
Integrase Inhibitor Prodrugs: Approaches to Enhancing the Anti-HIV Activity of β-Diketo Acids

Abstract
:1. Introduction

2. Discovery of Small Molecule Inhibitors of HIV-1 Integrase
2.1. Beta-Diketo Acids as HIV Integrase Inhibitors
Integrase Inhibition Protocol
2.2. Anti-HIV Activity Protocols
2.2.1. Anti-HIV Evaluation in Fresh Human PBMCs
2.2.2. Anti-HIV Assays in MAGI Cells
2.2.3. Anti-HIV Assays in GHOST X4/R5 Cells
2.3. Anti-HIV Activity of Selected Aryl and Heteroaryl Diketo Acid Integrase Inhibitors

| Compound | (ST) IC50, μM * | Anti-HIV Data EC50 or EC95 (μM) * and Cell Line | Cytotoxicity CC50 or CC95 (μM) * and Therapeutic Index (TI) * | References |
|---|---|---|---|---|
 1 L-708,906 | 0.10 3.5 | EC50, 2.0 H-9 Cells EC50, 5.5 MT-4 Cells | CC50, 88.3 (TI = 16) | [26,34] |
 2 | 0.35 Mn2+ | EC50, 0.6 293T Cells | - | [36] |
 3 | <0.10 | EC95, 0.52 MT-4 Cells | - | [27] |
 4 | <0.10 | EC95, 1.11 MT-4 Cells | CC95, >50 (TI > 45) | [27] |
 5 | <0.10 | EC95, 0.10 MT-4 Cells | CC95, >50 (TI > 500) | [27] |
 6 | 1.53 Mn2+ | EC50, 2.1 293T Cells | CC50, >50 (TI > 24) | [27] |
 7 | 2.4 Mn2+ | EC50, 5 293T Cells | CC50, >50 (TI > 10) | [44] |
 8 L-731,988 | 0.17 | EC50, 1.0 H-9 EC95, 9.6 MT-4 Cells | - | [26,27] |
 9 | 7.0 | EC50, 1.5 MT-4/KB Cells | 61 TI = 41 | [45] |
 10 5ClTEP | 0.65 Mn2+ | - | - | [36] |
 11 (S-1360) | 0.02 | EC50, 0.14 PBMC | CC50, 110 (TI = 786) | [32,33] |
2.4. HIV Integrase Inhibitors with Bis-Diketo Acid Structures

| Compound | 3′P (IC50, μM) * Mn2+ Assay | ST (IC50, μM) Mn2+ Assay | Anti-HIV-1 Data EC50 (μM), * CEM-SS Cells (Except 12, H-9 Cells) uH | CC50 (μM) (TI = CC50/EC50) * |
|---|---|---|---|---|
| 12 | 0.2 | 0.01 | 4.3 | >200 (TI > 47) |
| 13 | 2.5 | 0.3 | 0.8 | 11 (TI = 14) |
| 14 | 5.8 | 0.2 | >5 | 5 (TI < 1) |
| 15 | 5.3 | 0.2 | >9 | 9 (TI < 1) |
| 16 | 4.8 | 0.7 | >12 | 12 (TI < 1) |
| 17 | 32 | 2.7 | 16 | 124 (TI = 8) |
| 18 | 1.8 | 0.3 | 39 | >200 (TI > 5) |
| 19 | 7.3 | 0.4 | >200 | >200 (TI = 1) |
| 20 | 1.7 | 1.9 | >200 | >200 (TI = 1) |
| 21 | 1.8 | 0.2 | 17 | 81 (TI = 5) |
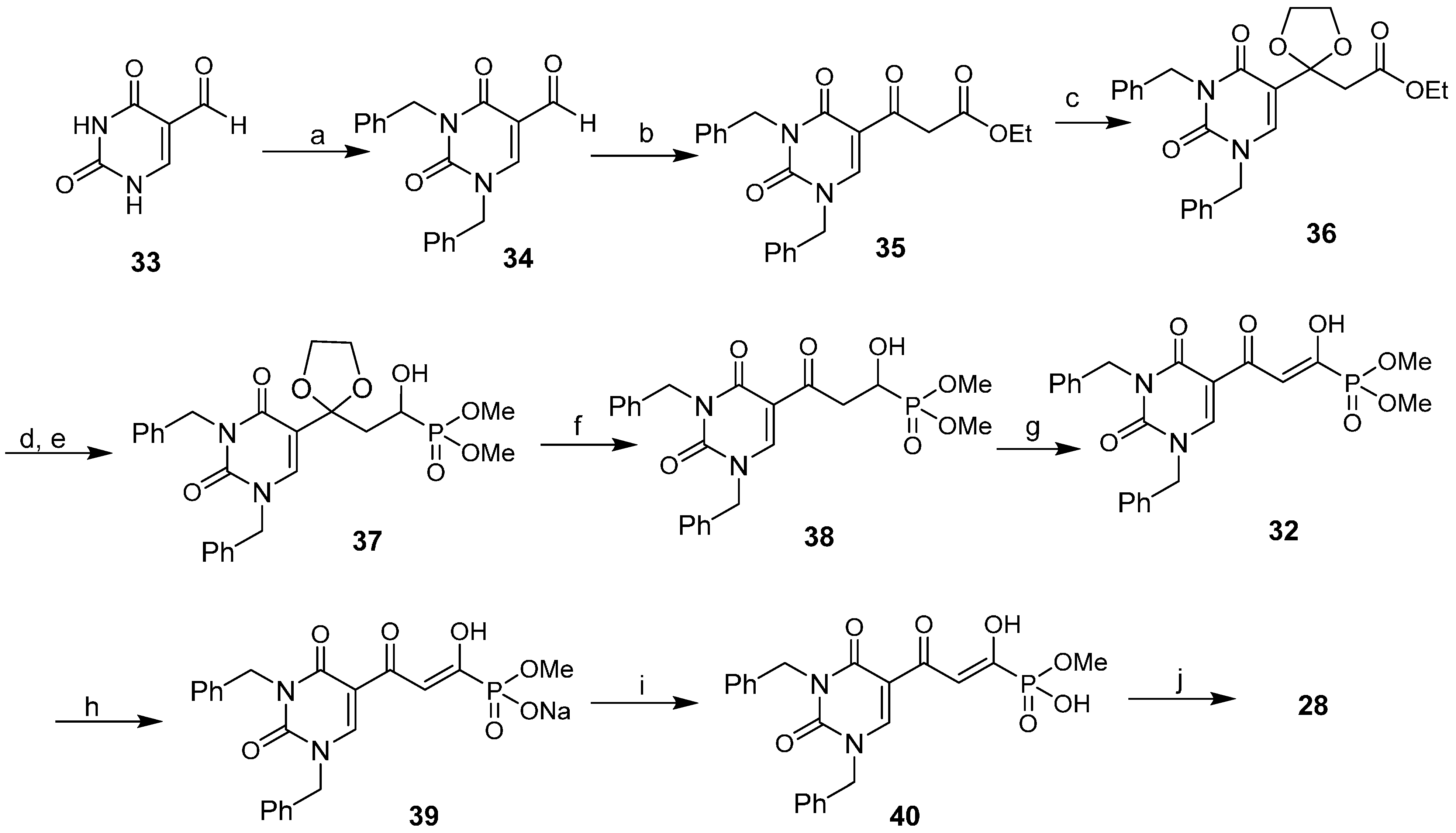
2.5. Discovery of Diketo Carboxylic Acids with Nucleobase Scaffolds

| Compound | Integrase Inhibition Data IC50 (μM) * | Cell Lines & HIV Isolates | EC50 * | CC50 * | TI * |
|---|---|---|---|---|---|
 22 | 3.7 (3′P) 0.2 (ST) Mn2+ Assay | PBMC HIV-1TEKI HIV-1NL4-3 | 50 nM <20 nM | >200 μM >200 μM | >4000 >10,000 |
 23 | 4.1 (3′P) <0.6 (ST) Mn2+Assay | GHOST X4/R5 HIV-1TEKI HIV-1NL4-3 | 0.85 μM 0.24 μM | >200 μM >200 μM | >235 >833 |
 24 | 10 (3′P) 0.5 (ST) Mn2+ Assay | PBMC HIV-1TEKI HIV-1NL4-3 | 2.63 μM 12.2 μM | >200 μM >200 μM | >76 >16.4 |
 25 | 17 (3′P) 3 (ST) Mn2+ Assay | PBMC HIV-1TEKI HIV-1NL4-3 | 2.50 μM 2.20 μM | >79.4 μM >79.4 μM | 31.8 36.1 |
 26 | 62 (3′P) 51 (ST) Mn2+ Assay | PBMC HIV-1TEKI HIV-1NL4-3 | 151 μM 24.8 μM | >200 μM >200 μM | >1.3 >8.1 |
 27 | 100 (3′P) 10 (ST) Mn2+ Assay | GHOST X4/R5 HIV-1TEKI HIV-1NL4-3 | 5.26 μM 7.64 μM | 97.7μM 97.7 μM | 18.6 12.8 |

2.6. Diketo Phosphonic Acids with Nucleobase Scaffolds


| Compounds | HIV-1 Isolate (Cell Line) | EC50 * | CC50 * | TI * |
|---|---|---|---|---|
| 28 | HIV-1TEKI (PBMC) | 4.0 μM | >200 μM | >50 |
| HIV-1NL4-3(PBMC) | 3.2 μM | >200 μM | >62 | |
| 29 | HIV-1TEKI (PBMC) | 8.1 μM | 46.9 μM | 5.8 |
| HIV-1NL4-3(PBMC) | 11.8 μM | 46.9 μM | 4.0 |

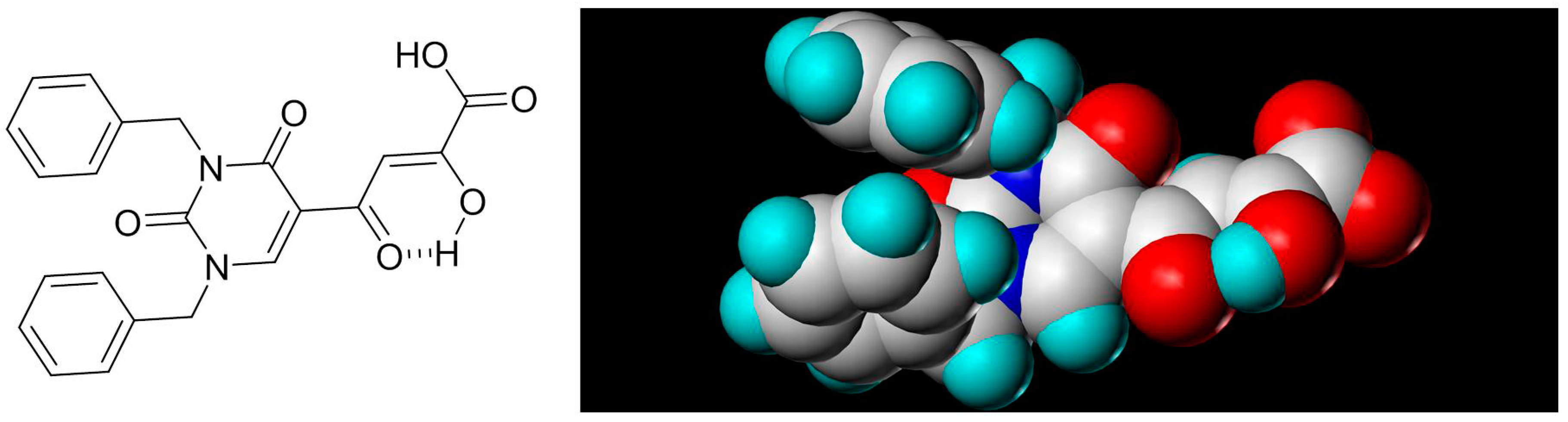
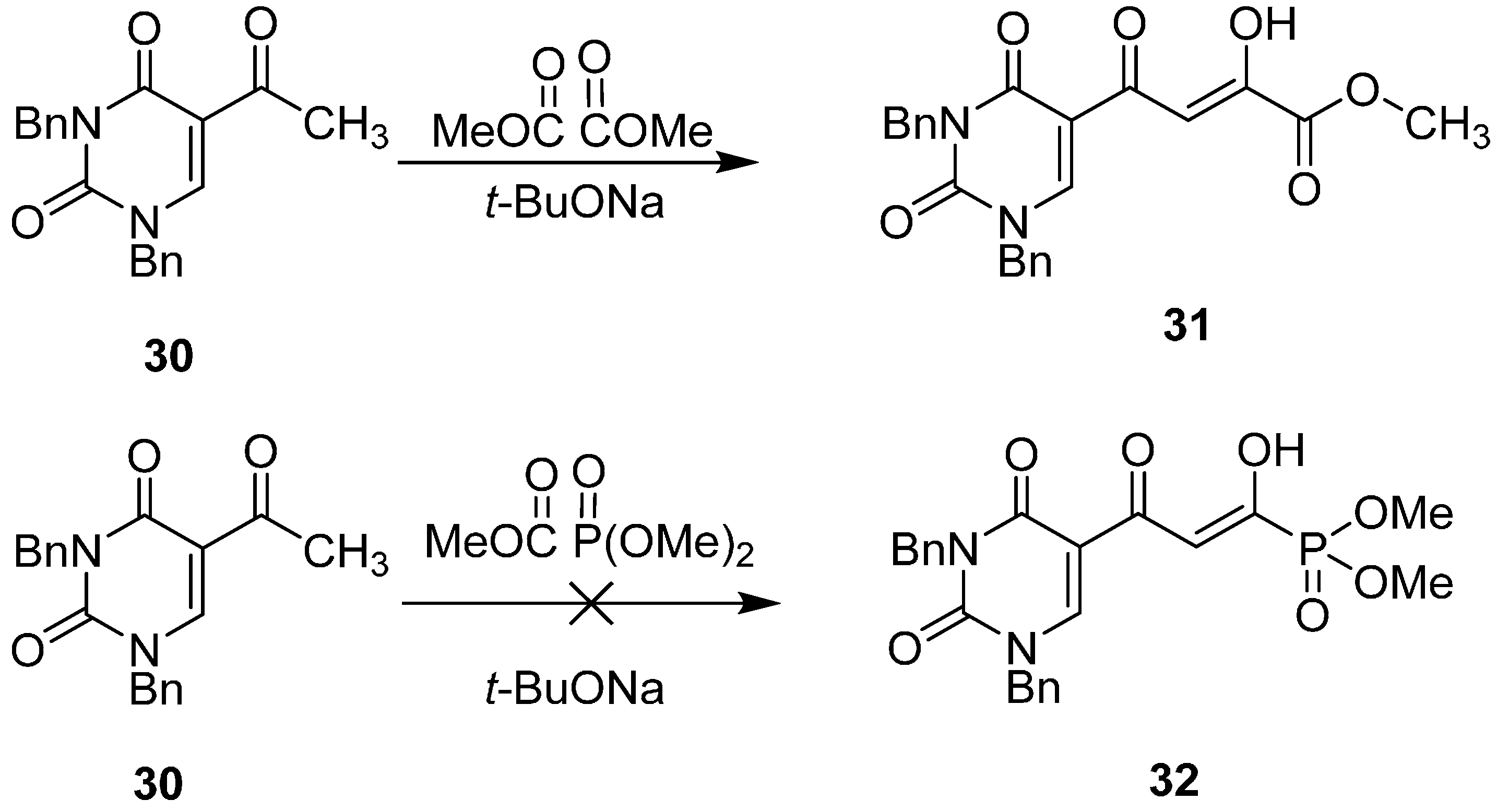
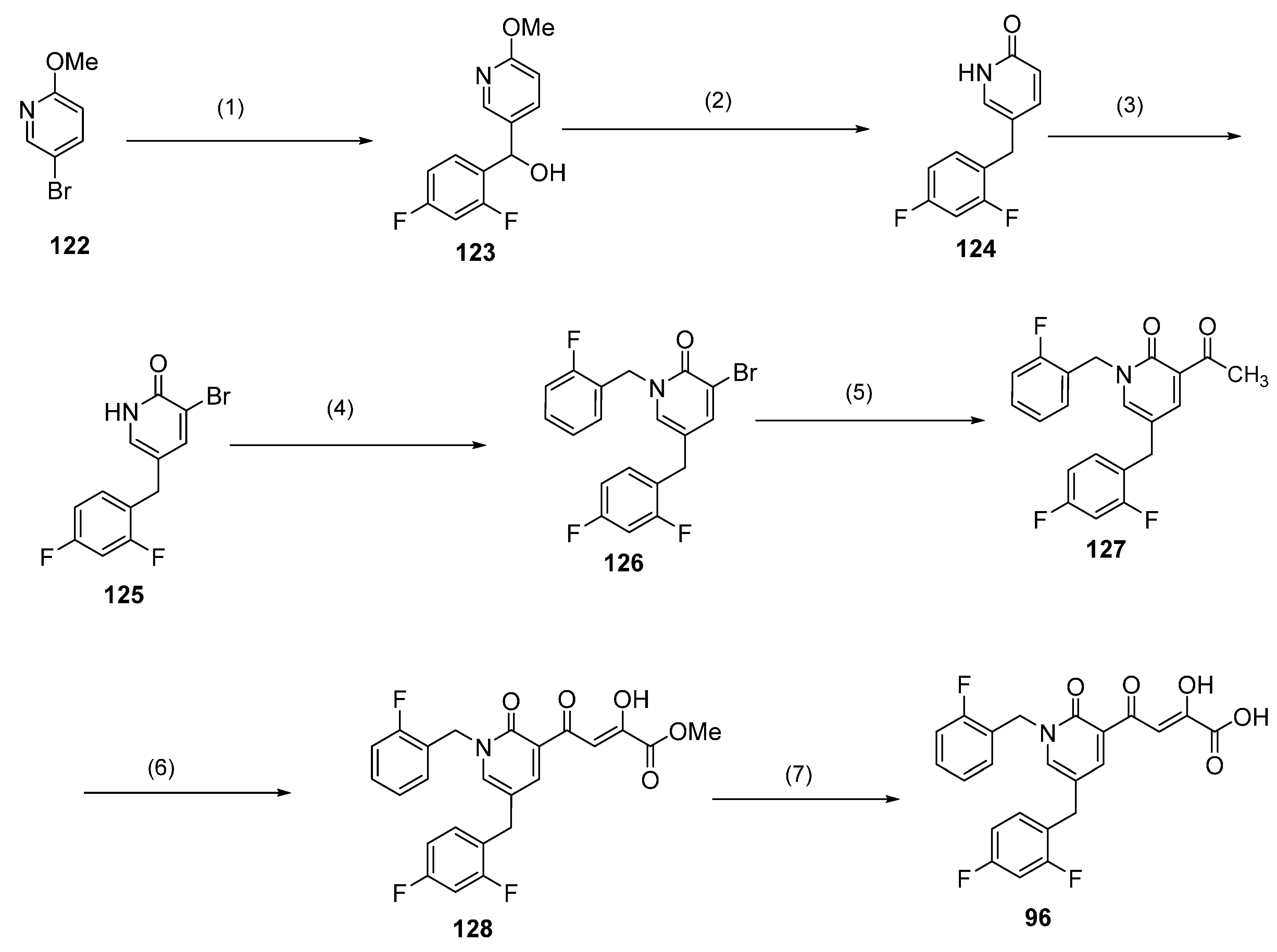
2.7. Diketo Carboxylic Acids Constructed on Pyridinone Scaffold


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Aryl Ring A | Aryl Ring B | EC50 (nM) a,b | ||||
|---|---|---|---|---|---|---|---|
| o | m | p | o | m | p | ||
| 41 | H | H | H | H | H | H | 2100 |
| 42 | F | H | H | H | H | H | 900 |
| 43 | H | H | F | H | H | H | 1000 |
| 44 | F | H | F | H | H | H | 600 |
| 45 | F | H | H | F | H | H | 700 |
| 46 | H | H | OMe | H | H | H | 1800 |
| 47 | H | H | F | H | H | F | 800 |
| 48 | H | H | Me | H | H | F | 1600 |
| 49 | H | Cl | F | H | H | H | 800 |
| 50 | H | H | OMe | H | H | OMe | >5000 |
| 51 | F | H | F | F | H | F | 300 |
| 52 | H | F | H | H | H | H | 2000 |
| 53 | H | Cl | H | H | Cl | H | 1100 |
| 54 | H | Cl | F | H | Cl | H | 1200 |
| 55 | F | H | H | H | H | F | 700 |
| 56 | H | Cl | F | H | Cl | F | 2400 |
| 57 | H | H | Me | H | Cl | F | >5000 |
| 58 | H | H | Me | H | Cl | H | 2500 |
| 59 | H | H | Me | F | H | H | 700 |
| 60 | H | H | F | H | Cl | F | >5000 |
| 61 | F | Cl | F | H | H | H | 600 |
| 62 | H | H | F | H | Cl | H | 1400 |
| 63 | 2,6-diF | H | H | H | H | F | 1400 |
| 64 | H | H | F | F | H | H | 800 |
| 65 | F | Cl | H | F | Cl | H | 2400 |
| 66 | H | H | Cl | H | Cl | H | 2500 |
| 67 | H | Cl | H | H | Cl | H | 1400 |
| 68 | F | Cl | H | H | Me | H | 1400 |
| 69 | F | H | F | F | H | H | 600 |
| 70 | H | Me | H | H | Cl | F | 2200 |
| 71 | F | Cl | H | H | H | H | 700 |
| 72 | F | 5-Cl | H | H | H | H | 900 |
| 73 | F | Cl | H | H | Cl | H | 1400 |
| 74 | F | H | F | H | Cl | F | 1400 |
| 75 | H | Cl | H | H | H | F | 1200 |
| 76 | H | Cl | H | F | H | H | 700 |
| 77 | 2,6-di-F | H | H | H | Cl | F | 5100 |
| 78 | 2,6-di-F | H | H | H | H | H | 1500 |
| 79 | H | H | H | F | H | F | 800 |
| 80 | F | H | H | H | Cl | F | 2200 |
| 81 | H | F | H | F | H | H | 1900 |
| 82 | F | H | H | H | Cl | H | 1900 |
| 83 | F | F | H | H | H | H | 900 |
| 82 | H | Me | H | F | H | H | 900 |
| 85 | F | F | H | H | H | F | 700 |
| 86 | H | H | CN | F | H | H | 1100 |
| 87 | F | F | H | F | H | H | 800 |
| 88 | H | F | H | H | H | F | 1000 |
| 89 | F | H | F | Cl | H | H | 800 |
| 90 | H | Cl | H | F | Cl | H | 1500 |
| 91 | H | H | F | F | H | F | 600 |
| 92 | H | Me | H | F | H | F | 700 |
| 93 | H | H | H | H | COOH | H | >5000 |
| 94 | H | H | H | Me | H | F | 1300 |
| 95 | H | H | F | Me | H | F | 1000 |
| 96 | F | H | H | F | H | F | 500 |
| 97 | F | H | H | Me | H | F | 1300 |
| 98 | H | Cl | F | F | H | H | 790 |
| 99 | F | H | F | H | H | F | 420 |
| 100 | F | Cl | H | F | H | H | 500 |

 , (Ph3P)2PdCl2/DMF, b. 1N HCl (91%); (6) a. t-BuONa/THF, MeO2CCO2Me, b. 1N HCl (77%); (7) 1N HCl/dioxane (78%).
, (Ph3P)2PdCl2/DMF, b. 1N HCl (91%); (6) a. t-BuONa/THF, MeO2CCO2Me, b. 1N HCl (77%); (7) 1N HCl/dioxane (78%).
, (Ph3P)2PdCl2/DMF, b. 1N HCl (91%); (6) a. t-BuONa/THF, MeO2CCO2Me, b. 1N HCl (77%); (7) 1N HCl/dioxane (78%).
, (Ph3P)2PdCl2/DMF, b. 1N HCl (91%); (6) a. t-BuONa/THF, MeO2CCO2Me, b. 1N HCl (77%); (7) 1N HCl/dioxane (78%).
3. Prodrug Design, Synthesis and Antiviral Evaluation
3.1. Anti-HIV Diketo Acid Prodrug Development

| Prodrug Derivatives of Integrase Inhibitors | Prodrug Anti-HIV EC50 (MAGI Cell) * | Diketo Acid Anti-HIV EC50 (MAGI Cell) * |
|---|---|---|
 101 | 300 nM | 2100 nM (Compound 41) |
 102 | >5000 nM | 710 nM (Compound 98) |
 103 | >5000 nM | 710 nM (Compound 98) |
 104 | 1900 nM | 710 nM (Compound 98) |
 105 | 5300 nM | 710 nM (Compound 98) |
 106 | 1600 nM | 710 nM (Compound 98) |
 107 | 40 nM | 700 nM (Compound 55) |
 108 | 3900 nM | 700 nM (Compound 55) |
 109 | 50 nM | 300nM (Compound 51) |
 110 | >5000 nM | 700 nM (Compound 71) |
 111 | 15 nM | 600 nM (Compound91) |
 112 | 400 nM | 420 nM (Compound 99) |
 113 | 100 nM | 1300 nM (Compound 94) |
 114 | 110 nM | 1300 nM (Compound 97) |
 115 | 226 nM | 1000 nM (Compound 95) |
 116 | 9 nM | 500 nM (Compound 96) |
 117 | 176 nM | 800 nM (Compound 64) |
 118 | 1200 nM | 500 nM (Compound 100) |
 119 | 400 nM | 600 nM (Compound 69) |
 120 | 4400 nM | 1400 nM (Compound 73) |
 121 | 2700 nM | 700 nM (Compound 71) |
| Prodrug | Anti-HIV EC50 (MAGI Cell Data) * |
|---|---|
| 107 | 40 nM |
| 109 | 50 nM |
| 111 | 15 nM |
| 116 | 9 nM |
| Raltegravir (positive control compound) | 6 nM |
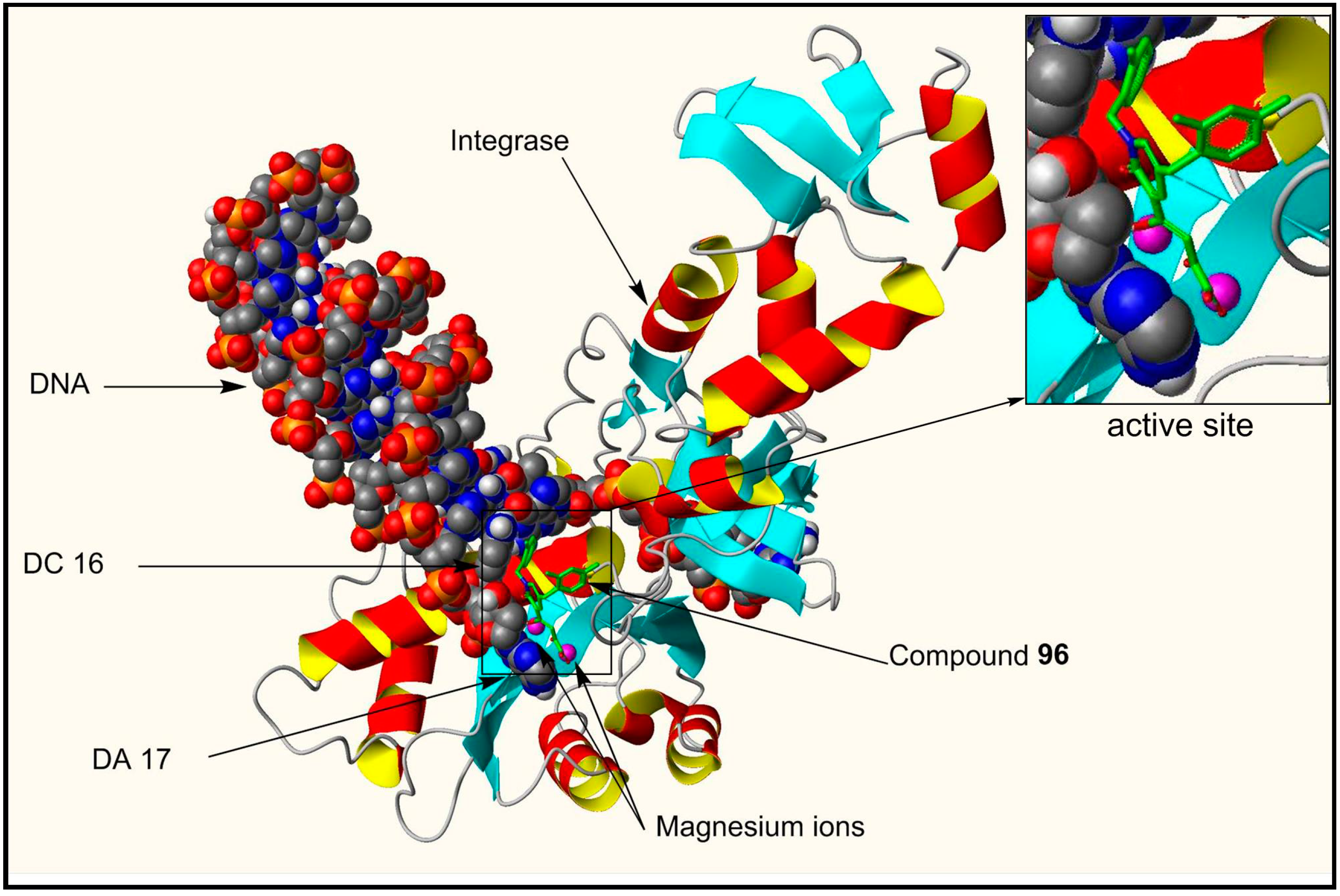
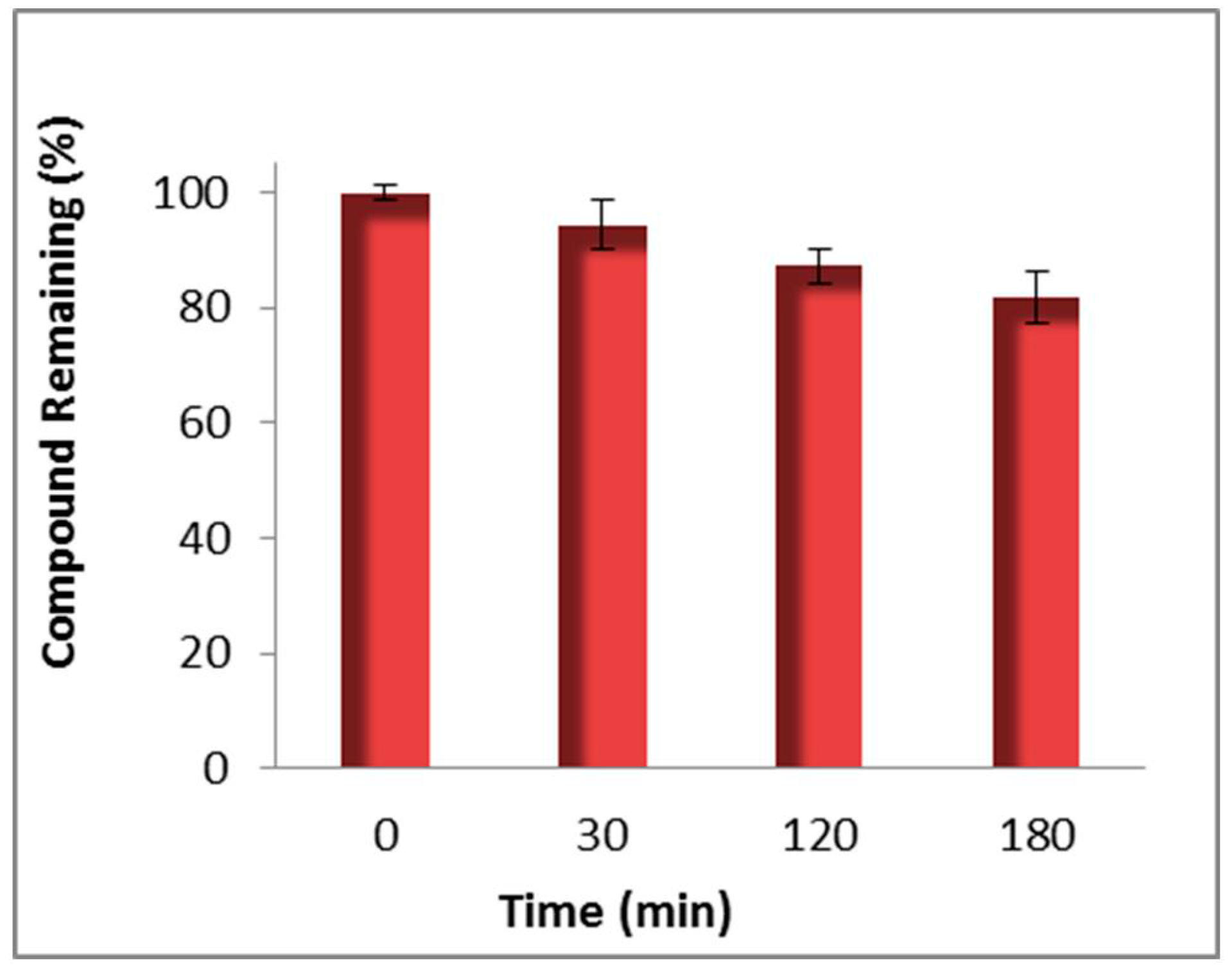
3.2. Stability, Metabolism and CYP and UGT Drug Interaction Studies on Compound 96

| CYP450 Isozyme | Substrate (Stock Solution) | Conc. (µM ) | Protein (mg/mL) | Incubation (min) | IC50 Data (µM) * |
|---|---|---|---|---|---|
| CYP3A4 | Testosterone (50 mM ) | 100 | 0.3 | 30 | >200 µM |
| CYP3A4 | Triazolam (50 mM) | 200 | 0.4 | 30 | >200 µM |
| CYP2D6 | Dextromethorphan (50 mM) | 200 | 2.0 | 60 | >200 µM |
| CYP2C8 | Amodiaquine (5 mM) | 200 | 0.4 | 30 | >65 µM |
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Frankel, A.D.; Young, J.A.T. HIV-1: Fifteen proteins and an RNA. Annu. Rev. Biochem. 1998, 67, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Trono, D.; Van Lint, C.; Rouzioux, C.; Verdin, E.; Barre-Sinoussi, F.; Chun, T.W.; Chomont, N. HIV persistence and the prospect of long-term drug-free remissions for HIV-infected individuals. Science 2010, 329, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Chi, G. HIV integrase inhibitors as therapeutic agents in AIDS. Rev. Med. Virol. 2007, 17, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Chi, G.; Ptak, R.; Neamati, N. HIV integrase inhibitors with nucleobase scaffolds: Discovery of a highly potent anti-HIV agent. J. Med. Chem. 2006, 49, 445–447. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Johnson, A.A.; Marchand, C. Integrase inhibitors to treat HIV/AIDS. Nat. Rev. Drug Discov. 2005, 4, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Nair, V. Novel inhibitors of HIV integrase: The discovery of potential anti-HIV therapeutic agents. Front. Med. Chem. Online 2005, 2, 3–20. [Google Scholar] [CrossRef]
- Summa, V.; Petrocchi, A.; Bonelli, F.; Crescenzi, B.; Donghi, M.; Ferrara, M.; Fiore, F.; Gardelli, C.; Paz, O.G.; Hazuda, D.J.; et al. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J. Med. Chem. 2008, 51, 5843–5855. [Google Scholar] [CrossRef] [PubMed]
- Laufer, R.; Paz, O.G.; di Marco, A.; Bonelli, F.; Monteagudo, E.; Summa, V.; Rowley, M. Quantitative prediction of human clearance guiding the development of raltegravir (MK-0518, Isentress) and related HIV integrase inhibitors. Drug Metab. Dispos. 2009, 37, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Garvey, E.P.; Johns, B.A.; Gartland, M.J.; Foster, S.A.; Miller, W.H.; Ferris, R.G.; Hazen, R.J.; Underwood, M.R.; Boros, E.E.; Thompson, J.B.; et al. The naphthyridinone GSK364735 is a novel, potent human immunodeficiency virus type 1 integrase inhibitor and antiretroviral. Antimicrob. Agents Chemother. 2008, 52, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Min, S.; Song, I.; Borland, J.; Chen, S.G.; Lou, Y.; Fujiwara, T.; Piscitelli, S.C. Pharmacokinetics and safety of S/GSK1349572, a next-generation HIV integrase inhibitor, in healthy volunteers. Antimicrob. Agents Chemother. 2010, 54, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Klibanov, O.M. Elvitegravir, an oral HIV integrase inhibitor, for the potential treatment of HIV infection. Curr. Opin. Investig. Drugs 2009, 10, 190–200. [Google Scholar] [PubMed]
- Asante-Appiah, E.; Skalka, A.M. HIV-1 integrase: Structural organization, conformational changes, and catalysis. Adv. Virus Res. 1999, 52, 351–369. [Google Scholar] [PubMed]
- Esposito, D.; Craigie, R. HIV integrase structure and function. Adv. Virus Res. 1999, 52, 319–333. [Google Scholar] [PubMed]
- Liao, C.Z.; Marchand, C.; Burke, T.R.; Pommier, Y.; Nicklaus, M.C. Authentic HIV-1 integrase inhibitors. Future Med. Chem. 2010, 2, 1107–1122. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 2010, 464, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Nair, V. HIV integrase as a target for antiviral chemotherapy. Rev. Med. Virol. 2002, 12, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Moss, D.M.; Neary, M.; Owen, A. The role of drug transporters in the kidney: Lessons from tenofovir. Front. Pharmacol. 2014, 5, 248. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. A Cutting-edge view on the current state of antiviral drug development. Med. Res. Rev. 2013, 33, 1249–1277. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, S.J.; Schinazi, R.F. Prodrug strategies for improved efficacy of nucleoside antiviral inhibitors. Curr. Opin. HIV AIDS 2013, 8, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Gulick, R. HIV treatment 2020: What will it look like? J. Int. AIDS Soc. 2014, 17. [Google Scholar] [CrossRef] [PubMed]
- Walji, A.M.; Sanchez, R.I.; Clas, S.D.; Nofsinger, R.; Ruiz, M.L.; Li, J.; Bennet, A.; John, C.; Bennett, D.J.; Sanders, J.M.; et al. Discovery of MK-8970: An acetal carbonate prodrug of Raltegravir with enhanced colonic absorption. Chem. Med. Chem. 2015, 10, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Sofia, M.J. Prodrugs in the treatment of viral diseases. RSC Drug Discov. Ser. 2013, 32, 421–450. [Google Scholar]
- De Clercq, E. New approaches toward anti-HIV chemotherapy. J. Med. Chem. 2005, 48, 1297–1313. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, A.; Uchida, H.; Neamati, N.; Sunder, S.; Jaworska-Maslanka, M.; Wickstrom, E.; Zeng, F.; Jones, R.A.; Mandes, R.F.; Chenault, H.K.; et al. Probing interactions between viral DNA and human immunodeficiency virus type 1 integrase using dinucleotides. Mol. Pharmacol. 1997, 51, 567–575. [Google Scholar] [PubMed]
- Taktakishvili, M.; Neamati, N.; Pommier, Y.; Pal, S.; Nair, V. Recognition and inhibition of HIV integrase by novel dinucleotides. J. Am. Chem. Soc. 2000, 122, 5671–5677. [Google Scholar] [CrossRef]
- Hazuda, D.J.; Felock, P.; Witmer, M.; Wolfe, A.; Stillmock, K.; Grobler, J.A.; Espeseth, A.; Gabryelski, L.; Schleif, W.; Blau, C.; et al. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science 2000, 287, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Wai, J.S.; Egbertson, M.S.; Payne, L.S.; Fisher, T.E.; Embrey, M.W.; Tran, L.O.; Melamed, J.Y.; Langford, H.M.; Guare, J.P.; Zhuang, L.G.; et al. 4-aryl-2,4-dioxobutanoic acid inhibitors of HIV-1 integrase and viral replication in cells. J. Med. Chem. 2000, 43, 4923–4926. [Google Scholar] [CrossRef] [PubMed]
- Marchand, C.; Zhang, X.C.; Pais, G.C.G.; Cowansage, K.; Neamati, N.; Burke, T.R.; Pommier, Y. Structural determinants for HIV-1 integrase inhibition by beta-diketo acids. J. Biol. Chem. 2002, 277, 12596–12603. [Google Scholar] [CrossRef] [PubMed]
- Hazuda, D.J.; Young, S.D.; Guare, J.P.; Anthony, N.J.; Gomez, R.P.; Wai, J.S.; Vacca, J.P.; Handt, L.; Motzel, S.L.; Klein, H.J.; et al. Integrase inhibitors and cellular immunity suppress retroviral replication in rhesus macaques. Science 2004, 305, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Grobler, J.A.; Stillmock, K.; Hu, B.H.; Witmer, M.; Felock, P.; Espeseth, A.S.; Wolfe, A.; Egbertson, M.; Bourgeois, M.; Melamed, J.; et al. Diketo acid inhibitor mechanism and HIV-1 integrase: Implications for metal binding in the active site of phosphotransferase enzymes. Proc. Natl. Acad. Sci. USA 2002, 99, 6661–6666. [Google Scholar] [CrossRef] [PubMed]
- Goldgur, Y.; Craigie, R.; Cohen, G.H.; Fujiwara, T.; Yoshinaga, T.; Fujishita, T.; Sugimoto, H.; Endo, T.; Murai, H.; Davies, D.R. Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: A platform for antiviral drug design. Proc. Natl. Acad. Sci. USA 1999, 96, 13040–13043. [Google Scholar] [CrossRef] [PubMed]
- Billich, A. S-1360 Shionogi-GlaxoSmithKline. Curr. Opin. Investig. Drugs 2003, 4, 206–209. [Google Scholar] [PubMed]
- Yoshinaga, T.; Sato, A.; Fujishita, T. S-1360: In vitro activity of a new HIV-1 integrase inhibitor in clinical development. In Proceeding of the 9th Conference on Retroviruses and Opportunistic Infections, Seattle, WA, USA, 24–28 February 2002; p. 55.
- Pannecouque, C.; Pluymers, W.; Van Maele, B.; Tetz, V.; Cherepanov, P.; de Clercq, E.; Witvrouw, M.; Debyser, Z. New class of HIV integrase inhibitors that block viral replication in cell culture. Curr. Biol. 2002, 12, 1169–1177. [Google Scholar] [CrossRef]
- Pluymers, W.; Pais, G.; Van Maele, B.; Pannecouque, C.; Fikkert, V.; Burke, T.R.; de Clercq, E.; Witvrouw, M.; Neamati, N.; Debyser, Z. Inhibition of human immunodeficiency virus type 1 integration by diketo derivatives. Antimicrob. Agents Chemother. 2002, 46, 3292–3297. [Google Scholar] [CrossRef] [PubMed]
- Pais, G.C.G.; Zhang, X.C.; Marchand, C.; Neamati, N.; Cowansage, K.; Svarovskaia, E.S.; Pathak, V.K.; Tang, Y.; Nicklaus, M.; Pommier, Y.; et al. Structure activity of 3-aryl-1,3-diketo-containing compounds as HIV-1 integrase inhibitors. J. Med. Chem. 2002, 45, 3184–3194. [Google Scholar] [CrossRef] [PubMed]
- Bacchi, A.; Carcelli, M.; Compari, C.; Fisicaro, E.; Pala, N.; Rispoli, G.; Rogolino, D.; Sanchez, T.W.; Sechi, M.; Neamati, N. HIV-1 IN strand transfer chelating inhibitors: A focus on metal binding. Mol. Pharm. 2011, 8, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, A.; Neamati, N.; Sundar, S.; Owen, J.; Pommier, Y. Retroviral integrase: A novel target in antiviral drug development and basic in vitro assays with purified enzyme in. In Antiviral Methods and Protocols; Kinchington, D., Schinazi, R., Eds.; Humana: Totowa, NJ, USA, 1999; Volume 24, pp. 327–338. [Google Scholar]
- Marchand, C.; Johnson, A.A.; Karki, R.G.; Pais, G.C.G.; Zhang, X.C.; Cowansage, K.; Patel, T.A.; Nicklaus, M.C.; Burke, T.R.; Pommier, Y. Metal-dependent inhibition of HIV-1 integrase by beta-diketo acids and resistance of the soluble double-mutant (F185K/C280S). Mol. Pharmacol. 2003, 64, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Beare, K.D.; Coster, M.J.; Rutledge, P.J. Diketoacid inhibitors of HIV-1 integrase: From L-708,906 to raltegravir and beyond. Curr. Med. Chem. 2012, 19, 1177–1192. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Grant, G.H.; Richards, W.G. Binding modes of diketo-acid inhibitors of HIV-1 integrase: A comparative molecular dynamics simulation study. J. Mol. Graph. Model. 2011, 29, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Vandegraaff, N.; Kumar, R.; Hocking, H.; Burke, T.R.; Mills, J.; Rhodes, D.; Burrell, C.J.; Li, P. Specific inhibition of human immunodeficiency virus type 1 (HIV-1) integration in cell culture: Putative inhibitors of HIV-1 integrase. Antimicrob. Agents Chemother. 2001, 45, 2510–2516. [Google Scholar] [CrossRef] [PubMed]
- Rosemond, M.J.C.; St John-Williams, L.; Yamaguchi, T.; Fujishita, T.; Walsh, J.S. Enzymology of a carbonyl reduction clearance pathway for the HIV integrase inhibitor, S-1360: Role of human liver cytosolic aldo-keto reductases. Chem. Biol. Interact. 2004, 147, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.C.; Pais, G.C.G.; Svarovskaia, E.S.; Marchand, C.; Johnson, A.A.; Karki, R.G.; Nicklaus, M.C.; Pathak, V.K.; Pommier, Y.; Burke, T.R. Azido-containing aryl beta-diketo acid HIV-1 integrase inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 1215–1219. [Google Scholar] [CrossRef]
- Costi, R.; Di Santo, R.; Artico, M.; Roux, A.; Ragno, R.; Massa, S.; Tramontano, E.; La Colla, M.; Loddo, R.; Marongiu, M.E.; et al. 6-aryl-2,4-dioxo-5-hexenoic acids, novel integrase inhibitors active against HIV-1 multiplication in cell-based assays. Bioorg. Med. Chem. Lett. 2004, 14, 1745–1749. [Google Scholar] [CrossRef] [PubMed]
- Di Santo, R.; Costi, R.; Roux, A.; Artico, M.; Lavecchia, A.; Marinelli, L.; Novellino, E.; Palmisano, L.; Andreotti, M.; Amici, R.; et al. Novel bifunctional quinolonyl diketo acid derivatives as HIV-1 integrase inhibitors: Design, synthesis, biological activities, and mechanism of action. J. Med. Chem. 2006, 49, 1939–1945. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.Q.; Jiang, X.H.; Dayam, R.; Sanchez, T.; Shoemaker, R.; Sei, S.; Neamati, N. Rational design and synthesis of novel dimeric diketoacid-containing inhibitors of HIV-1 integrase: Implication for binding to two metal ions on the active site of integrase. J. Med. Chem. 2004, 47, 2561–2573. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.F.; Jiang, X.H.; Sanchez, T.; Zhang, H.S.; Dayam, R.; Neamati, N.; Long, Y.Q. Novel dimeric aryldiketo containing inhibitors of HIV-1 integrase: Effects of the phenyl substituent and the linker orientation. Bioorg. Med. Chem. 2008, 16, 7777–7787. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Nair, V. Novel HIV integrase inhibitors with anti-HIV activity: Insights into integrase inhibition from docking studies. Antivir. Chem. Chemother. 2006, 17, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Chi, G.C.; Nair, V.; Semenova, E.; Pommier, Y. A novel diketo phosphonic acid that exhibits specific, strand-transfer inhibition of HIV integrase and anti-HIV activity. Bioorg. Med. Chem. Lett. 2007, 17, 1266–1269. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Uchil, V.; Neamati, N. beta-Diketo acids with purine nucleobase scaffolds: Novel, selective inhibitors of the strand transfer step of HIV integrase. Bioorg. Med. Chem. Lett. 2006, 16, 1920–1923. [Google Scholar] [CrossRef] [PubMed]
- Kukhar, V.P.; Hudson, H.R. Aminophosphonic and Aminophosphinic Acids, Chemistry and Biological Activity; John Wiley and Sons, Ltd: Chichester, UK, 2000. [Google Scholar]
- Sekine, M.; Kume, A.; Nakajima, M.; Hata, T. A new method for acylation of enolates by means of dialkyl acylphosphonates as acylating agents. Chem. Lett. 1981, 10, 1087–1090. [Google Scholar] [CrossRef]
- Holmquist, C.R.; Roskamp, E.J. A selective method for the direct conversion of aldehydes into beta-keto-esters with ethyl diazoacetate catalyzed by Tin(II) Chloride. J. Org. Chem. 1989, 54, 3258–3260. [Google Scholar] [CrossRef]
- Karaman, R.; Goldblum, A.; Breuer, E.; Leader, H. Acylphosphonic acids and methyl hydrogen acylphosphonates-physical and chemical-properties and theoretical calculations. J. Chem. Soc. Perkin Trans. 1 1989, 4, 765–774. [Google Scholar] [CrossRef]
- Seo, B.I.; Uchil, V.R.; Okello, M.; Mishra, S.; Ma, X.H.; Nishonov, M.; Shu, Q.N.; Chi, G.C.; Nair, V. Discovery of a potent HIV integrase inhibitor that leads to a prodrug with significant anti-HIV activity. ACS Med. Chem. Lett. 2011, 2, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Urban, J.J.; Vontersch, R.L.; Famini, G.R. Effect of fluorine substitution on phenol acidities in the gas-phase and in aqueous-solution—A computational study using continuum solvation models. J. Org. Chem. 1994, 59, 5239–5245. [Google Scholar] [CrossRef]
- Resnati, G. Synthesis of chiral and bioactive fluoroorganic compounds. Tetrahedron 1993, 49, 9385–9445. [Google Scholar] [CrossRef]
- DiMagno, S.G.; Sun, H.R. The strength of weak interactions: Aromatic fluorine in drug design. Curr. Top. Med. Chem. 2006, 6, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.; Maertens, G.N.; Cherepanov, P. 3′-Processing and strand transfer catalysed by retroviral integrase in crystallo. EMBO J. 2012, 31, 3020–3028. [Google Scholar] [CrossRef] [PubMed]
- Boros, E.E.; Burova, S.A.; Erickson, G.A.; Johns, B.A.; Koble, C.S.; Kurose, N.; Sharp, M.J.; Tabet, E.A.; Thompson, J.B.; Toczko, M.A. A scaleable synthesis of methyl 3-amino-5-(4-fluorobenzyl)-2-pyridinecarboxylate. Org. Process Res. Dev. 2007, 11, 899–902. [Google Scholar] [CrossRef]
- Singh, B.; Bacon, E.R.; Lesher, G.Y.; Robinson, S.; Pennock, P.O.; Bode, D.C.; Pagani, E.D.; Bentley, R.G.; Connell, M.J.; Hamel, L.T.; et al. Novel and potent Adenosine 3′,5′-cyclic phosphate phosphodiesterase-III inhibitors-Thiazolo[4,5-B][1,6]naphthyridin-2-ones. J. Med. Chem. 1995, 38, 2546–2550. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P. Reactions of 2-aminopyridine with benzyl-chloride—benzylation of pyridine ring. Pol. J. Chem. 1984, 58, 959–960. [Google Scholar]
- Nair, V.; Turner, G.A.; Chamberlain, S.D. Novel approaches to functionalized nucleosides via palladium-catalyzed cross coupling with organostannanes. J. Am. Chem. Soc. 1987, 109, 7223–7224. [Google Scholar] [CrossRef]
- Nair, V.; Turner, G.A.; Buenger, G.S.; Chamberlain, S.D. new methodologies for the synthesis of C-2 functionalized hypoxanthine nucleosides. J. Org. Chem. 1988, 53, 3051–3057. [Google Scholar] [CrossRef]
- Jiang, X.H.; Song, L.D.; Long, Y.Q. Highly efficient preparation of aryl beta-diketo acids with tert-butyl methyl oxalate. J. Org. Chem. 2003, 68, 7555–7558. [Google Scholar] [CrossRef] [PubMed]
- Uchil, V.; Seo, B.; Nair, V. A novel strategy to assemble the beta-diketo acid pharmacophore of HIV integrase inhibitors on purine nucleobase scaffolds. J. Org. Chem. 2007, 72, 8577–8579. [Google Scholar] [CrossRef] [PubMed]
- Kassahun, K.; McIntosh, I.; Cui, D.; Hreniuk, D.; Merschman, S.; Lasseter, K.; Azrolan, N.; Iwamoto, M.; Wagner, J.A.; Wenning, L.A. Metabolism and disposition in humans of raltegravir (MK-0518), an Anti-AIDS drug targeting the human immunodeficiency virus 1 integrase enzyme. Drug Metab. Dispos. 2007, 35, 1657–1663. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd ed.; Kluwer Academic/Plenum: New York, NY, USA, 2005. [Google Scholar]
- Baranczewski, P.; Stanczak, A.; Sundberg, K.; Svensson, R.; Wallin, A.; Jansson, J.; Garberg, P.; Postlind, H. Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development. Pharmacol. Rep. 2006, 58, 453–472. [Google Scholar] [PubMed]
- Chauret, N.; Gauthier, A.; Martin, J.; NicollGriffith, D.A. In vitro comparison of cytochrome P450-mediated metabolic activities in human, dog, cat, and horse. Drug Metab. Dispos. 1997, 25, 1130–1136. [Google Scholar] [PubMed]
- Williams, J.A.; Hyland, R.; Jones, B.C.; Smith, D.A.; Hurst, S.; Goosen, T.C.; Peterkin, V.; Koup, J.R.; Ball, S.E. Drug-drug interactions for UDP-glucuronosyltransferase substrates: A pharmacokinetic explanation for typically observed low exposure (AUC(i)/AUC) ratios. Drug Metab. Dispos. 2004, 32, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Okello, M.O.; Mishra, S.; Nishonov, M.; Mankowski, M.K.; Russell, J.D.; Wei, J.; Hogan, P.A.; Ptak, R.G.; Nair, V. A novel anti-HIV active integrase inhibitor with a favorable in vitro cytochrome P-450 and uridine 5´-diphospho-glucuronosyltransferase metabolism profile. Antivir. Res. 2013, 98, 365–372. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nair, V.; Okello, M. Integrase Inhibitor Prodrugs: Approaches to Enhancing the Anti-HIV Activity of β-Diketo Acids. Molecules 2015, 20, 12623-12651. https://doi.org/10.3390/molecules200712623
Nair V, Okello M. Integrase Inhibitor Prodrugs: Approaches to Enhancing the Anti-HIV Activity of β-Diketo Acids. Molecules. 2015; 20(7):12623-12651. https://doi.org/10.3390/molecules200712623
Chicago/Turabian StyleNair, Vasu, and Maurice Okello. 2015. "Integrase Inhibitor Prodrugs: Approaches to Enhancing the Anti-HIV Activity of β-Diketo Acids" Molecules 20, no. 7: 12623-12651. https://doi.org/10.3390/molecules200712623
APA StyleNair, V., & Okello, M. (2015). Integrase Inhibitor Prodrugs: Approaches to Enhancing the Anti-HIV Activity of β-Diketo Acids. Molecules, 20(7), 12623-12651. https://doi.org/10.3390/molecules200712623