1); 52.3 (ArCH2NH); 63.9 (C-2ʹ, C-3ʹ, Ar); 64.0 (C-2ʹ, C-3ʹ, Ar); 77.2 (OC(CH3)3); 116.4 (C-7ʹ, Ar); 116.5 (C-8ʹ, Ar); 120.6 (C-5ʹ, Ar); 134.0 (C-6ʹ, Ar); 141.9 (C-4aʹ, C-8aʹ, Ar); 143.0 (C-4aʹ, C-8aʹ, Ar); 155.5 (C=O).
3.1.4. Standard Procedure for the Preparation of Hydrochloride Salts 6a–f after Deprotection of N-Boc Monoprotected Diamines 5a–f
In a 100 mL two-necked round-bottomed flask provided with a magnetic stirrer and condenser, N-Boc monoprotected diamine 5 (12.1 mmol, 1 equiv.) was solubilized in 1,4-dioxane (80 mL) at room temperature under vigorous stirring for 10 min. To this homogeneous solution was added dropwise for 1 h a solution of 6 M HCl (20 mL). The reaction mixture was stirred during 4 h at 25 °C and was concentrated in a rotary evaporator under reduced pressure for elimination of volatile compounds. To the crystallized crude reaction mixture was added 60 mL of anhydrous Et2O and after triturating, the insoluble salt 6 was collected by filtration, on a Büchner funnel (porosity N°4) then washed with 6 × 10 mL of Et2O. The desired salt 6 was dried under high vacuum (10−2 Torr) at 25 °C for 1 h that gave a yellowish or white powder and was further used without purification.
N-1-Phenylmethyl-butane-1,4-diamine hydrochloride (6a). Compound 6a was prepared in 84% yield (2.18 g) from (4-phenylmethylamino-butyl)-carbamic acid tert-butyl ester 5a (3.37 g, 12.1 mmol) according to the standard procedure as yellowish powder. Mp > 260 °C. 1H-NMR (DMSO-d6) δ: 1.57–1.67 (m, 2H, CH2, H-3); 1.70–1.81 (m, 2H, CH2, H-2); 2.79 (t, 2H, J = 7.3 Hz, CH2NH, H-4); 2.89 (t, 2H, J = 7.5 Hz, CH2NH2, H-1); 4.10 (s, 2H, CH2NH, H-4); 7.4–7.45 (m, 3H, H-3ʹ, H-4ʹ, H-5ʹ, Ar); 7.58–7.62 (m, 2H, H-2ʹ, H-6ʹ, Ar); 8.19 (br s, 2H, NH2); 9.57 (br s, 1H, NH). 13C-NMR (DMSO-d6) δ: 22.3 (C-3); 24.0 (C-2); 38.0 (C-4); 45.5 (C-1); 49.7 (ArCH2NH); 128.5 (C-3ʹ, C-5ʹ, Ar); 128.8 (C-4ʹ, Ar); 130.1 (C-2ʹ, C-6ʹ, Ar); 132.0 (C-1ʹ, Ar).
N-1-(4-Methoxy-phenylmethyl)-butane1,4-diamine hydrochloride (6b). Compound 6b was prepared in 97% yield (2.87 g) from (4-methoxyphenylmethylamino-butyl)-carbamic acid tert-butyl ester 5b (3.73 g, 12.1 mmol) according to the standard procedure as pale brownish powder. Mp > 260 °C. 1H-NMR (DMSO-d6) δ: 1.57–1.66 (m, 2H, H-3); 1.69–1.79 (m, 2H, H-2); 2.77–2.84 (m, 4H, H-1, H-4); 3.76 (s, 3H, OCH3); 4.03 (s, 2H, ArCH2NH); 6.97 (d, 2H, J = 8.7 Hz, H-2ʹ, H-6ʹ, Ar); 7.51 (d, 2H, J = 8.7 Hz, H-3ʹ, H-5ʹ, Ar), 8.15 (br s, 2H, NH2); 9.41 (br s, 1H, NH). 13C-NMR (DMSO-d6) δ: 22.3 (C-3); 24.0 (C-2); 38.0 (C-4); 45.3 (C-1); 49.2 (ArCH2NH); 55.2 (OCH3); 113.9 (C-2ʹ, C-6ʹ, Ar); 123.7 (C-1ʹ, Ar); 131.6 (C-3ʹ, C-5ʹ, Ar); 159.6 (C-4ʹ, Ar).
N-1-(2-Chloro-phenylmethyl)-butane1,4-diamine hydrochloride (6c). Compound 6c was prepared in 98% yield (2.95 g) from (2-chlorophenylmethylamino-butyl)-carbamic acid tert-butyl ester 5c (3.79 g, 12.1 mmol) according to the standard procedure as pale brownish powder. Mp = 206–208 °C. 1H-NMR (DMSO-d6) δ: 1.60–1.69 (m, 2H, H-3); 1.74–1.84 (m, 2H, H-2); 2.73–2.84 (m, 2H, CH2NH, H-4); 2.93–3.02 (m, 2H, CH2NH2, H-1); 4.23 (t, 2H, J = 5.9 Hz, ArCH2NH); 7.39–7.48 (m, 2H, H-4ʹ, H-5ʹ, Ar); 7.52–7.56 (m, 1H, H-6ʹ, Ar); 7.81–7.85 (m, 1H, H-3ʹ, Ar); 8.18 (br s, 2H, NH2, 2H); 9.68 (br s, 1H, NH). 13C-NMR (DMSO-d6) δ: 22.3 (C-3); 24.0 (C-2); 38.0 (C-4); 46.1 (C-1); 46.7 (ArCH2NH); 127.5 (C-5ʹ, Ar); 129.5 (C-4ʹ, Ar); 129.9 (C-1ʹ, Ar); 130.7 (C-6ʹ, Ar); 131.9 (C-3ʹ, Ar); 133.5 (C-2ʹ, Ar).
N-1-(3-Methoxy-phenylmethyl)-butane-1,4-diamine hydrochloride (6d). Compound 6d was prepared in 98% yield (2.98 g) from [4-(3-methoxy-phenylmethylamino)-butyl]-carbamic acid tert-butyl ester 5d (3.73 g, 12.1 mmol) according to the standard procedure as pale brownish powder. Mp = 190–192 °C. 1H-NMR (DMSO-d6) δ: 1.58–1.67 (m, 2H, H-3); 1.71–1.81 (m, 2H, H-2); 2.78 (t, 2H, J = 7.3 Hz, CH2NH, H-4); 2.87 (t, 2H, J = 7.6 Hz, CH2NH2, H-1); 3.77 (s, 3H, OCH3); 4.08 (t, 2H, J = 5.6 Hz, ArCH2NH); 6.94–6.97 (m, 1H, H-5ʹ, Ar); 7.12–7.14 (m, 1H, H-6ʹ, Ar); 7.29–7.35 (m, 2H, H-2ʹ, H-4ʹ, Ar); 8.18 (br s, 2H, NH2); 9.58 (br s, 1H, NH). 13C-NMR (DMSO-d6) δ: 22.3 (C-3); 24.0 (C-2); 38.0 (C-4); 45.5 (C-1); 49.7 (ArCH2NH); 55.2 (OCH3); 114.5 (C-5ʹ, Ar); 115.5 (C-6ʹ, Ar); 122.0 (C-4ʹ, Ar); 129.6 (C-2ʹ, Ar); 133.4 (C-1ʹ, Ar); 159.2 (C-3ʹ, Ar).
N-1-Benzo[1,3]dioxol-5-ylmethyl-butane-1,4-diamine hydrochloride (6e). Compound 6e was prepared in 72% yield (2.25 g) from {4-[(benzo[1,3]dioxol-5-ylmethyl)-amino]-butyl}-carbamic acid tert-butyl ester 5e (3.90 g, 12.1 mmol) according to the standard procedure as pale brownish powder. Mp > 260 °C. 1H-NMR (DMSO-d6) δ: 1.58–1.67 (m, 2H, H-3); 1.71–1.81 (m, 2H, H-2); 2.78 (t, 2H, J = 7.3 Hz, CH2NH, H-4); 2.87 (t, 2H, J = 7.6 Hz, CH2NH2, H-1); 3.77 (s, 3H, OCH3); 4.08 (t, 2H, J = 5.6 Hz, ArCH2NH); 6.94–6.97 (m, 1H, H-5ʹ, Ar); 7.12–7.14 (m, 1H, H-6ʹ, Ar); 7.29–7.35 (m, 2H, H-2ʹ, H-4ʹ, Ar); 8.18 (br s, 2H, NH2); 9.58 (br s, 1H, NH). 13C-NMR (DMSO-d6) δ: 22.3 (C-3); 24.0 (C-2); 38.0 (C-4); 45.2 (C-1); 49.4 (ArCH2NH); 101.2 (C-2ʹ, Ar); 108.2 (C-6ʹ, Ar); 110.4 (C-7ʹ, Ar); 124.1 (C-4ʹ, Ar); 125.4 (C-5ʹ, Ar); 147.2 (C-3aʹ, C-7aʹ, Ar); 147.6 (C-3aʹ, C-7aʹ, Ar).
N-1-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-butane-1,4-diamine hydrochloride (6f). Compound 6e was prepared in 92% yield (3.04 g) from {4-[(2,3-dihydro-benzo[1,4]dioxin-6-ylmethyl)-amino]-butyl}-carbamic acid tert-butyl ester 5f (4.07 g, 12.1 mmol) according to the standard procedure as pale brownish powder. Mp = 239–241 °C. 1H-NMR (DMSO-d6) δ: 1.57–1.66 (m, 2H, H-3); 1.68–1.78 (m, 2H, H-2); 2.73–2.82 (m, 4H, H-1, H-4); 3.97 (t, 2H, J = 5.4 Hz, ArCH2NH); 4.24 (s, 4H, OCH2CH2O); 6.87 (d, 1H, J = 8.2 Hz, H-7ʹ, Ar); 7.01–7.04 (m, 1H, H-8ʹ, Ar); 7.15 (m, 1H, H-5ʹ, Ar); 8.18 (br s, 2H, NH2); 9.43 (br s, 1H, NH). 13C-NMR (DMSO-d6) δ: 22.3 (C-3); 24.0 (C-2); 38.0 (C-4); 45.2 (C-1); 49.1 (ArCH2NH); 64.0 (C-2ʹ, Ar); 64.1 (C-3ʹ, Ar); 117.0 (C-7ʹ, Ar); 119.0 (C-8ʹ, Ar); 123.2 (C-5ʹ, Ar); 124.7 (C-6ʹ, Ar); 143.1 (C-4aʹ, C-8aʹ, Ar); 143.8 (C-4aʹ, C-8aʹ, Ar).
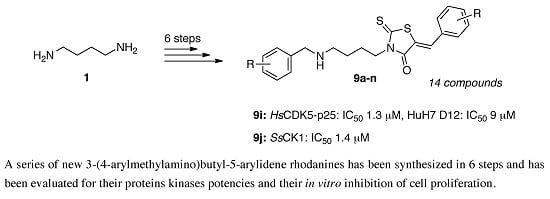
3.1.5. Standard Procedure for the Preparation of N-Substituted 5-Arylidene Rhodanine Derivatives 9a–f under Microwave Irradiation Using “One-Pot Two-steps” Protocol
In a 10 mL glass tube were placed successively bis-(carboxymethyl)trithiocarbonate 7 (0.11 g, 0.50 mol, 1 equiv.), dimethoxyethane (2 mL), triethylamine (135 mL, 101 mg, 1 mmol, 2 equiv. or 202 mL, 152 mg, 1.5 mmol, 3 equiv.) and hydrochloride salt 6 (0.5 mmol, 1 equiv.). The glass tube was sealed with a snap cap and placed in the Monowave® 300 Anton Paar microwave cavity (p = 800 Watt). The reaction mixture was irradiated at 90 °C for 15–60 min. under vigorous magnetic stirring. After microwave dielectric heating, the crude reaction mixture was allowed to cool down at room temperature, aldehyde 3 (0.5 mol, 1 equiv.) was added to the cooled reaction mixture which was immediately submitted to microwave irradiation at 110 °C for 5–30 min. After cooling down to room temperature, the volatile compounds of the reaction mixture were removed in a rotary evaporator under reduced pressure. To the crude reaction mixture was added 4 mL of MeOH and after triturating, the insoluble product 9 was collected by filtration on a Büchner funnel (porosity N°4), washed with deionized water (5 mL), triturated and mixing with ethanol (3 × 5 mL) during 3 h, washed successively with ethanol (5 mL) and anhydrous ether (5 × 2 mL), then dried under high vacuum (10−2 Torr) at 25 °C for 1 h. The desired compound 9 was eventually purified by recrystallization in EtOH after control by 1H-NMR in solution of CDCl3/TFA (98:2).
(5Z)3-[4-(4-Methoxyphenylmethylamino)butyl]-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one (9a). According to the standard procedure, compound 9a was prepared in 10% yield (23 mg) from N-1-(4-methoxy-phenylmethyl)-butane-1,4-diamine hydrochloride 6b (122.3 mg, 0.5 mmol, 1 equiv.), triethylamine (202 μL, 152 mg, 1.5 mmol, 3 equiv.) and piperonaldehyde 3e (75.1 mg, 0.5 mmol, 1 equiv.) after 30 min. at 90 °C, followed by a second reaction time of 30 min. at 110 °C (for condensation step with 3e), which gave 9a as a yellowish powder. Mp = 170–229 °C (decomposition). 1H-NMR (CDCl3/TFA 98:2) δ: 1.61–1.69 (m, 4H, CH2, H-2ʹʹ, H-3ʹʹ); 3.05–3.11 (m, 2H, ArCH2NH); 3.73 (s, 3H, OCH3); 4.00–4.08 (m, 4H, CH2, H-1ʹʹ, H-4ʹʹ), 5.97 (s, 2H, CH2, H-2ʹ); 6.80–7.16 (m, 7H, H-2ʹʹʹ, H-3ʹʹʹ, H-5ʹʹʹ, H-6ʹʹʹ, H-4ʹ, H-6ʹ, H-7ʹ, Ar); 7.55 (br s, 1H, NH); 7.61 (s, 1H, HC=C). 13C-NMR (CDCl3/TFA 98:2) δ: 23.1 (C-2ʹʹ, 3ʹʹ); 23.8 (C-3ʹʹ, 2ʹʹ); 44.2 (C-4ʹʹ, C-1ʹʹ); 47.0 (ArCH2NH); 52.1 (C-1ʹʹ, C-4ʹʹ); 55.5 (OCH3); 102.2 (C-2ʹ); 109.4 (C-2ʹʹʹ, C-6ʹʹʹ); 109.5 (C-3ʹʹʹ, C-5ʹʹʹ); 115.0 (C-6ʹ); 120.0 (C-1ʹʹʹ); 127.5 (C=CH); 128.2 (C-7ʹ); 131.2 (C-4ʹ); 135.0 (C=CH); 148.8 (C-5ʹ); 148.9 (C-4ʹʹʹ); 150.6 (C-3ʹa); 150.9 (C-7ʹa); 169.4 (C=O, C-4); 192.9 (C=S, C-2). HRMS, m/z: 457.1256 found (calculated for C23H25N2O4S2 [M + H]+ requires 457.1253).
(5Z)3-[4-(4-Methoxyphenylmethylamino)butyl]-5-(4-methoxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one (9b). According to the standard procedure, compound 9b was prepared in 6% yield (13.3 mg) from N-1-(4-methoxy-phenylmethyl)-butane-1,4-diamine hydrochloride 6b (122.3 mg, 0.5 mmol, 1 equiv.), triethylamine (202 μL, 152 mg, 1.5 mmol, 3 equiv.) and 4-methoxybenzaldehyde 3b (68.1 mg, 0.5 mmol, 1 equiv.) after 30 min. at 90 °C, followed by a second reaction time of 30 min. at 110 °C (for condensation step with 3b), which gave 9b as a yellowish powder. Mp = 253–255 °C. 1H-NMR (CDCl3/TFA 98:2) δ: 1.67–1.73 (m, 4H, CH2, H-2ʹʹ, H-3ʹʹ); 3.07–3.13 (m, 2H, ArCH2NH); 3.81 (s, 6H, ArOCH3); 4.04–4.11 (s, 4H, CH2, H-1ʹʹ, H-4ʹʹ), 6.94 (dd, 4H, J = 8.8 Hz, H-2ʹ, H-6ʹ, H-2ʹʹʹ, H-6ʹʹʹ, Ar); 7.40 (d, 4H, J = 8.8 Hz, H-3ʹ, H-5ʹ, H-3ʹʹʹ, H-5ʹʹʹ, Ar); 7.60 (br s, 1H, NH); 7.67 (s, 1H, CH=). 13C-NMR (CDCl3/TFA 98:2) δ: 24.3 (C-2ʹʹ, C-3ʹʹ); 44.0 (C-1ʹʹ, C-4ʹʹ); 46.8 (ArCH2NH); 55.6 (C-4ʹ, OCH3, C-4ʹʹʹ, OCH3); 115.1 (C-2ʹ, C-6ʹ, C-2ʹʹʹ, C-6ʹʹʹ); 119.6 (C-1ʹ, C-1ʹʹʹ); 126.0 (C=); 133.1 (C-3ʹ, C-5ʹ, C-3ʹʹʹ, C-5ʹʹʹ); 130.5 (C-5ʹ, C-5ʹʹʹ); 134.6 (CH=); 162.0 (C-4ʹ, C-4ʹʹʹ); 169.0 (C=O, C-4); 193.2 (C=S, C-2). HRMS, m/z: 443.1463 found (calculated for C23H27N2O3S2 [M + H]+ requires 443.1460).
(5Z)3-[4-(4-Methoxyphenylmethylamino)butyl]-5-(2,3-dihydro-1,4-benzodioxin-6-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one (9c). According to the standard procedure, compound 9c was prepared in 5% yield (11.8 mg) from N-1-(4-methoxy-phenylmethyl)-butane-1,4-diamine hydrochloride 6b (122.3 mg, 0.5 mmol, 1 equiv.), triethylamine (202 μL, 152 mg, 1.5 mmol, 3 equiv.) and 2,3-dihydro-1,4-benzodioxin-6-carboxaldehyde 3f (82.1 mg, 0.5 mmol, 1 equiv.) after 30 min. at 90 °C, followed by a second reaction time of 30 min. at 110 °C (for condensation step with 3f), which gave 9c as a yellowish powder. Mp = 255–261 °C. 1H-NMR (CDCl3/TFA 98:2) δ: 1.68 (s, 4H, CH2, H-2ʹʹ, H-3ʹʹ); 3.03–3.08 (m, 2H, ArCH2NH); 3.73 (s, 3H, OCH3); 4.07 (s, 4H, CH2, H-1ʹʹ, H-4ʹʹ), 4.20–4.25 (m, 2H, CH2, H-2ʹ, H-3ʹ); 6.84–6.97 (m, 7H, H-5ʹ, H-7ʹ, H-8ʹ, H-2ʹʹʹ, H-3ʹʹʹ, H-5ʹʹʹ, H-6ʹʹʹ, Ar); 7.58 (s, 1H, CH=); 7.60 (br s, 1H, NH). 13C-NMR (CDCl3/TFA 98:2) δ: 24.2 (C-2ʹʹ, C-3ʹʹ); 44.1 (C-4ʹʹ, C-1ʹʹ); 47.1 (ArCH2NH); 55.0 (OCH3); 64.2 (C-2ʹ); 64.8 (C-3ʹ); 118.4 (C-5ʹ, C-7ʹ, C-8ʹ); 119.7 (C-2ʹʹʹ, C-6ʹʹʹ); 120.3 (C=); 125.7 (C-3ʹʹʹ, C-5ʹʹʹ); 126.8 (C-6ʹ); 135.0 (CH=); 143.9 (C-8ʹa); 146.7 (C-4ʹa); 169.5 (C=O, C-4); 193.2 (C=S, C-2). HRMS, m/z: 471.1412 found (calculated for C24H27N2O4S2 [M + H]+ requires 471.1415).
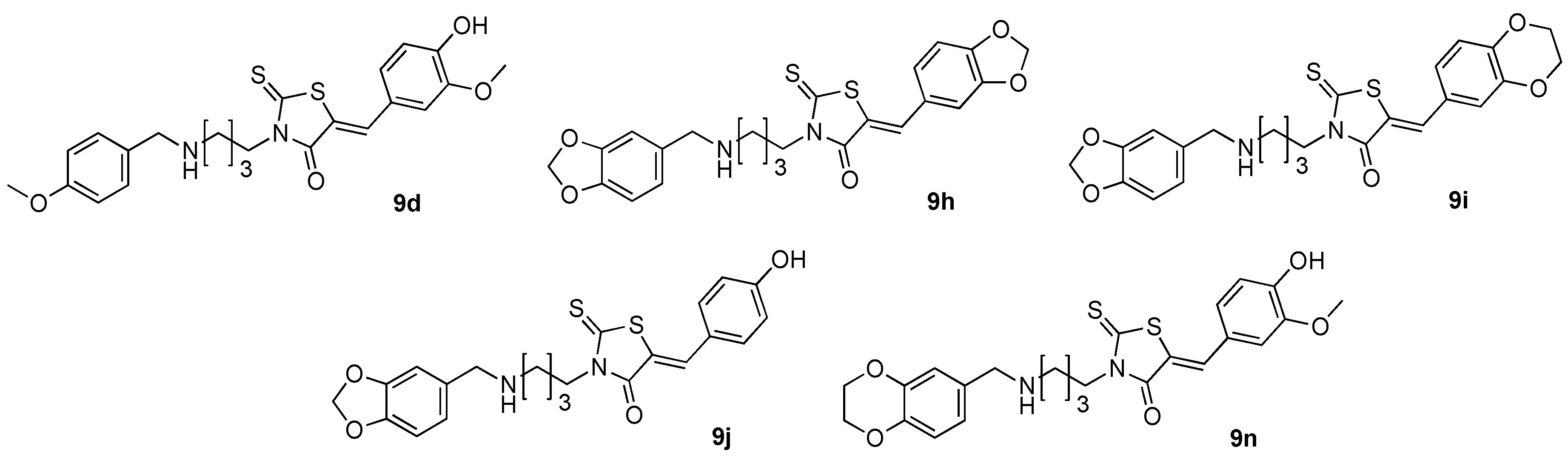
(5Z)3-[4-(4-Methoxyphenylmethylamino)butyl]-5-(4-hydroxy-3-methoxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one (9d). According to the standard procedure, compound 9d was prepared in 30% yield (68.8 mg) from N-1-(4-methoxy-phenylmethyl)-butane-1,4-diamine hydrochloride 6b (122.3 mg, 0.5 mmol, 1 equiv.), triethylamine (202 μL, 152 mg, 1.5 mmol, 3 equiv.) and 4-hydroxy-3-methoxybenzaldehyde 3g (76.1 mg, 0.5 mmol, 1 equiv.) after 30 min. at 90 °C, followed by a second reaction time of 30 min. at 110 °C (for condensation step with 3g), which gave 9d as a yellowish powder. Mp = 191–193 °C. 1H-NMR (CDCl3/TFA 98:2) δ: 1.69 (m, 4H, CH2, H-2ʹʹ, H-3ʹʹ); 3.06–3.07 (m, 2H, ArCH2NH); 3.72 (s, 3H, C-4ʹʹʹ, OCH3); 3.86 (s, 3H, C-3ʹ, OCH3); 4.04–4.08 (m, 4H, CH2, H-1ʹʹ, H-4ʹʹ), 6.81–7.17 (m, 7H, H-2ʹ, H-5ʹ, H-6ʹ, H-2ʹʹʹ, H-3ʹʹʹ, H-5ʹʹʹ, H-6ʹʹʹ, Ar); 7.55 (br s, 1H, NH); 7.60 (s, 1H, CH=C). 13C-NMR (CDCl3/TFA 98:2) δ: 23.1 (C-2ʹʹ, C-3ʹʹ); 23.9 (C-2ʹʹ, C-3ʹʹ); 43.1 (C-1ʹʹ, C-4ʹʹ); 47.0 (ArCH2NH); 55.5 (ArOCH3); 56.1 (ArOCH3); 112.6 (C-6ʹ); 115.0 (C-2ʹʹʹ, C-6ʹʹʹ); 115.6 (C-2ʹ); 118.9 (C-1ʹʹʹ); 121.2 (C=); 125.9 (C-1ʹ); 126.8 (C-5ʹ); 131.2 (C-3ʹʹʹ, C-5ʹʹʹ); 135.9 (CH=); 147.2 (C-4ʹʹʹ); 148.9 (C-3ʹ); 160.8 (C-4ʹ); 169.4 (C=O, C-4); 193.1 (C=S, C-2). HRMS, m/z: 459.1412 found (calculated for C23H27N2O4S2 [M + H]+ requires 459.1416).
(5Z)3-[4-(3-Methoxyphenylmethylamino)butyl]-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one (9e). According to the standard procedure, compound 9e was prepared in 21% yield (48.0 mg) from N-1-(3-methoxy-phenylmethyl)-butane-1,4-diamine hydrochloride 6d (122.3 mg, 0.5 mmol, 1 equiv.), triethylamine (202 μL, 152 mg, 1.5 mmol, 3 equiv.) and piperonaldehyde 3e (75.1 mg, 0.5 mmol, 1 equiv.) after 15 min. at 90 °C, followed by a second reaction time of 15 min. at 110 °C (for condensation step with 3e), which gave 9e as a yellowish powder. Mp = 178–246 °C (decomposition). 1H-NMR (CDCl3/TFA 98:2) δ: 1.82 (s, 4H, CH2, H-2ʹʹ, H-3ʹʹ); 3.19–3.22 (m, 2H, ArCH2NH); 3.83 (s, 3H, OCH3); 4.13-4.23 (m, 4H, CH2, H-1ʹʹ, H-4ʹʹ), 6.10 (s, 2H, CH2, H-2ʹ); 6.92–7.28 (m, 6H, H-2ʹʹʹ, H-4ʹʹʹ, H-6ʹʹʹ, H-4ʹ, H-6ʹ, H-7ʹ, Ar), 7.33–7.38 (m, 1H, H-5ʹʹʹ, Ar), 7.55 (br s, 1H, NH); 7.70 (s, 1H, CH=). 13C-NMR (CDCl3/TFA 98:2) δ: 23.1 (C-2ʹʹ, C-3ʹʹ); 23.9 (C-3ʹʹ, C-2ʹʹ); 42.9 (C-4ʹʹ, C-1ʹʹ); 46.8 (ArCH2NH); 52.1 (C-1ʹʹ, C-4ʹʹ); 55.3 (OCH3); 102.1 (C-2ʹ); 109.3 (C-6ʹʹʹ); 109.4 (C-5ʹʹʹ); 114.9 (C-4ʹʹʹ); 115.9 (C-2ʹʹʹ); 119.8 (C-1ʹʹʹ); 121.8 (C-6ʹ); 127.4 (C=); 127.6 (C-5ʹ); 127.9 (C-7ʹ); 130.7 (C-4ʹ); 134.5 (CH=); 148.8 (C-7ʹa); 150.5 (C-3ʹa); 160.3 (C-3ʹʹʹ); 168.5 (C=O, C-4); 193.1 (C=S, C-2). HRMS, m/z: 457.1259 found (calculated for C23H25N2O4S2 [M + H]+ requires 457.1257).
(5Z)3-[4-(3-Methoxyphenylmethylamino)butyl]-5-(2,3-dihydro-1,4-benzodioxin-6-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one (9f). According to the standard procedure, compound 9c was prepared in 9% yield (21.2 mg) from N-1-(3-methoxy-phenylmethyl)-butane-1,4-diamine hydrochloride 6d (122.3 mg, 0.5 mmol, 1 equiv.), triethylamine (202 μL, 152 mg, 1.5 mmol, 3 equiv.) and 2,3-dihydro-1,4-benzodioxin-6-carboxaldehyde 3f (82.1 mg, 0.5 mmol, 1 equiv.) after 15 min. at 90 °C, followed by a second reaction time of 15 min. at 110 °C (for condensation step with 3f), which gave 9f as a yellowish powder. Mp = 247–251 °C. 1H-NMR (CDCl3/TFA 98:2) δ: 1.84 (s, 4H, CH2, H-2ʹʹ, H-3ʹʹ); 3.04–3.12 (m, 2H, ArCH2NH); 3.95 (s, 3H, OCH3); 4.23 (s, 4H, CH2, H-1ʹʹ, H-4ʹʹ); 4.37–4.40 (m, 4H, H-2ʹ, H-3ʹ); 6.99–7.28 (m, 6H, H-2ʹʹʹ, H-4ʹʹʹ, H-5ʹʹʹ, H-6ʹʹʹ, H-5ʹ, H-7ʹ, H-8ʹ, Ar); 7.60 (br s, 1H, NH); 7.74 (s, 1H, CH=). 13C-NMR (CDCl3/TFA 98:2) δ: 24.1 (C-2ʹʹ, C-3ʹʹ); 44.1 (C-1ʹʹ, C-4ʹʹ); 47.3 (ArCH2NH); 64.2 (C-2ʹ); 64.8 (C-3ʹ); 118.5 (C-5ʹ, C-7ʹ, C-8ʹ); 119.8 (C-4ʹʹʹ, C-6ʹʹʹ); 125.9 (C-2ʹʹʹ, C-5ʹʹʹ); 126.8 (C=); 135.3 (CH=); 143.8 (C-8ʹa); 146.6 (C-4ʹa); 168.5 (C=O, C-4); 193.3 (C=S, C-2). HRMS, m/z: 471.1412 found (calculated for C24H27N2O4S2 [M + H]+ requires 471.1415).
(5Z)3-[4-(3-Methoxyphenylmethylamino)butyl]-5-(3-methoxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one (9g). According to the standard procedure, compound 9g was prepared in 5% yield (11.1 mg) from N-1-(3-methoxy-phenylmethyl)-butane-1,4-diamine hydrochloride 6d (122.3 mg, 0.5 mmol, 1 equiv.), triethylamine (202 μL, 152 mg, 1.5 mmol, 3 equiv.) and 3-methoxybenzaldehyde 3d (68.1 mg, 0.5 mmol, 1 equiv.) after 30 min. at 90 °C, followed by a second reaction time of 30 min at 110 °C (for condensation step with 3d), which gave 9g as a yellowish powder. Mp = 225–227 °C. 1H-NMR (CDCl3/TFA 98:2) δ: 1.73 (s, 4H, CH2, H-2ʹʹ, H-3ʹʹ); 3.06–3.10 (m, 2H, ArCH2NH); 3.80 (s, 6H, ArOCH3); 4.11 (s, 4H, CH2, H-1ʹʹ, H-4ʹʹ), 6.93–6.96 (m, 4H, H-2ʹ, H-6ʹ, H-2ʹʹʹ, H-6ʹʹʹ, Ar); 7.04 (d, 2H, J = 7.7 Hz, H-4ʹ, H-4ʹʹʹ, Ar), 7.32 (m, 2H, H-5ʹ, H-5ʹʹʹ, Ar), 7.55 (br s, 1H, NH); 7.66 (s, 1H, CH=). 13C-NMR (CDCl3/TFA 98:2) δ: 24.3 (C-2ʹʹ, C-3ʹʹ); 44.0 (C-1ʹʹ, C-4ʹʹ); 47.1 (ArCH2NH); 55.5 (ArOCH3); 115.3 (C-6ʹ, C-6ʹʹʹ); 117.3 (C-2ʹ, C-2ʹʹʹ); 123.1 (C=); 123.6 (C-4ʹ, C-4ʹʹʹ); 130.5 (C-5ʹ, C-5ʹʹʹ); 134.0 (CH=); 134.5 (C-1ʹ, C-1ʹʹʹ); 160.0 (C-3ʹ, C-3ʹʹʹ); 168.5 (C=O, C-4); 193.3 (C=S, C-2). HRMS, m/z: 443.1463 found (calculated for C23H27N2O3S2 [M + H]+ requires 443.1465).
(5Z)3-[4-(1,3-Benzodioxol-5-ylmethylamino)butyl]-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one (9h). According to the standard procedure, compound 9h was prepared in 59% yield (139 mg) from N-1- benzo[1,3]dioxol-5-ylmethyl-butane-1,4-diamine hydrochloride 6e (129.4 mg, 0.5 mmol, 1 equiv.), triethylamine (135 μL, 101 mg., 1 mmol, 2 equiv.) and piperonaldehyde 3e (75.1 mg, 0.5 mmol, 1 equiv.) after 15 min. at 90 °C, followed by a second reaction time of 15 min. at 110 °C (for condensation step with 3e), which gave 9h as a yellowish powder. Mp = 245–256 °C. 1H-NMR (CDCl3/TFA 98:2) δ: 1.74 (s, 4H, CH2, H-2ʹʹ, H-3ʹʹ); 3.04–3.05 (m, 2H, ArCH2NH); 4.05–4.07 (m, 4H, CH2, H-1ʹʹ, H-4ʹʹ); 5.91 (s, 2H, CH2, H-2ʹʹʹ); 6.00 (s, 2H, CH2, H-2ʹ); 6.72–7.01 (m, 6H, H-4ʹ, H-6ʹ, H-7ʹ, H-4ʹʹʹ, H-6ʹʹʹ, H-7ʹʹʹ, Ar); 7.58 (s, 1H, CH=); 7.65 (br s, 1H, NH);. 13C-NMR (CDCl3/TFA 98:2) δ: 23.1 (C-2ʹʹ, C-3ʹʹ); 24.0 (C-3ʹʹ); 43.1 (C-1ʹʹ, C-4ʹʹ); 46.5 (ArCH2NH); 51.9 (C-4ʹʹ, C-1ʹʹ); 101.8 (C-2ʹ, C-2ʹʹʹ); 102.2 (C-2ʹʹʹ, C-2ʹ); 109.0 (C-6ʹʹʹ); 109.3 (C-6ʹ); 109.4 (C-7ʹʹʹ); 109.8 (C-7ʹ); 119.8 (C-5ʹʹʹ); 122.6 (C=); 124.3 (C-4ʹʹʹ); 127.4 (C-4ʹ); 128.1 (C-5ʹ); 134.9 (CH=); 148.6 (C-7ʹa); 148.9 (C-3ʹa); 149.2 (C-7ʹʹʹa); 150.6 (C-3ʹʹʹa); 168.9 (C=O, C-4); 193.0 (C=S, C-2). HRMS, m/z: 471.1048 found (calculated for C23H23N2O5S2 [M + H]+ requires 471.1049).
(5Z)3-[4-(1,3-Benzodioxol-5-ylmethylamino)butyl]-5-(2,3-dihydro-1,4-benzodioxin-6-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one (9i). According to the standard procedure, compound 9i was prepared in 59% yield (143 mg) from N-1-benzo[1,3]dioxol-5-ylmethyl-butane-1,4-diamine hydrochloride 6e (129.4 mg, 0.5 mmol, 1 equiv.), triethylamine (135 μL, 101 mg., 1 mmol, 2 equiv.) and 2,3-dihydro-1,4-benzodioxin-6-carboxaldehyde 3f (82.1 mg, 0.5 mmol, 1 equiv.) after 15 min. at 90 °C, followed by a second reaction time of 15 min. at 110 °C (for condensation step with 3f), which gave 9i as a yellowish powder. Mp = 253–257 °C. 1H-NMR (CDCl3/TFA 98:2) δ: 1.68 (s, 4H, CH2, H-2ʹʹ, H-3ʹʹ); 3.05–3.06 (m, 2H, ArCH2NH); 4.03–4.04 (m, 4H, CH2, H-1ʹʹ, H-4ʹʹ); 4.20–4.25 (m, 4H, CH2, H-2ʹ, H-3ʹ); 5.89 (s, 2H, CH2, H-2ʹʹʹ); 6.70 (s, 3H, H-4ʹʹʹ, H-6ʹʹʹ, H-7ʹʹʹ, Ar); 6.85–6.96 (m, 3H, H-5ʹ, H-7ʹ, H-8ʹ, Ar); 7.50 (br s, 1H, NH); 7.55 (s, 1H, CH=). 13C-NMR (CDCl3/TFA 98:2) δ: 23.1 (C-2ʹʹ, C-3ʹʹ); 23.8 (C-3ʹʹ, C-2ʹʹ); 43.1 (C-1ʹʹ, C-4ʹʹ); 47.0 (ArCH2NH); 52.4 (C4ʹʹ, C-1ʹʹ); 64.2 (C-2ʹ, C-3ʹ); 64.8 (C-3ʹ, C-2ʹ); 101.8 (C-2ʹʹʹ); 109.1 (C-6ʹʹʹ); 109.5 (C-7ʹʹʹ); 118.5 (C-7ʹ); 119.7 (C-5ʹʹʹ); 119.8 (C-8ʹ); 122.3 (C=); 124.1 (C-4ʹʹʹ); 125.9 (C-5ʹ); 126.6 (C-6ʹ); 135.5 (CH=); 144.0 (C-7ʹʹʹa); 146.9 (C-3ʹʹʹa); 148.6 (C-8ʹa); 149.4 (C-4ʹa); 169.5 (C=O, C-4); 193.2 (C=S, C-2). HRMS, m/z: 485.1205 found (calculated for C24H25N2O5S2 [M + H]+ requires 485.1203).
(5Z)3-[4-(1,3-Benzodioxol-5-ylmethylamino)butyl]-5-(4-hydroxy-benzylidene)-2-thioxo-1,3-thiazolidin-4-one (9j). According to the standard procedure, compound 9j was prepared in 34% yield (75.2 mg) from N-1-benzo[1,3]dioxol-5-ylmethyl-butane-1,4-diamine hydrochloride 6e (129.4 mg, 0.5 mmol, 1 equiv.), triethylamine (135 μL, 101 mg, 1 mmol, 2 equiv.) and 4-hydroxybenzaldehyde 3h (61.1 mg, 0.5 mmol, 1 equiv.) after 15 min. at 90 °C, followed by a second reaction time of 15 min. at 110 °C (for condensation step with 3h), which gave 9i as a orange powder. Mp = 93–124 °C (decomposition). 1H-NMR (CDCl3/TFA 98:2) δ: 1.59–1.73 (m, 4H, CH2, H-2ʹʹ, H-3ʹʹ); 3.01–3.13 (m, 2H, ArCH2NH); 4.00–4.05 (m, 4H, CH2, H-1ʹʹ, H-4ʹʹ); 5.88 (s, 2H, CH2, H-2ʹʹʹ); 6.66–6.69 (m, H-4ʹʹʹ, H-6ʹʹʹ, H-7ʹʹʹ, Ar); 6.86 (dd, 2H, J = 8.7 Hz, H-2ʹ, H-6ʹ, Ar); 7.32 (dd, 2H, J = 8.7 Hz, H-3ʹ, H-5ʹ, Ar); 7.55 (br s, 1H, NH); 7.60 (s, 1H, CH=, 1H). 13C-NMR (CDCl3/TFA 98:2) δ: 22.5 (C-2ʹʹ, C-3ʹʹ); 23.2 (C-3ʹʹ, C-2ʹʹ); 42.5 (C-1ʹʹ, C-4ʹʹ); 46.4 (ArCH2NH); 51.9 (C-4ʹʹ, C-1ʹʹ); 101.2 (C-2ʹʹʹ); 108.5 (C-6ʹʹʹ); 108.8 (C-7ʹʹʹ); 116.1 (C-2ʹ, C-6ʹ); 118.6 (C-5ʹʹʹ); 121.7 (C=); 123.4 (C-4ʹʹʹ); 125.6 (C-1ʹ); 132.9 (C-3ʹ, C-5ʹ); 134.8 (CH=); 148.0 (C-7ʹʹʹa); 148.8 (C-3ʹʹʹa); 157.6 (C-4ʹ); 169.0 (C=O, C-4); 192.6 (C=S, C-2). HRMS, m/z: 443.1099 found (calculated for C22H23N2O4S2 [M + H]+ requires 443.1095).
(5Z)3-[4-(1,3-Benzodioxol-5-ylmethylamino)butyl]-5-(4-hydroxy-3-methoxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one (9k). According to the standard procedure, compound 9k was prepared in 46% yield (109 mg) from N-1-benzo[1,3]dioxol-5-ylmethyl-butane-1,4-diamine hydrochloride 6e (129.4 mg, 0.5 mmol, 1 equiv.), triethylamine (202 μL, 152 mg, 1.5 mmol, 3 equiv.) and 4-hydroxy-3-methoxybenzaldehyde 3g (76.1 mg, 0.5 mmol, 1 equiv.) after 30 min. at 90 °C, followed by a second reaction time of 30 min. at 110 °C (for condensation step with 3g), which gave 9k as a red powder. Mp = 200–203 °C. 1H-NMR (CDCl3/TFA 98:2) δ: 1.71 (s, 4H, CH2, H-2ʹʹ, H-3ʹʹ); 3.05–3.06 (m, 2H, ArCH2NH); 3.88 (s, 3H, OCH3); 4.04 (s, 4H, CH2, H-1ʹʹ, H-4ʹʹ); 5.91 (s, 2H, CH2, H-2ʹʹʹ); 6.74 (s, 3H, H-4ʹʹʹ, H-6ʹʹʹ, H-7ʹʹʹ, Ar); 6.87–7.02 (m, 3H, H-2ʹ, H-5ʹ, H-6ʹ, Ar); 7.58 (s, 1H, CH=); 7.65 (br s, 1H, NH). 13C-NMR (CDCl3/TFA 98:2) δ: 23.1 (C-2ʹʹ, C-3ʹʹ); 23.9 (C-3ʹʹ, C-2ʹʹ); 42.9 (C-1ʹʹ, C-4ʹʹ); 46.6 (ArCH2NH); 52.1 (C-4ʹʹ, C-1ʹʹ); 56.1 (OCH3); 101.8 (C-2ʹʹʹ); 109.0 (C-6ʹʹʹ); 109.6 (C-7ʹʹʹ); 112.3 (C-6ʹ); 115.5 (C-2ʹ); 119.1 (C-5ʹʹʹ); 122.7 (C=); 124.0 (C-5ʹ); 125.8 (C-1ʹ); 126.5 (C-4ʹʹʹ); 134.9 (CH=); 147.1 (C-3ʹ); 148.6 (C-7ʹʹʹa); 148.8 (C-4ʹ); 149.2 (C-3ʹʹʹa); 168.5 (C=O, C-4); 193.2 (C=S, C-2). HRMS, m/z: 473.1205 found (calculated for C23H25N2O5S2 [M + H]+ requires 473.1206).
(5Z)-[4-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethylamino)butyl]-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one (9l). According to the standard procedure, compound 9l was prepared in 15% yield (36.3 mg) from N-1-(2,3-dihydro-benzo[1,4]dioxin-6-ylmethyl)-butane-1,4-diamine hydrochloride 6f (136.4 mg, 0.5 mmol, 1 equiv.), triethylamine (202 μL, 152 mg, 1.5 mmol, 3 equiv.) and piperonaldehyde 3e (75.1 mg, 0.5 mmol, 1 equiv.) after 15 min. at 90 °C, followed by a second reaction time of 15 min. at 110 °C (for condensation step with 3e), which gave 9l as a yellowish powder. Mp = 172–233 °C (decomposition). 1H-NMR (CDCl3/TFA 98:2) δ: 1.66–1.69 (m, 4H, CH2, H-2ʹʹ, H-3ʹʹ); 3.03–3.10 (m, 2H, ArCH2NH); 4.03–4.07 (m, 4H, CH2, H-1ʹʹ, H-4ʹʹ); 4.16–4.19 (m, 4H, CH2, H-2ʹʹʹ, H-3ʹʹʹ); 5.97 (s, 2H, CH2, H-2ʹ); 6.66–7.00 (m, 6H, H-4ʹ, H-6ʹ, H-7ʹ, H-5ʹʹʹ, H-7ʹʹʹ, H-8ʹʹʹ, Ar); 7.55 (br s, 1H, NH); 7.59 (s, 1H, CH=). 13C-NMR (CDCl3/TFA 98:2) δ: 23.8 (C-2ʹʹ, C-3ʹʹ); 24.2 (C-3ʹʹ); 43.1 (C-1ʹʹ, C-4ʹʹ); 47.0 (ArCH2NH); 52.1 (C-4ʹʹ, C-1ʹʹ); 64.2 (C-2ʹʹʹ); 64.4 (C-3ʹʹʹ); 102.2 (C-2ʹ); 109.4 (C-7ʹʹʹ); 109.5 (C-8ʹʹʹ); 118.5 (C-5ʹʹʹ); 119.5 (C-6ʹʹʹ); 122.0 (C=); 122.9 (C-6ʹ); 127.3 (C-5ʹ); 128.2 (C-7ʹ); 135.0 (C-4ʹ); 135.5 (CH=); 143.9 (C-4ʹʹʹa); 145.1 (C-8ʹʹʹa); 150.6 (C-3ʹa); 150.8 (C-7ʹa); 168.5 (C=O, C-4); 193.0 (C=S, C-2). HRMS, m/z: 485.1205 found (calculated for C24H25N2O5S2 [M + H]+ requires 485.1206).
(5Z)3-[4-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethylamino)butyl]-5-(2,3-dihydro-benzo[1,4]dioxin-6-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one (9m). According to the standard procedure, compound 9m was prepared in 7% yield (17.5 mg) from N-1-(2,3-dihydro-benzo[1,4]dioxin-6-ylmethyl)-butane-1,4-diamine hydrochloride 6f (136.4 mg, 0.5 mmol, 1 equiv.), triethylamine (202 μL, 152 mg, 1.5 mmol, 3 equiv.) and 2,3-dihydro-1,4-benzodioxin-6-carboxaldehyde 3f (82.1 mg, 0.5 mmol, 1 equiv.) after 15 min. at 90 °C, followed by a second reaction time of 15 min. at 110 °C (for condensation step with 3f), which gave 9m as a yellowish powder. Mp = 170–243 °C (decomposition). 1H-NMR (CDCl3/TFA 98:2) δ: 1.64–1.69 (m, 4H, CH2, H-2ʹʹ, H-3ʹʹ); 3.04–3.10 (m, 2H, ArCH2NH); 4.03–4.08 (m, 4H, CH2, H-1ʹʹ, H-4ʹʹ); 4.16–4.23 (m, 8H, CH2, H-2ʹ, H-3ʹ, H-2ʹʹʹ, H-3ʹʹʹ); 6.75–7.57 (m, 6H, H-5ʹ, H-7ʹ, H-8ʹ, H-5ʹʹʹ, H-7ʹʹʹ, H-8ʹʹʹ, Ar); 7.60 (br s, 1H, NH); 8.77 (s, 1H, CH=). 13C-NMR (CDCl3/TFA 98:2) δ: 24.2 (C-2ʹʹ, C-3ʹʹ); 44.0 (C-1ʹʹ, C-4ʹʹ); 64.2 (C-2ʹ, C-2ʹʹʹ); 64.8 (C-3ʹ, C-3ʹʹʹ); 118.4 (C-7ʹ, C-7ʹʹʹ); 119.6 (C-8ʹ, C-8ʹʹʹ); 120.3 (C=); 125.6 (C-5ʹ, C-5ʹʹʹ); 126.8 (C-6ʹ, C-6ʹʹʹ); 134.5 (CH=); 144.0 (C-8ʹa, C-8ʹʹʹa); 146.6 (C-4ʹa, C-4ʹʹʹa); 166.6 (C=O, C-4); 182.5 (C=S, C-2). HRMS, m/z: 499.1364 found (calculated for C25H27N2O5S2 [M + H]+ requires 499.1366).
(5Z)3-[4-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethylamino)butyl]-5-(4-hydroxy-3-methoxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one (9n). According to the standard procedure, compound 9n was prepared in 34% yield (82.7 mg) from N-1-(2,3-dihydro-benzo[1,4]dioxin-6-ylmethyl)-butane-1,4-diamine hydrochloride 6f (136.4 mg, 0.5 mmol, 1 equiv.), triethylamine (202 μL, 152 mg, 1.5 mmol, 3 equiv.) and 4-hydroxy-3-methoxybenzaldehyde 3g (76.1 mg, 0.5 mmol, 1 equiv.) after 30 min. at 90 °C, followed by a second reaction time of 30 min. at 110 °C (for condensation step with 3g), which gave 9n as a red powder. Mp = 202–204 °C. 1H-NMR (CDCl3/TFA 98:2) δ: 1.69 (s, 4H, CH2, H-2ʹʹ, H-3ʹʹ); 3.06 (s, 2H, ArCH2NH); 3.86 (s, 3H, OCH3); 4.03 (s, 4H, CH2, H-1ʹʹ, H-4ʹʹ); 4.16 (s, 4H, CH2, H-2ʹʹʹ, H-3ʹʹʹ); 6.67–7.03 (m, 6H, H-2ʹ, H-5ʹ, H-6ʹ, H-5ʹʹʹ, H-7ʹʹʹ, H-8ʹʹʹ, Ar); 7.60 (s, 1H, CH=). 13C-NMR (CDCl3/TFA 98:2) δ: 23.1 (C-2ʹʹ or 3ʹʹ); 23.8 (C-3ʹʹ ou C-2ʹʹ); 43.1 (C-1ʹʹ ou 4ʹʹ); 47.0 (ArCH2NH); 52.1 (C-4ʹʹ, C-1ʹʹ); 56.1 (OCH3); 64.4 (C-2ʹʹʹ, C-3ʹʹʹ); 64.4 (C-3ʹʹʹ, C-2ʹʹʹ); 112.6 (C-6ʹ); 115.6 (C-2ʹ); 118.4 (C-7ʹʹʹ); 118.6 (C-8ʹʹʹ); 119.0 (C-6ʹʹʹ); 122.0 (C=); 122.9 (C-5ʹʹʹ); 125.9 (C-1ʹ); 126.8 (C-5ʹ); 135.9 (CH=); 143.9 (C-8ʹʹʹa); 145.1 (C-3ʹa); 147.2 (C-4ʹʹʹa); 148.9 (C-4ʹ);169.4 (C=O, C-4); 193.1 (C=S, C-2). HRMS, m/z: 487.1361 found (calculated for C24H27N2O5S2 [M + H]+ requires 487.1359).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
 Notes: a Reaction realized in a tube (sealed with a snap cap) under microwave irradiation (μω) with the Monowave® 300 Anton-Paar reactor. b Isolated yield. c Reaction temperature: 90 °C for the preparation of 8 (1st period of microwave irradiation). d Reaction temperature: 110 °C for condensation reaction after addition of 3 (2nd period of microwave irradiation).
Notes: a Reaction realized in a tube (sealed with a snap cap) under microwave irradiation (μω) with the Monowave® 300 Anton-Paar reactor. b Isolated yield. c Reaction temperature: 90 °C for the preparation of 8 (1st period of microwave irradiation). d Reaction temperature: 110 °C for condensation reaction after addition of 3 (2nd period of microwave irradiation).