
Separation and Identification of Four New Compounds with Antibacterial Activity from Portulaca oleracea L.

Abstract
:1. Introduction
2. Results and Discussion

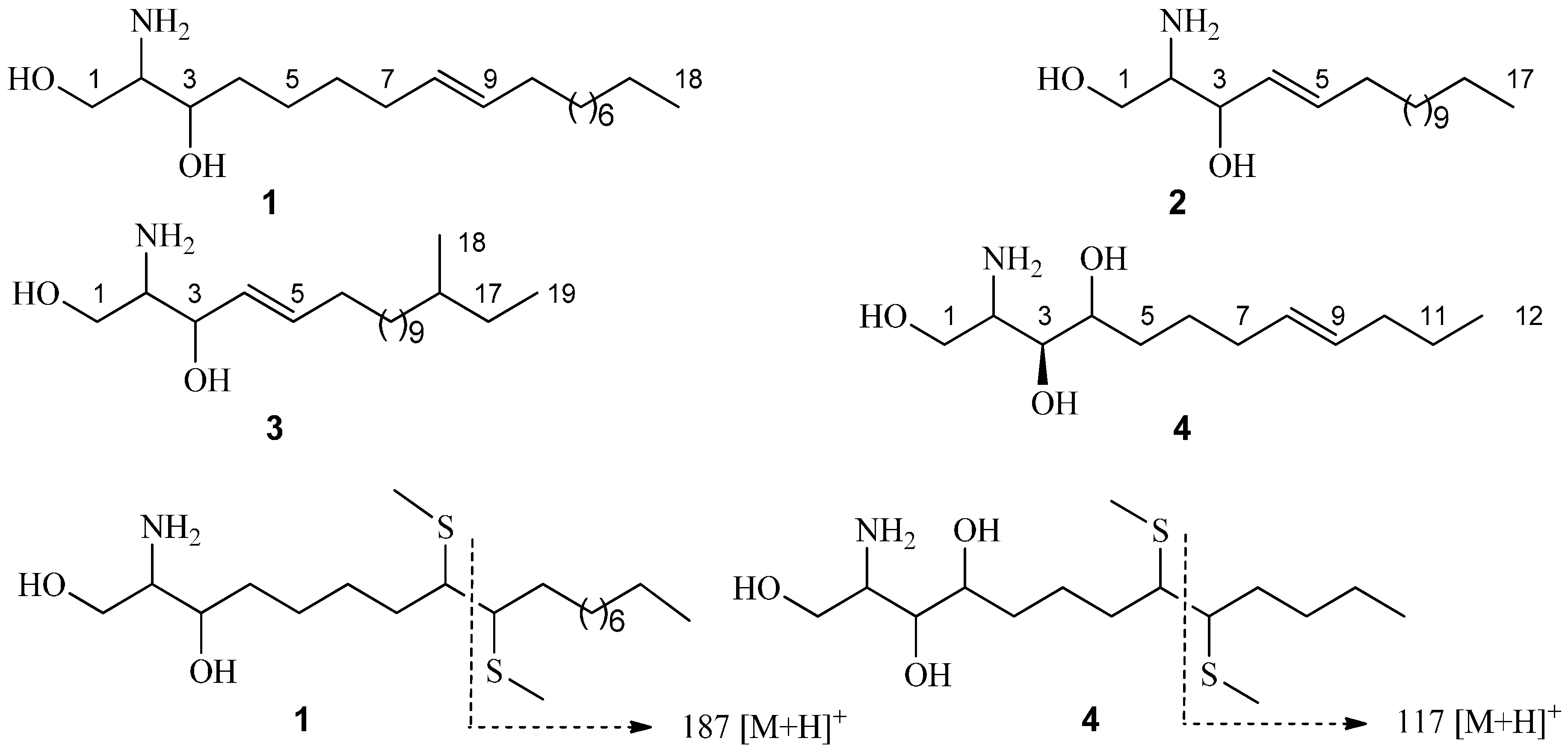{kind=link}
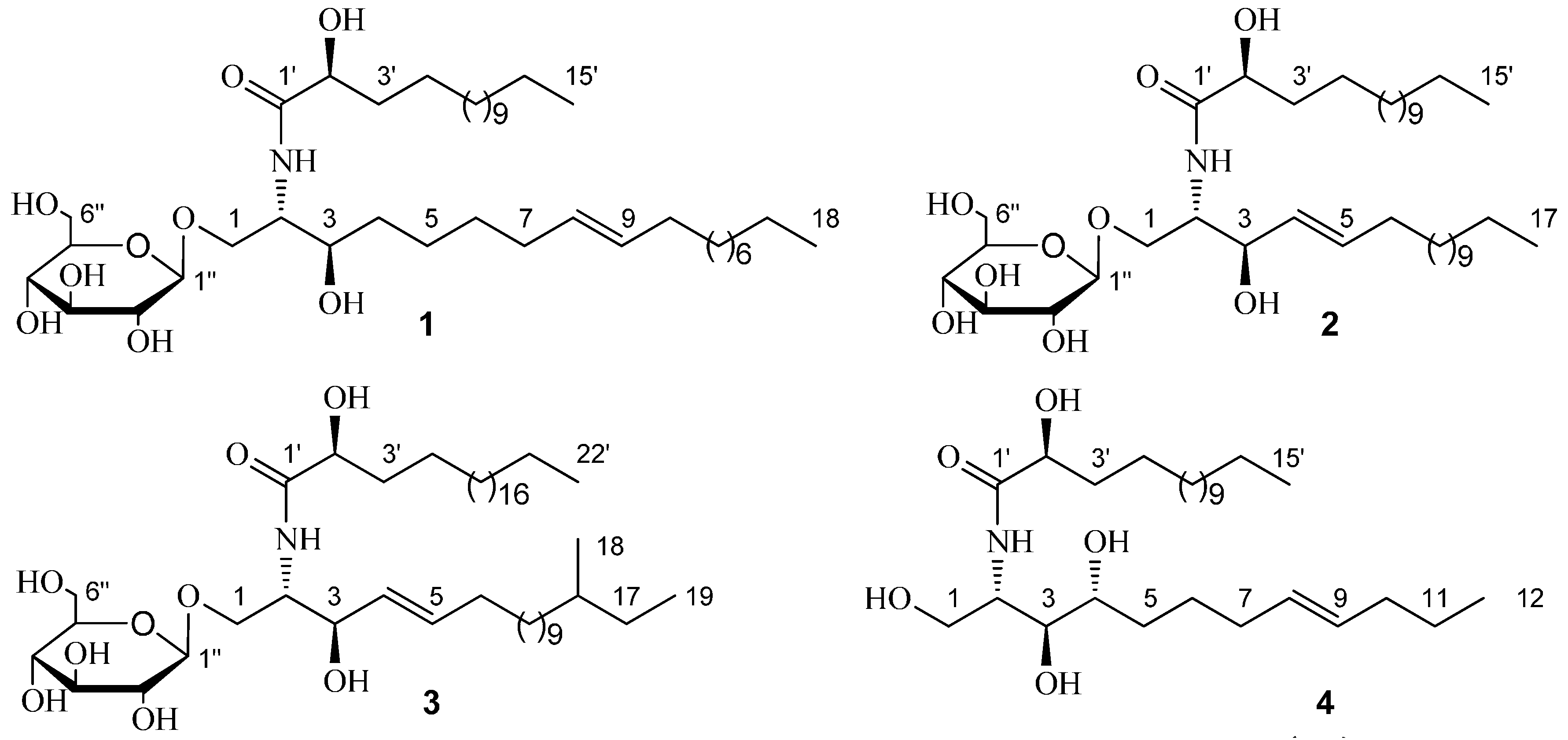{kind=link}
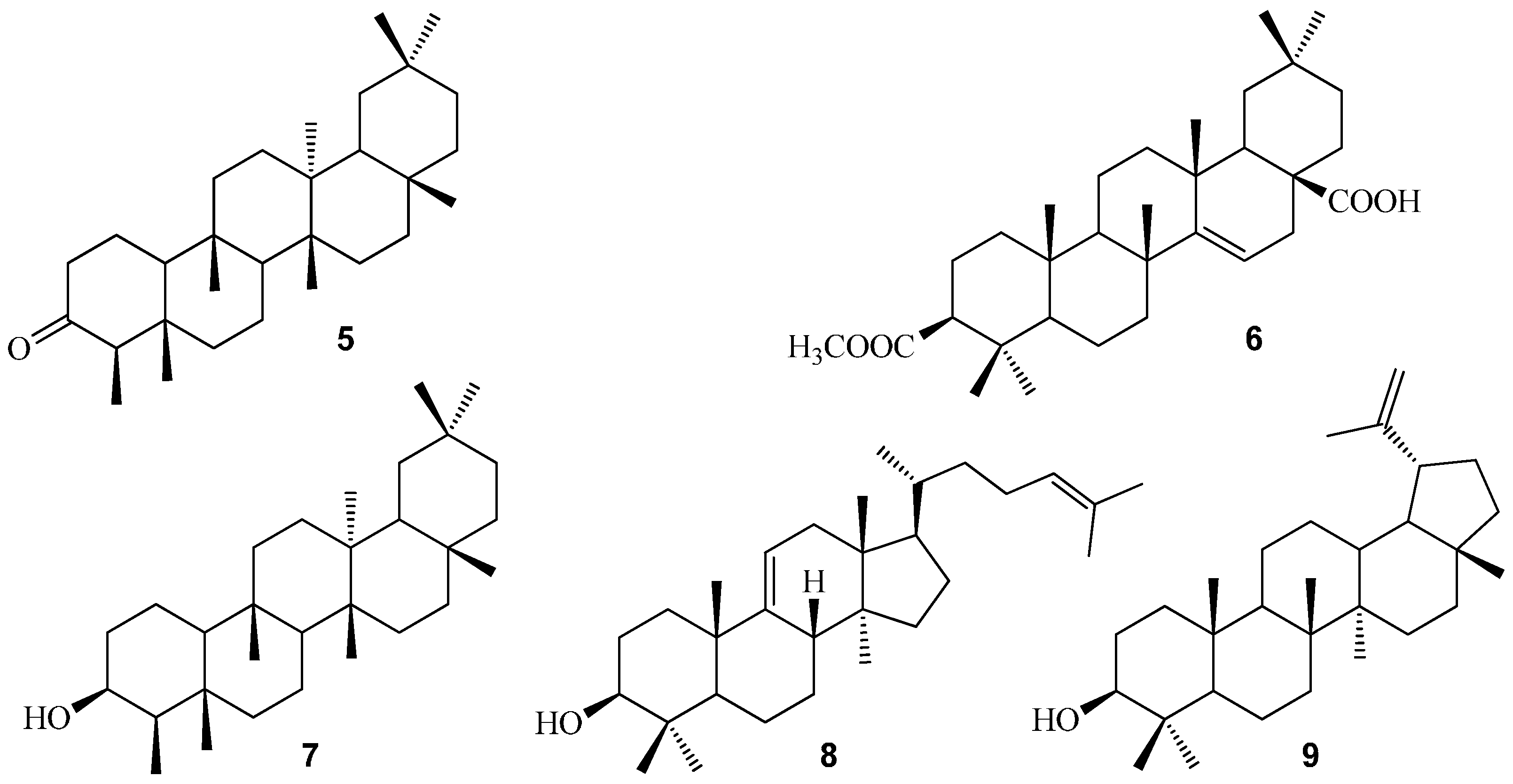{kind=link}
| 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|
| H | δH (J, Hz) | H | δH (J, Hz) | H | δH (J, Hz) | H | δH (J, Hz) |
| NH | 8.40, d (8.4) | NH | 8.36, d (8.0) | NH | 8.36, d (8.0) | NH | 8.55, d (8.0) |
| 1 | 4.35, dd (11.6, 5.6) 4.73, m | 1 | 4.22, m 4.72, m | 1 | 4.22, m 4.72, m | 1 | 4.49, dd (11.0, 4.6) 4.41, dd (11.0, 4.8) |
| 2 | 4.60, m | 2 | 4.78, m | 2 | 4.78, m | 2 | 5.10, m |
| 3 | 4.22, m | 3 | 4.76, m | 3 | 4.76, m | 3 | 4.34, dd (4.8, 6.4) |
| 4–6 | 1.15–1.40, brs | 4 | 5.86, m | 4 | 5.86, m | 4 | 4.27, m |
| 7 | 2.10, m | 5 | 5.98, m | 5 | 5.98, m | 5 | 2.22, 1.96, m |
| 8 | 5.46, m | 6 | 2.05, m | 6 | 2.05, m | 6 | 1.67, m |
| 9 | 5.46, m | 7–16 | 1.16–1.42, brs | 7–17 | 1.16–1.42, brs | 7 | 2.01, m |
| 10 | 2.02, m | 17 | 0.88, t (6.4) | 18 | 0.88, d (7.4) | 8 | 5.52, m |
| 11–17 | 1.15–1.40, brs | 2′ | 4.60, m | 19 | 0.86, t (6.4) | 9 | 5.52, m |
| 18 | 0.85, t (6.4) | 3′ | 1.86, m | 2′ | 4.60, m | 10 | 1.89, m |
| 2′ | 4.70, m | 4′ | 1.73, m 1.16–1.42, brs | 3′ | 1.86, m | 11 | 1.26–1.38, brs |
| 3′ | 1.86, m | 5′–14′ | 1.16–1.42, brs | 4′ | 1.73, m 1.16–1.42, brs | 12 | 0.88, t (6.8) |
| 4′ | 1.73, m 1.15–1.40, brs | 15′ | 0.88, t, 6.4 | 5′–21′ | 1.16–1.42, brs | 2′ | 4.60, dd (7.6, 3.2) |
| 5′–14′ | 1.15–1.40, brs | 1′′ | 4.90, d, (7.6) | 22′ | 0.88, t (6.4) | 3′ | 2.18, 2.02, m |
| 15′ | 0.85, t (6.4) | 2′′ | 4.01, m | 1′′ | 4.90, d, (7.6) | 4′ | 1.96, 1.73, m |
| 1′′ | 4.90, d (7.6) | 3′′ | 4.22, m | 2′′ | 4.01, m | 5′–14′ | 1.26–1.38, brs |
| 2′′ | 3.91, m | 4′′ | 4.22, m | 3′′ | 4.22, m | 15′ | 0.88, t (6.8) |
| 3′′ | 4.20, m | 5′′ | 3.89, m | 4′′ | 4.22, m | ||
| 4′′ | 4.03, m | 6′′ | 4.34, 4.49, m | 5′′ | 3.89, m | ||
| 5′′ | 4.12, m | 6′′ | 4.34, 4.49, m | ||||
| 6′′ | 4.18, 4.52, m | ||||||
| 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|
| C | δC | C | δC | C | δC | C | δC |
| 1 | 70.4, CH2 | 1 | 70.2, CH2 | 1 | 70.4, CH2 | 1 | 62.0, CH2 |
| 2 | 54.6, CH | 2 | 54.6, CH | 2 | 54.5, CH | 2 | 52.9, CH |
| 3 | 71.3, CH | 3 | 72.4, CH | 3 | 72.4, CH | 3 | 76.8, CH |
| 4–6 | 29.5–30.4, CH2 | 4 | 131.6, CH | 4 | 131.7, CH | 4 | 72.4, CH |
| 7 | 33.2 | 5 | 132.8, CH | 5 | 132.8, CH | 5 | 33.8, CH2 |
| 8 | 130.2, CH | 6 | 34.2, CH2 | 6 | 34.2, CH2 | 6 | 27.6, CH2 |
| 9 | 130.7, CH | 7–14 | 29.6–30.4, CH2 | 7–15 | 29.6–30.5, CH2 | 7 | 33.3, CH2 |
| 10 | 32.1, CH2 | 15 | 32.1, CH2 | 16 | 35.6, CH | 8 | 130.6, CH |
| 11–15 | 29.5–30.4, CH2 | 16 | 22.8, CH2 | 17 | 30.6, CH2 | 9 | 130.6, CH |
| 16 | 32.1, CH2 | 17 | 14.2, CH3 | 18 | 19.6, CH3 | 10 | 33.0, CH2 |
| 17 | 22.9, CH2 | 18 | 19 | 11.7, CH3 | 11 | 30.4–29.5, CH2 | |
| 18 | 14.3, CH3 | 1′ | 175.8, C | 1′ | 175.8, C | 12 | 14.3, CH3 |
| 1′ | 175.5, C | 2′ | 72.6, CH | 2′ | 72.6, CH | 1′ | 175.1, C |
| 2′ | 72.4, CH | 3′ | 35.7, CH2 | 3′ | 35.7, CH2 | 2′ | 72.8, CH |
| 3′ | 35.7, CH2 | 4′ | 26.3, CH2 | 4′ | 26.3, CH2 | 3′ | 35.6, CH2 |
| 4′ | 26.3, CH2 | 5′–12′ | 29.6–30.4, CH2 | 5′–19′ | 29.6–30.4, CH2 | 4′ | 26.8, CH2 |
| 5′–12′ | 29.5–30.4, CH2 | 13′ | 32.1, CH2 | 20′ | 32.1, CH2 | 5′–12′ | 30.4–29.5, CH2 |
| 13′ | 32.1, CH2 | 14′ | 22.8, CH2 | 21′ | 22.8, CH2 | 13′ | 32.1, CH2 |
| 14′ | 22.9, CH2 | 15′ | 14.2, CH3 | 22′ | 14.2, CH3 | 14′ | 22.8, CH2 |
| 15′ | 14.3, CH3 | 1′′ | 105.6, CH | 1′′ | 105.6, CH | 15′ | 14.3, CH3 |
| 1′′ | 105.7, CH | 2′′ | 75.1, CH | 2′′ | 75.1, CH | ||
| 2′′ | 75.2, CH | 3′′ | 78.6, CH | 3′′ | 78.6, CH | ||
| 3′′ | 78.5, CH | 4′′ | 71.5, CH | 4′′ | 71.5, CH | ||
| 4′′ | 71.6, CH | 5′′ | 78.6, CH | 5′′ | 78.6, CH | ||
| 5′′ | 78.6, CH | 6′′ | 62.6, CH2 | 6′′ | 62.6, CH2 | ||
| 6′′ | 62.7, CH2 | ||||||



| Strains | MICs (SD = 0) | MBCs (SD = 0) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 * | 2 * | 3 * | 4 * | Amoxicillin | 1 | 2 | 3 | 4 | Amoxicillin | |
| E. coli | 0.1875 | 0.1875 | 0.1875 | 0.375 | 2.34 × 10−2 | 0.25 | 0.25 | 0.25 | 0.50 | 3.12 × 10−2 |
| S. aureus | 0.1875 | 0.1875 | 0.1875 | 0.375 | 1.17 × 10−2 | 0.50 | 0.50 | 0.50 | 0.50 | 1.56 × 10−2 |
| S. flexneri | 0.1875 | 0.1875 | 0.1875 | 0.1875 | 5.85 × 10−3 | 0.25 | 0.25 | 0.25 | 0.25 | 3.90 × 10−3 |
| S. typhi | 0.1875 | 0.1875 | 0.1875 | 0.375 | 1.17 × 10−2 | 0.25 | 0.25 | 0.25 | 0.50 | 7.80 × 10−3 |
3. Experimental Section
3.1. General
3.2. Bacterial Strains and the Preparation of Inoculums
3.3. Plant Materials
3.4. Extraction and Isolation
3.5. Methanolysis of 1–4
3.6. Dimethyl Disulfide Derivative of LCBs from 1 and 4
3.7. Antibacterial Test in Vitro
3.7.1. Compounds 1–9 Serial Dilution
3.7.2. Determination of MICs and MBCs
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Liu, X.F.; Zheng, C.G.; Shi, H.G.; Tang, G.S.; Wang, W.Y.; Zhou, J.; Dong, L.W. Ethanol extract from Portulaca oleracea L. attenuated acetaminophen-induced mice liver injury. Am. J. Transl. Res. 2015, 7, 309–318. [Google Scholar] [PubMed]
- Zhou, Y.X.; Xin, H.L.; Rahman, K.; Wang, S.J.; Peng, C.; Zhang, H. Portulaca oleracea L.: A review of phytochemistry and pharmacological effects. Biomed. Res. Int. 2015, 2015, 925631. [Google Scholar] [CrossRef] [PubMed]
- Behravan, J.; Mosafa, F.; Soudmand, N.; Taghiabadi, E.; Razavi, B.M.; Karimi, G. Protective effects of aqueous and ethanolic extracts of Portulaca oleracea L. aerial parts on H2O2-induced DNA damage in lymphocytes by comet assay. J. Acupunct. Meridian Stud. 2011, 4, 193–197. [Google Scholar] [PubMed]
- Xin, H.L.; Hou, Y.H.; Xu, Y.F.; Yue, X.Q.; Li, M.; Lu, J.C.; Ling, C.Q. Portulacerebroside A: New cerebroside from Portulaca oleracea L. Chin. J. Nat. Med. 2008, 6, 401–403. [Google Scholar] [CrossRef]
- Elkhayat, E.S.; Ibrahim, S.R.; Aziz, M.A. Portulene, a new diterpene from Portulaca oleracea. J. Asian Nat. Prod. Res. 2008, 10, 1039–1043. [Google Scholar] [PubMed]
- Chan, B.C.; Han, X.Q.; Lui, S.L.; Wong, C.W.; Wang, T.B.; Cheung, D.W.; Cheng, S.W.; Ip, M.; Han, S.Q.; Yang, X.S.; et al. Combating against methicillin-resistant Staphylococcus aureus—Two fatty acids from Purslane ( Portulaca oleracea L.) exhibit synergistic effects with erythromycin. J. Pharm. Pharmacol. 2015, 67, 107–116. [Google Scholar] [PubMed]
- Noreen, S.; Hussain, I.; Tariq, M.I.; Ijaz, B.; Iqbal, S.; Qamar-Ul, Z.; Ashfaq, U.A.; Husnain, T. Portulaca oleracea L. as a prospective candidate inhibitor of hepatitis C virus NS3 serine protease. Viral Immunol. 2015, 28, 282–289. [Google Scholar] [PubMed]
- Al-Sheddi, E.S.; Farshori, N.N.; Al-Oqail, M.M.; Musarrat, J.; Al-Khedhairy, A.A.; Siddiqui, M.A. Portulaca oleracea seed oil exerts cytotoxic effects on human liver cancer (HepG2) and human lung cancer (A-549) cell lines. Asian Pac. J. Cancer Prev. 2015, 16, 3383–3387. [Google Scholar] [PubMed]
- Chan, K.; Islam, M.W.; Kamil, M.; Radhakrishnan, R.; Zakaria, M.N.; Habibullah, M.; Attas, A. The analgesic and anti-inflammatory effects of Portulaca oleracea L. subsp. sativa (haw.) Celak. J. Ethnopharmacol. 2000, 73, 445–451. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Zakaria, M.N.; Islam, M.W.; Chen, H.B.; Kamil, M.; Chan, K.; Al-Attas, A. Neuropharmacological actions of Portulaca oleraceae L. v. sativa (hawk). J. Ethnopharmacol. 2001, 76, 171–176. [Google Scholar] [CrossRef]
- Abdel Moneim, A.E. The neuroprotective effects of purslane (Portulaca oleracea) on rotenone-induced biochemical changes and apoptosis in brain of rat. Neurol. Disord. Drug Targets 2013, 12, 830–841. [Google Scholar] [CrossRef]
- Kim, J.Y.; Oh, H.M.; Kwak, S.C.; Cheon, Y.H.; Lee, M.S.; Rho, M.C.; Oh, J. Purslane suppresses osteoclast differentiation and bone resorbing activity via inhibition of Akt/GSK3 β-c-Fos-NFATc1 signaling in vitro and prevents lipopolysaccharide-induced bone loss in vivo. Biol. Pharm. Bull. 2015, 38, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, H.; Kawashima, K.; Sakagami, M.; Shimomura, M.; Ohashi, K.; Kitagawa, I. Sphingolipids and glycerolipids. Ι.: Chemical structures and ionophoretic activities of soyacerebrosides Ι and ΙΙ form soybean. Chem. Pharm. Bull. 1990, 38, 2933–2938. [Google Scholar] [CrossRef] [PubMed]
- Costantino, V.; Fattorusso, E.; Mangoni, A. Glycolipids from sponges. Part 9: Plakoside C and D, two further prenylated glycosphingolipids from the marine sponge Ectyoplasia ferox. Tetrahedron 2000, 56, 5953–5957. [Google Scholar] [CrossRef]
- Kang, S.S.; Kim, J.S.; Son, K.H.; Kim, H.P.; Chang, H.W. Cyclooxygenase-2 inhibitory cerebrosides from Phytolaccae Radix. Chem. Pharm. Bull. 2001, 49, 321–323. [Google Scholar] [CrossRef] [PubMed]
- Ling, T.J.; Xia, T.; Wan, X.C.; Li, D.X.; Wei, X.Y. Cerebrosides from the Roots of Serratula chinensis. Molecules 2006, 11, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Wada, N.; Onaka, H.; Matsubara, R.; Isobe, R.; Inagaki, M.; Higuchi, R. Constituents of holothuroidea, isolation of ante-iso type regio isomer on long chain base moiety of glucocerebroside from the sea cucumber Holothuria leucospilota. Chem. Pharm. Bull. 2005, 53, 788–791. [Google Scholar] [CrossRef] [PubMed]
- Satoshi, K.; Kazufumi, N.; Masanori, I.; Ryuichi, H. Isolation and structure determination of six glucocerebrosides from the starfish Luidia maculate. Chem. Pharm. Bull. 2002, 50, 1091–1096. [Google Scholar]
- Yang, N.Y.; Ren, D.C.; Duan, J.A.; Xu, X.H.; Xie, N.; Tian, L.J. Ceramides and cerebrosides from Ligusticum chuanxiong Hort. Helv. Chim. Acta 2009, 92, 291–297. [Google Scholar] [CrossRef]
- Chen, C.; Wei, G.; Zhu, H.; Guo, Y.; Li, X.N.; Zhang, J.; Liu, Y.; Yao, G.; Luo, Z.; Xue, Y.; et al. A new 3,4- seco- oleanane-type triterpenoid with an unusual enedione moiety from Hypericum ascyron. Fitoterapia 2015, 103, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Supaluk, P.; Saowapa, S.; Apilak, W.; Ratana, L.; Somsak, R.; Virapong, P. Bioactive metabolites from Spilanthes acmella Murr. Molecules 2009, 14, 850–867. [Google Scholar]
- Ding, H.W.; Li, F.F.; Song, S.J. Chemical constituents from Portulaca oleracea L. J. Shenyang Pharm. Univ. 2009, 26, 878–881. [Google Scholar]
- Tsai, P.W.; Kathlia, A.; Castro-Cruz, D.; Shen, C.C.; Chiou, C.T.; Ragasa, C.Y. Chemical constituents of Artocarpus camansi. Pharmacogn. J. 2013, 5, 80–82. [Google Scholar] [CrossRef]
- Rabia, R.; Zaitoon, I.; Sajid, A.; Muhammad, N.; Muhammad, Y.K.; Jamshed, I. Identification of highly potent and selective α-Glucosidase inhibitors with antiglycation potential, isolated from Rhododendron arboretum. R. Nat. Prod. 2015, 9, 262–266. [Google Scholar]
- Li, N.; Di, L.; Gao, W.C.; Wang, K.J.; Zu, L.B. Cytotoxic iridoids from the roots of Patrinia scabra. J. Nat. Prod. 2012, 75, 1723–1728. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Li, N.; Zu, L.B.; Wang, K.J.; Zhao, Y.X.; Wang, Z. Three new iridoid glucosides from the roots of Patrinia scabra. Bull. Korean Chem. Soc. 2011, 32, 3251–3254. [Google Scholar] [CrossRef]
- Lin, S.M.; Molan, P.C.; Cursons, R.T. The in vitro susceptibility of Campylobacter spp. to the antibacterial effect of manuka honey. Eur. J. Clin. Microbiol. 2009, 28, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.M.; Molan, P.C.; Cursons, R.T. The controlled in vitro susceptibility of gastrointestinal pathogens to the antibacterial effect of manuka honey. Eur. J. Clin. Microbiol. 2011, 30, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds of Portulacerebroside B–D and Portulaceramide A are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, X.; Li, J.; Liu, B.; Zhang, N.; Liu, H. Separation and Identification of Four New Compounds with Antibacterial Activity from Portulaca oleracea L. Molecules 2015, 20, 16375-16387. https://doi.org/10.3390/molecules200916375
Lei X, Li J, Liu B, Zhang N, Liu H. Separation and Identification of Four New Compounds with Antibacterial Activity from Portulaca oleracea L. Molecules. 2015; 20(9):16375-16387. https://doi.org/10.3390/molecules200916375
Chicago/Turabian StyleLei, Xia, Jianmin Li, Bin Liu, Ning Zhang, and Haiyang Liu. 2015. "Separation and Identification of Four New Compounds with Antibacterial Activity from Portulaca oleracea L." Molecules 20, no. 9: 16375-16387. https://doi.org/10.3390/molecules200916375
APA StyleLei, X., Li, J., Liu, B., Zhang, N., & Liu, H. (2015). Separation and Identification of Four New Compounds with Antibacterial Activity from Portulaca oleracea L. Molecules, 20(9), 16375-16387. https://doi.org/10.3390/molecules200916375