
Isocyanide-Based Multicomponent Reactions for the Synthesis of Heterocycles

Abstract
:
1. Introduction
1.1. Passerini Reaction






1.2. Ugi Reaction



1.3. Nitrilium Trapping by an External Nucleophile



2. Intramolecular Trapping of Nitrilium Ion
2.1. Trapping by a Carboxylic Acid

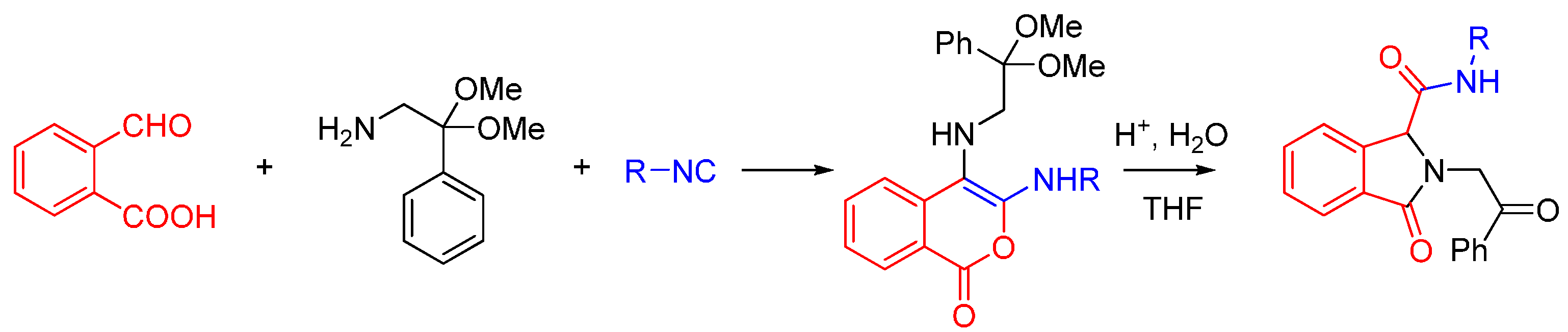
2.2. Trapping by Amide

2.3. Trapping by an Imine


2.4. Trapping by a Secondary Amine

2.5. Trapping by Amine


2.6. Trapping by Enamide

2.7. Trapping by Hydrazide

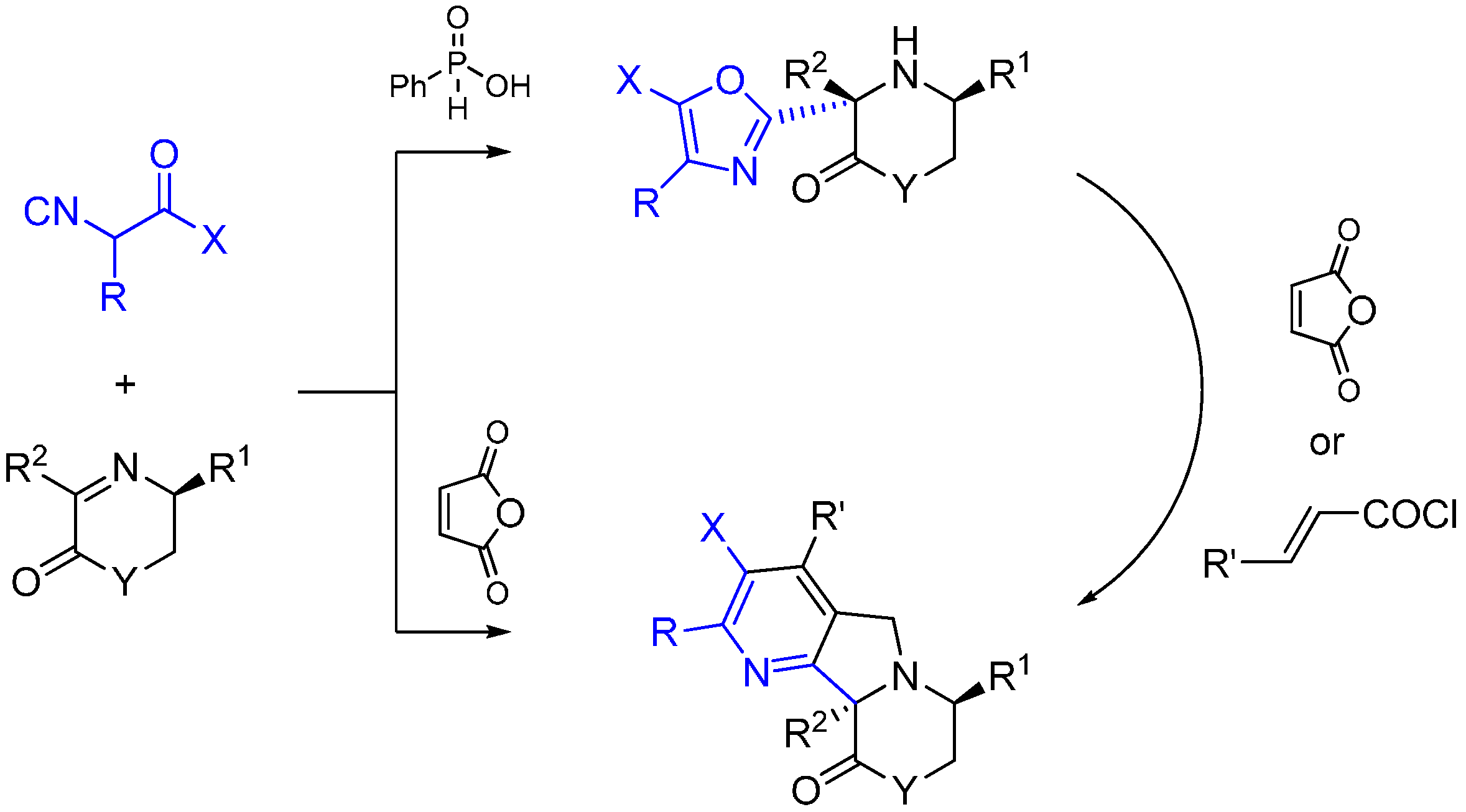
2.8. Trapping by Activated Aryl Carbon



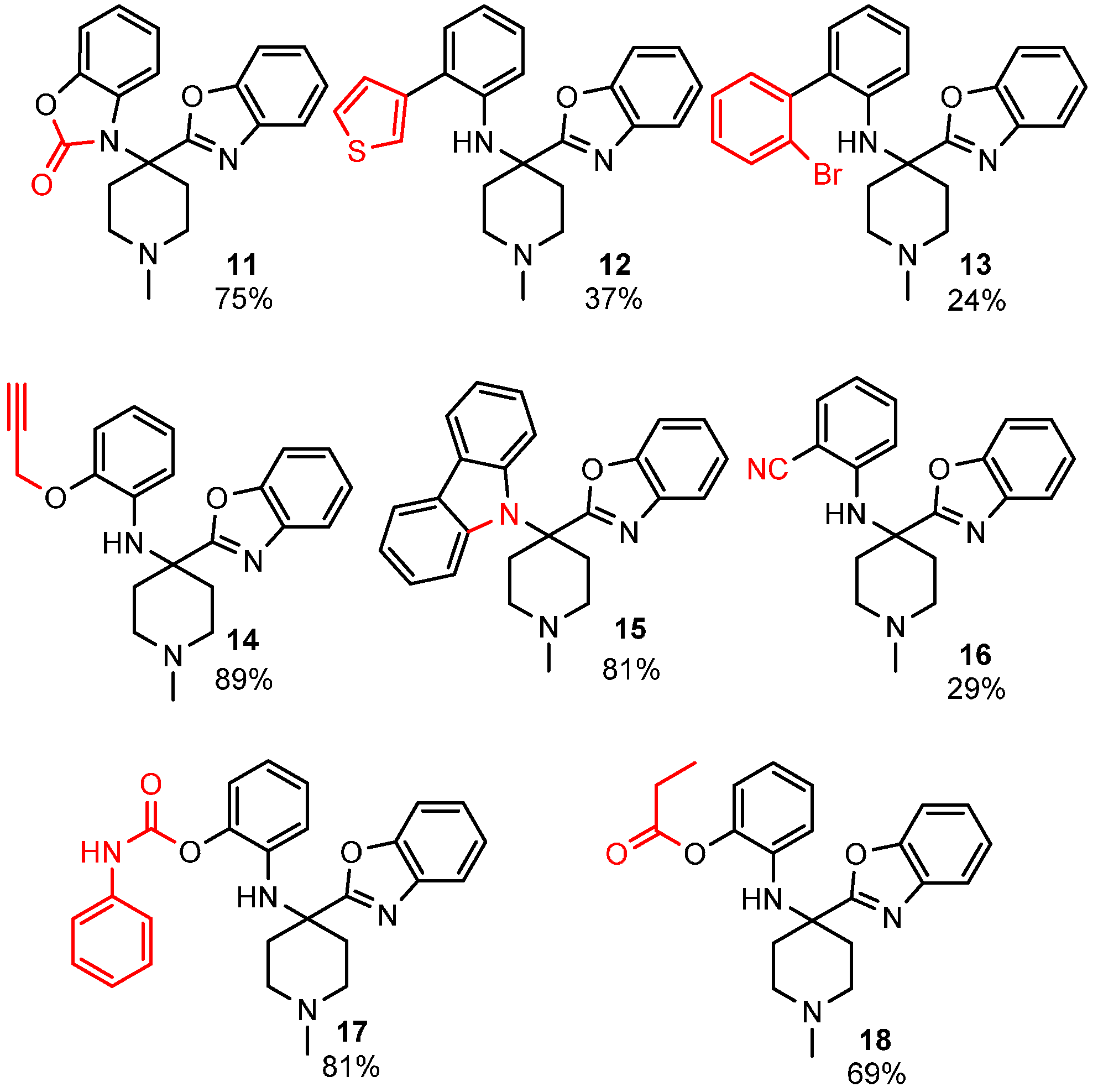
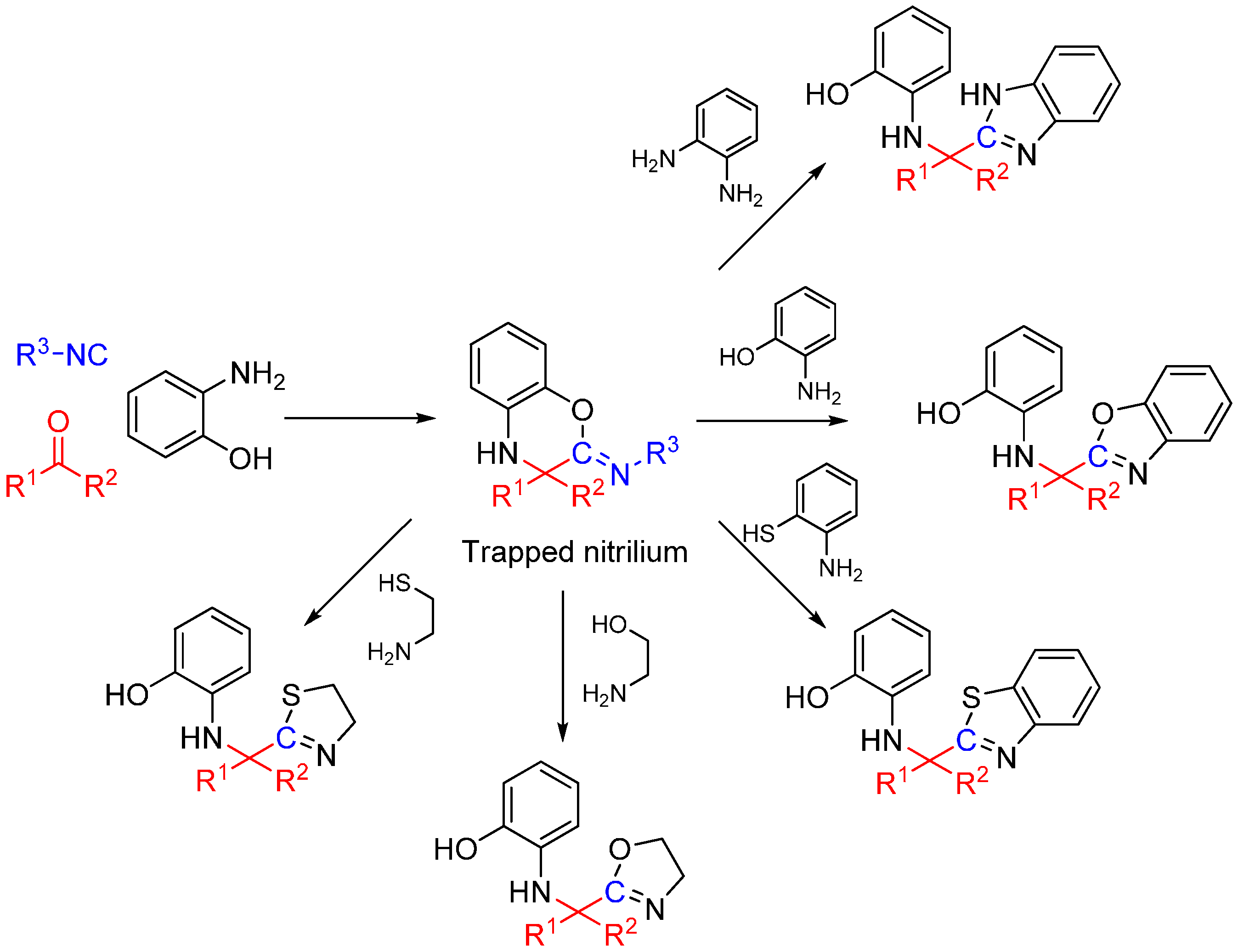
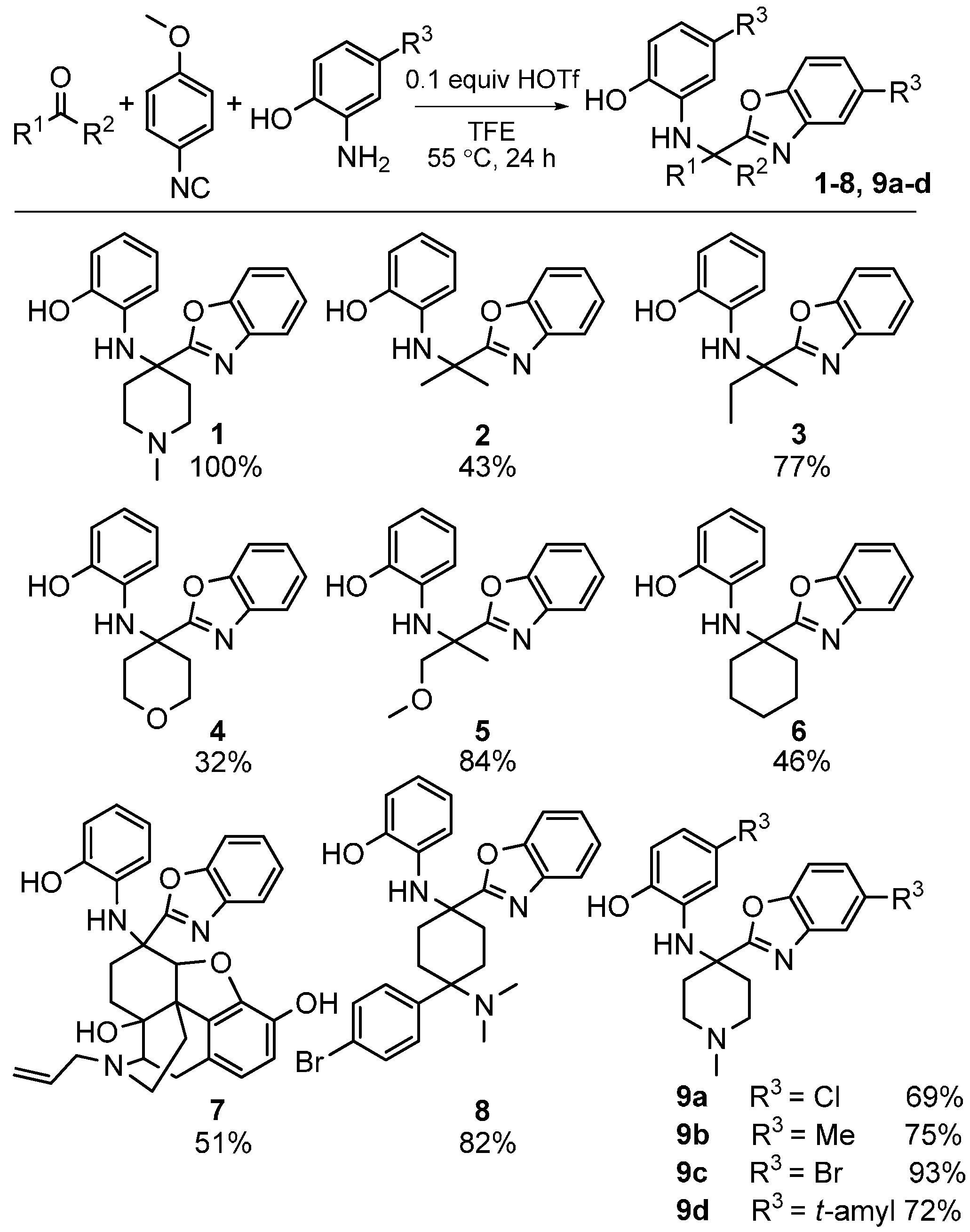
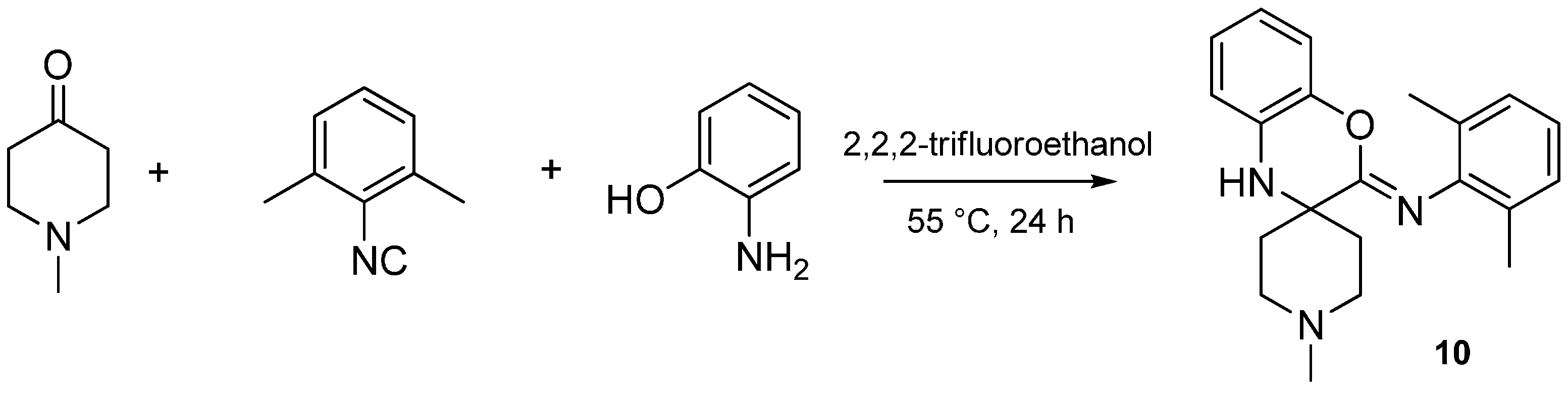
3. Intramolecular Nitrilium Trapping by Aminophenols






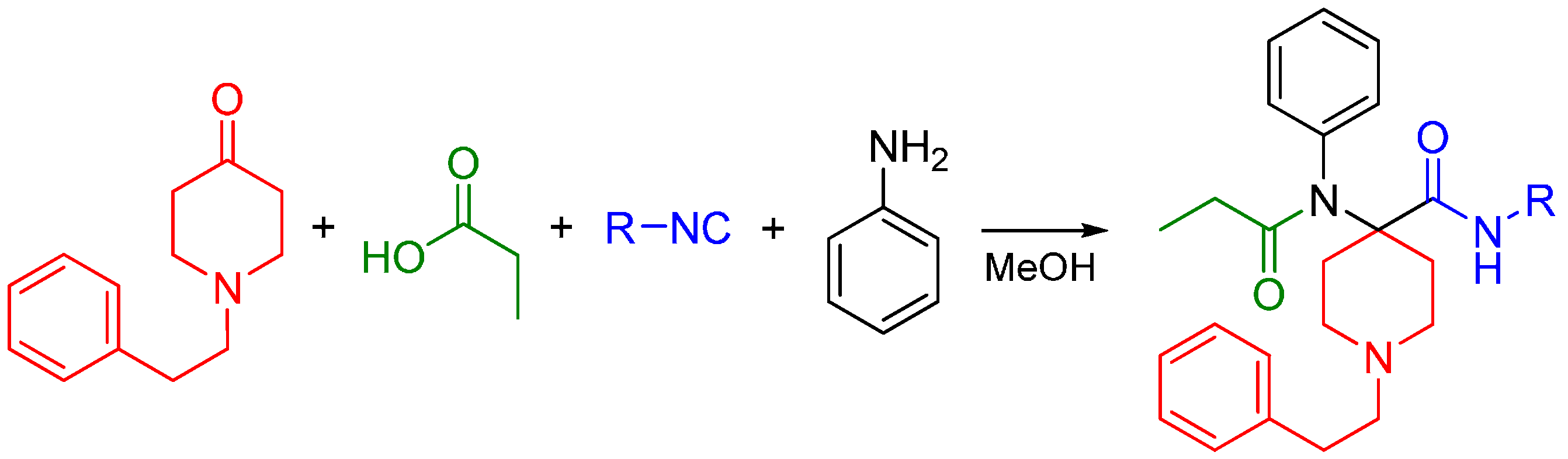
4. Synthesis of Opioids by the Ugi Reaction




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
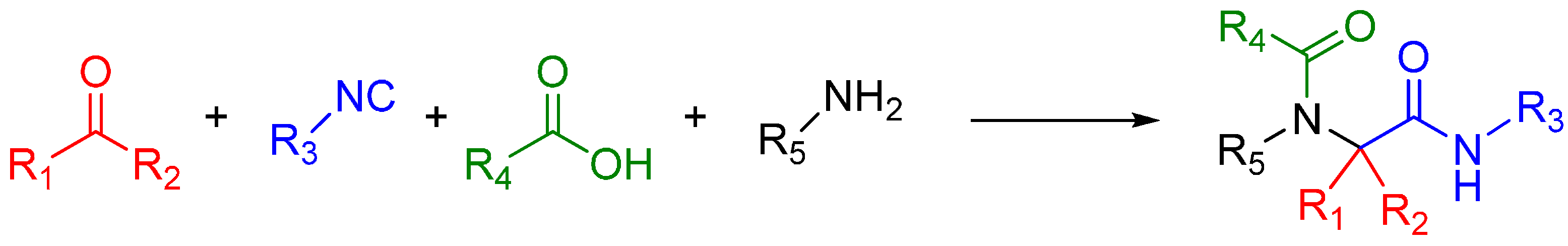{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd. | MOR-CHO | DOR-CHO | KOR-CHO | ED50 (mg/kg) |
|---|---|---|---|---|
| 20 | 10.3 ± 5.1 | >100 | 87.6 ± 29 | 0.78 ± 0.26 |
| 21 | 29.4 ± 15 | 90.7 ± 23 | >100 | 9.92 ± 0.08 |
| 22 | 0.84 ± 0.34 | 2.65 ± 0.32 | 0.44 ± 0.05 | 3.10 ± 0.19 |
| 23 | 2.66 ± 1.3 | 8.90 ± 7.7 | >100 | 10.0 ± 0.00 |
| 24 | 21.1 ± 11 | 87.9 ± 4.8 | >100 | >10 |
| 25 | 2.73 ± 2.2 | 71.2 ± 8.7 | >100 | 1.09 ± 0.05 |
| 26 | 27.0 ± 20 | 27.0 ± 3.6 | >100 | >10 |
| 27 | 25.0 ± 9.8 | 8.83 ± 0.63 | >100 | >10 |
| 28 | >100 | >100 | >100 | >10 |
| 29 | >100 | >100 | >100 | >10 |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ugi, I.; Meyr, R.; Isonitrile, V. Erweiterter anwendungsbereich der passerini-reaktion. Chem. Ber. 1961, 94, 2229–2233. [Google Scholar] [CrossRef]
- Andreana, P.R.; Liu, C.C.; Schreiber, S.L. Stereochemical control of the passerini reaction. Org. Lett. 2004, 6, 4231–4233. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Fan, Y. Catalytic, enantioselective α-additions of isocyanides: Lewis base catalyzed passerini-type reactions. J. Org. Chem. 2005, 70, 9667–9676. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-X.; Wang, M.-X.; Wang, D.-X.; Zhu, J. Catalytic enantioselective passerini three-component reaction. Angew. Chem. Int. Ed. 2008, 47, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Brioche, J.; Masson, G.; Zhu, J. Passerini three-component reaction of alcohols under catalytic aerobic oxidative conditions. Org. Lett. 2010, 12, 1432–1435. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Oguchi, T.; Taguchi, T. Direct alkylative passerini reaction of aldehydes, isocyanides, and free aliphatic alcohols catalyzed by indium(III) triflate. J. Org. Chem. 2009, 74, 3927–3929. [Google Scholar] [CrossRef] [PubMed]
- El Kaim, L.; Gizolme, M.; Grimaud, L. O-Arylative Passerini reactions. Org. Lett. 2006, 8, 5021–5023. [Google Scholar] [CrossRef] [PubMed]
- Sehlinger, A.; Kreye, O.; Meier, M.A.R. Tunable polymers obtained from Passerini multicomponent reaction derived acrylate monomers. Macromolecules 2013, 46, 6031–6037. [Google Scholar] [CrossRef]
- Paravidino, M.; Scheffelaar, R.; Schmitz, R.F.; de Kanter, F.J.J.; Groen, M.B.; Ruijter, E.; Orru, R.V.A. A flexible six-component reaction to access constrained depsipeptides based on a dihydropyridinone core. J. Org. Chem. 2007, 72, 10239–10242. [Google Scholar] [CrossRef] [PubMed]
- Jee, J.-A.; Song, S.; Rudick, J.G. Enhanced reactivity of dendrons in the Passerini three-component reaction. Chem. Commun. 2015, 51, 5456–5459. [Google Scholar] [CrossRef] [PubMed]
- Szymański, W.; Velema, W.A.; Feringa, B.L. Photocaging of carboxylic acids: A modular approach. Angew. Chem. Int. Ed. 2014, 53, 8682–8686. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, S.; Diestel, R.; Hinkelmann, B.; Muthukumar, Y.; Verma, R.P.; Sasse, F.; Jacob, C. Novel peptidomimetic compounds containing redox active chalcogens and quinones as potential anticancer agents. Eur. J. Med. Chem. 2012, 58, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, W.; Zwolinska, M.; Klossowski, S.; Młynarczuk-Biały, I.; Biały, Ł.; Issat, T.; Malejczyk, J.; Ostaszewski, R. Synthesis of novel, peptidic kinase inhibitors with cytostatic/cytotoxic activity. Bioorg. Med. Chem. 2014, 22, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Zha, Z.; Ploessl, K.; Lieberman, B.P.; Zhu, L.; Wise, D.R.; Thompson, C.B.; Kung, H.F. Synthesis of optically pure 4-fluoro-glutamines as potential metabolic imaging agents for tumors. J. Am. Chem. Soc. 2011, 133, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Velázquez, F.; Venkatraman, S.; Wu, W.; Blackman, M.; Prongay, A.; Girijavallabhan, V.; Shih, N.-Y.; Njoroge, F.G. Application of ring-closing metathesis for the synthesis of macrocyclic peptidomimetics as inhibitors of hcv ns3 protease. Org. Lett. 2007, 9, 3061–3064. [Google Scholar] [CrossRef] [PubMed]
- Soeta, T.; Kojima, Y.; Ukaji, Y.; Inomata, K. O-Silylative Passerini reaction: A new one-pot synthesis of α-siloxyamides. Org. Lett. 2010, 12, 4341–4343. [Google Scholar] [CrossRef] [PubMed]
- Soeta, T.; Matsuzaki, S.; Ukaji, Y. A one-pot o-sulfinative passerini/oxidation reaction: Synthesis of α-(sulfonyloxy)amide derivatives. J. Org. Chem. 2015, 80, 3688–3694. [Google Scholar] [CrossRef] [PubMed]
- Soeta, T.; Matsuzaki, S.; Ukaji, Y. A one-pot o-phosphinative passerini/pudovik reaction: Efficient synthesis of highly functionalized α-(phosphinyloxy)amide derivatives. Chem. Eur. J. 2014, 20, 5007–5012. [Google Scholar] [CrossRef] [PubMed]
- Sunderhaus, J.D.; Martin, S.F. Applications of multicomponent reactions to the synthesis of diverse heterocyclic scaffolds. Chem.-Eur. J. 2009, 15, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Paulvannan, K. Preparation of tricyclic nitrogen heterocycles via tandem four-component condensation/intramolecular Diels-Alder reaction. Tetrahedron Lett. 1999, 40, 1851–1854. [Google Scholar] [CrossRef]
- Koopmanschap, G.; Ruijter, E.; Orru, R.V.A. Isocyanide-based multicomponent reactions towards cyclic constrained peptidomimetics. Beilstein J. Org. Chem. 2014, 10, 544–598. [Google Scholar] [CrossRef] [PubMed]
- Bonnaterre, F.; Bois-Choussy, M.; Zhu, J. Rapid access to oxindoles by the combined use of an Ugi four-component reaction and a microwave-assisted intramolecular buchwald−hartwig amidation reaction. Org. Lett. 2006, 8, 4351–4354. [Google Scholar] [CrossRef] [PubMed]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Godet, T.; Bonvin, Y.; Vincent, G.; Merle, D.; Thozet, A.; Ciufolini, M.A. Titanium catalysis in the Ugi reaction of α-amino acids with aromatic aldehydes. Org. Lett. 2004, 6, 3281–3284. [Google Scholar] [CrossRef] [PubMed]
- Okandeji, B.O.; Gordon, J.R.; Sello, J.K. Catalysis of Ugi four component coupling reactions by rare earth metal triflates. J. Org. Chem. 2008, 73, 5595–5597. [Google Scholar] [CrossRef] [PubMed]
- Dömling, A.; Wang, W.; Wang, K. Chemistry and biology of multicomponent reactions. Chem. Rev. 2012, 112, 3083–3135. [Google Scholar] [CrossRef] [PubMed]
- Shaabani, A.; Keshipour, S.; Shaabani, S.; Mahyari, M. Zinc chloride catalyzed three-component Ugi reaction: Synthesis of n-cyclohexyl-2-(2-hydroxyphenylamino)acetamide derivatives. Tetrahedron Lett. 2012, 53, 1641–1644. [Google Scholar] [CrossRef]
- Pan, S.C.; List, B. Catalytic three-component Ugi reaction. Angew. Chem. Int. Ed. 2008, 47, 3622–3625. [Google Scholar] [CrossRef] [PubMed]
- Ruijter, E.; Scheffelaar, R.; Orru, R.V.A. Multicomponent reaction design in the quest for molecular complexity and diversity. Angew. Chem. Int. Ed. 2011, 50, 6234–6246. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.T.; R, S.B.; Lal, M.; Mir, M.H. Formation of unexpected [small alpha]-amino amidine through three-component “Ugi condensation reaction”. RSC Adv. 2012, 2, 5506–5509. [Google Scholar] [CrossRef]
- El Kaïm, L.; Grimaud, L.; Oble, J. Phenol Ugi–smiles systems: Strategies for the multicomponent n-arylation of primary amines with isocyanides, aldehydes, and phenols. Angew. Chem. Int. Ed. 2005, 44, 7961–7964. [Google Scholar] [CrossRef] [PubMed]
- El Kaim, L.; Gizolme, M.; Grimaud, L.; Oble, J. Direct access to heterocyclic scaffolds by new multicomponent Ugi−Smiles couplings. Org. Lett. 2006, 8, 4019–4021. [Google Scholar] [CrossRef] [PubMed]
- Dömling, A.; Ugi, I. Multicomponent reactions with isocyanides. Angew. Chem. Int. Ed. 2000, 39, 3168–3210. [Google Scholar] [CrossRef]
- Hulme, C.; Gore, V. “Multi-component reactions: Emerging chemistry in drug discovery” “from xylocain to crixivan”. Curr. Med. Chem. 2003, 10, 51–80. [Google Scholar] [CrossRef] [PubMed]
- Faggi, C.; García-Valverde, M.A.; Marcaccini, S.; Menchi, G. Isolation of Ugi four-component condensation primary adducts: A straightforward route to isocoumarins. Org. Lett. 2010, 12, 788–791. [Google Scholar] [CrossRef] [PubMed]
- Bonne, D.; Dekhane, M.; Zhu, J. Exploiting the dual reactivity of o-isocyanobenzamide: Three-component synthesis of 4-imino-4H-3,1-benzoxazines. Org. Lett. 2005, 7, 5285–5288. [Google Scholar] [CrossRef] [PubMed]
- Groebke, K.; Weber, L.; Mehlin, F. Synthesis of imidazo[1,2-a] annulated pyridines, pyrazines and pyrimidines by a novel three-component condensation. Synlett 1998, 6, 661–663. [Google Scholar] [CrossRef]
- Blackburn, C.; Guan, B.; Fleming, P.; Shiosaki, K.; Tsai, S. Parallel synthesis of 3-aminoimidazo[1,2-a]pyridines and pyrazines by a new three-component condensation. Tetrahedron Lett. 1998, 39, 3635–3638. [Google Scholar] [CrossRef]
- Bienaymé, H.; Bouzid, K. A new heterocyclic multicomponent reaction for the combinatorial synthesis of fused 3-aminoimidazoles. Angew. Chem. Int. Ed. 1998, 37, 2234–2237. [Google Scholar] [CrossRef]
- Hulme, C.; Lee, Y.-S. Emerging approaches for the syntheses of bicyclic imidazo[1,2-x]-heterocycles. Mol. Divers. 2008, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Devi, N.; Rawal, R.K.; Singh, V. Diversity-oriented synthesis of fused-imidazole derivatives via Groebke–Blackburn–Bienayme reaction: A review. Tetrahedron 2015, 71, 183–232. [Google Scholar] [CrossRef]
- Akritopoulou-Zanze, I.; Wakefield, B.D.; Gasiecki, A.; Kalvin, D.; Johnson, E.F.; Kovar, P.; Djuric, S.W. Scaffold oriented synthesis. Part 4: Design, synthesis and biological evaluation of novel 5-substituted indazoles as potent and selective kinase inhibitors employing heterocycle forming and multicomponent reactions. Bioorg. Med. Chem. Lett. 2011, 21, 1480–1483. [Google Scholar] [CrossRef] [PubMed]
- Baviskar, A.T.; Madaan, C.; Preet, R.; Mohapatra, P.; Jain, V.; Agarwal, A.; Guchhait, S.K.; Kundu, C.N.; Banerjee, U.C.; Bharatam, P.V. N-Fused imidazoles as novel anticancer agents that inhibit catalytic activity of topoisomerase IIα and induce apoptosis in g1/s phase. J. Med. Chem. 2011, 54, 5013–5030. [Google Scholar] [CrossRef] [PubMed]
- Shukla, N.M.; Salunke, D.B.; Yoo, E.; Mutz, C.A.; Balakrishna, R.; David, S.A. Antibacterial activities of groebke-blackburn-bienayme-derived imidazo[1,2-a]pyridin-3-amines. Bioorg. Med. Chem. Lett. 2012, 20, 5850–5863. [Google Scholar] [CrossRef] [PubMed]
- Burchak, O.N.; Mugherli, L.; Ostuni, M.; Lacapère, J.J.; Balakirev, M.Y. Combinatorial discovery of fluorescent pharmacophores by multicomponent reactions in droplet arrays. J. Am. Chem. Soc. 2011, 133, 10058–10061. [Google Scholar] [CrossRef] [PubMed]
- Elleder, D.; Baiga, T.; Russell, R.; Naughton, J.; Hughes, S.; Noel, J.; Young, J. Identification of a 3-aminoimidazo[1,2-a]pyridine inhibitor of hiv-1 reverse transcriptase. Virol. J. 2012, 9, 305. [Google Scholar] [CrossRef] [PubMed]
- Bode, M.L.; Gravestock, D.; Moleele, S.S.; van der Westhuyzen, C.W.; Pelly, S.C.; Steenkamp, P.A.; Hoppe, H.C.; Khan, T.; Nkabinde, L.A. Imidazo[1,2-a]pyridin-3-amines as potential HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. Lett. 2011, 19, 4227–4237. [Google Scholar] [CrossRef] [PubMed]
- Ganem, B. Strategies for innovation in multicomponent reaction design. Acc. Chem. Res. 2009, 42, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Shaabani, A.; Maleki, A.; Mofakham, H.; Khavasi, H.R. Novel isocyanide-based three-component synthesis of 3,4-dihydroquinoxalin-2-amine derivatives. J. Comb. Chem. 2008, 10, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Soeta, T.; Tamura, K.; Ukaji, Y. [5 + 1] cycloaddition of c,n-cyclic n′-acyl azomethine imines with isocyanides. Org. Lett. 2012, 14, 1226–1229. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.-H.; Wang, D.-X.; Zhao, L.; Zhu, J.; Wang, M.-X. Synthesis of substituted pyridines from cascade [1 + 5] cycloaddition of isonitriles to n-formylmethyl-substituted enamides, aerobic oxidative aromatization, and acyl transfer reaction. J. Amer. Chem. Soc. 2013, 135, 4708–4711. [Google Scholar] [CrossRef] [PubMed]
- Shutske, G.M.; Kapples, K.J.; Tomer, J.D.; Hrib, N.J.; Jurcak, J.J. Substituted-4-amino-3-pyridinols, a Process for Their Preparation and Their Use as Medicaments. EP0477903 A2, 1 April 1991. [Google Scholar]
- Hashimoto, T.; Kimura, H.; Kawamata, Y.; Maruoka, K. A catalytic asymmetric Ugi-type reaction with acyclic azomethine imines. Angew. Chem. Int. Ed. Engl. 2012, 51, 7279–7281. [Google Scholar] [CrossRef] [PubMed]
- Pirrung, M.C.; Ghorai, S. Versatile, fragrant, convertible isonitriles. J. Am. Chem. Soc. 2006, 128, 11772–11773. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Schneekloth, J.S.; Sorensen, E.J. A chemical synthesis of 11-methoxy mitragynine pseudoindoxyl featuring the interrupted ugi reaction. Chem. Sci. 2012, 3, 2849–2852. [Google Scholar] [CrossRef] [PubMed]
- Schneekloth, J.S.; Jimin, K., Jr.; Sorensen, E.J. An interrupted Ugi reaction enables the preparation of substituted indoxyls and aminoindoles. Tetrahedron 2009, 65, 3096–3101. [Google Scholar] [CrossRef] [PubMed]
- Kysil, V.; Tkachenko, S.; Khvat, A.; Williams, C.; Tsirulnikov, S.; Churakova, M.; Ivachtchenko, A. Tmscl-promoted isocyanide-based mcr of ethylenediamines: An efficient assembling of 2-aminopyrazine core. Tetrahedron Lett. 2007, 48, 6239–6244. [Google Scholar] [CrossRef]
- Xia, L.; Li, S.; Chen, R.; Liu, K.; Chen, X. Catalytic ugi-type condensation of α-isocyanoacetamide and chiral cyclic imine: Access to asymmetric construction of several heterocycles. J. Org. Chem. 2013, 78, 3120–3131. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharappa, A.P.; Badiger, S.E.; Dubey, P.K.; Panigrahi, S.K.; Manukonda, S.R. Design and synthesis of 2-substituted benzoxazoles as novel ptp1b inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 2579–2584. [Google Scholar] [CrossRef] [PubMed]
- Ulhaq, S.; Chinje, E.C.; Naylor, M.A.; Jaffar, M.; Stratford, I.J.; Threadgill, M.D. Heterocyclic analogues of l-citrulline as inhibitors of the isoforms of nitric oxide synthase (nos) and identification of nδ-(4,5-dihydrothiazol-2-yl)ornithine as a potent inhibitor. Bioorg. Med. Chem. Lett. 1999, 7, 1787–1796. [Google Scholar] [CrossRef]
- Horton, D.A.; Bourne, G.T.; Smythe, M.L. The combinatorial synthesis of bicyclic privileged structures or privileged substructures. Chem. Rev. 2003, 103, 893–930. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Hirao, A.; Imai, A.; Nakamura, H.; Murata, Y.; Ohashi, K.; Nakata, E. Novel non-peptide nociceptin/orphanin fq receptor agonist, 1-[1-(1-methylcyclooctyl)-4-piperidinyl]-2-[(3R)-3-piperidinyl]-1H-benzimidazole: Design, synthesis, and structure−activity relationship of oral receptor occupancy in the brain for orally potent antianxiety drug(1, 2). J. Med. Chem. 2009, 52, 610–625. [Google Scholar] [PubMed]
- Siracusa, M.A.; Salerno, L.; Modica, M.N.; Pittalà, V.; Romeo, G.; Amato, M.E.; Nowak, M.; Bojarski, A.J.; Mereghetti, I.; Cagnotto, A.; et al. Synthesis of new arylpiperazinylalkylthiobenzimidazole, benzothiazole, or benzoxazole derivatives as potent and selective 5-ht1a serotonin receptor ligands†. J. Med. Chem. 2008, 51, 4529–4538. [Google Scholar] [CrossRef] [PubMed]
- Varadi, A.; Palmer, T.C.; Notis, P.R.; Redel-Traub, G.N.; Afonin, D.; Subrath, J.J.; Pasternak, G.W.; Hu, C.; Sharma, I.; Majumdar, S. Three-component coupling approach for the synthesis of diverse heterocycles utilizing reactive nitrilium trapping. Org. Lett. 2014, 16, 1668–1671. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.V. Synthetic strategies towards benzoxazole ring systems: A review. Asian J. Chem. 2004, 16, 1241–1260. [Google Scholar] [CrossRef]
- Tempest, P.; Ma, V.; Thomas, S.; Hua, Z.; Kelly, M.G.; Hulme, C. Two-step solution-phase synthesis of novel benzimidazoles utilizing a udc (Ugi/de-Boc/cyclize) strategy. Tetrahedron Lett. 2001, 42, 4959–4962. [Google Scholar] [CrossRef]
- Boissarie, P.J.; Hamilton, Z.E.; Lang, S.; Murphy, J.A.; Suckling, C.J. A powerful palladium-catalyzed multicomponent process for the preparation of oxazolines and benzoxazoles. Org. Lett. 2011, 13, 6256–6259. [Google Scholar] [CrossRef] [PubMed]
- Spatz, J.H.; Bach, T.; Umkehrer, M.; Bardin, J.; Ross, G.; Burdack, C.; Kolb, J. Diversity oriented synthesis of benzoxazoles and benzothiazoles. Tetrahedron Lett. 2007, 48, 9030–9034. [Google Scholar] [CrossRef]
- El Kaim, L.; Grimaud, L. Beyond the ugi reaction: Less conventional interactions between isocyanides and iminium species. Tetrahedron 2009, 65, 2153–2171. [Google Scholar] [CrossRef]
- Vlaar, T.; Ruijter, E.; Maes, B.U.W.; Orru, R.V.A. Palladium-catalyzed migratory insertion of isocyanides: An emerging platform in cross-coupling chemistry. Angew. Chem. Int. Ed. 2013, 52, 7084–7097. [Google Scholar] [CrossRef] [PubMed]
- Mosberg, H.I.; Yeomans, L.; Anand, J.P.; Porter, V.; Sobczyk-Kojiro, K.; Traynor, J.R.; Jutkiewicz, E.M. Development of a bioavailable mu opioid receptor (mopr) agonist, delta opioid receptor (dopr) antagonist peptide that evokes antinociception without development of acute tolerance. J. Med. Chem. 2014, 57, 3148–3153. [Google Scholar] [CrossRef] [PubMed]
- Ananthan, S. Opioid ligands with mixed mu/delta opioid receptor interactions: An emerging approach to novel analgesics. AAPS J. 2006, 8, E118–E125. [Google Scholar] [CrossRef] [PubMed]
- Ananthan, S.; Kezar, H.S.; Carter, R.L.; Saini, S.K.; Rice, K.C.; Wells, J.L.; Davis, P.; Xu, H.; Dersch, C.M.; Bilsky, E.J.; et al. Synthesis, opioid receptor binding, and biological activities of naltrexone-derived pyrido- and pyrimidomorphinans. J. Med. Chem. 1999, 42, 3527–3538. [Google Scholar] [CrossRef] [PubMed]
- Schiller, P.W.; Fundytus, M.E.; Merovitz, L.; Weltrowska, G.; Nguyen, T.M.D.; Lemieux, C.; Chung, N.N.; Coderre, T.J. The opioid μ agonist/δ antagonist dipp-nh2[ψ] produces a potent analgesic effect, no physical dependence, and less tolerance than morphine in rats. J. Med. Chem. 1999, 42, 3520–3526. [Google Scholar] [CrossRef] [PubMed]
- Purington, L.C.; Pogozheva, I.D.; Traynor, J.R.; Mosberg, H.I. Pentapeptides displaying mu opioid receptor agonist and delta opioid receptor partial agonist/antagonist properties. J. Med. Chem. 2009, 52, 7724–7731. [Google Scholar] [CrossRef] [PubMed]
- Lowery, J.J.; Raymond, T.J.; Giuvelis, D.; Bidlack, J.M.; Polt, R.; Bilsky, E.J. In vivo characterization of mmp-2200, a mixed delta/mu opioid agonist, in mice. J. Pharmacol. Exp. Ther. 2011, 336, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Do Carmo, G.P.; Polt, R.; Bilsky, E.J.; Rice, K.C.; Negus, S.S. Behavioral pharmacology of the μ/δ opioid glycopeptide mmp2200 in rhesus monkeys. J. Pharmacol. Exp. Ther. 2008, 326, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Maguire, P.; Tsai, N.; Kamal, J.; Cometta-Morini, C.; Upton, C.; Loew, G. Pharmacological profiles of fentanyl analogs at mu, delta and kappa opiate receptors. Eur. J. Pharmacol. 1992, 213, 219–225. [Google Scholar] [CrossRef]
- Malaquin, S.; Jida, M.; Gesquiere, J.-C.; Deprez-Poulain, R.; Deprez, B.; Laconde, G. Ugi reaction for the synthesis of 4-aminopiperidine-4-carboxylic acid derivatives. Application to the synthesis of carfentanil and remifentanil. Tetrahedron Lett. 2010, 51, 2983–2985. [Google Scholar] [CrossRef]
- Pentel, P.R.; Portoghese, P.S.; Pravetoni, M.; Naour, M.C.P.L. Compositions and Methods of Treating Opioid Addiction. US20140093525 A1, 3 April 2014. [Google Scholar]
- Portoghese, P.; Eyup, A. Analgesic Conjugates. WO2014124317 A1, 14 August 2014. [Google Scholar]
- Váradi, A.; Palmer, T.C.; Haselton, N.; Afonin, D.; Subrath, J.J.; le Rouzic, V.; Hunkele, A.; Pasternak, G.W.; Marrone, G.F.; Borics, A.; et al. Synthesis of carfentanil amide opioids using the ugi multicomponent reaction. ACS Chem. Neurosci. 2015, 6, 1570–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srulevitch, D.B.; Lien, E.J. Design, synthesis and sar of analgesics. Prog. Clin. Biol. Res. 1989, 291, 377–381. [Google Scholar] [PubMed]
- Srulevitch, D.B.; Lien, E.J. 4-phenylamidopiperidines: Synthesis, pharmacological testing and sar analysis. Acta Pharm. Jugosl. 1991, 41, 89–106. [Google Scholar]
- Kolesnikov, Y.A.; Pick, C.G.; Ciszewska, G.; Pasternak, G.W. Blockade of tolerance to morphine but not to kappa opioids by a nitric oxide synthase inhibitor. Proc. Nat. Acad. Sci. USA 1993, 90, 5162–5166. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Váradi, A.; Palmer, T.C.; Notis Dardashti, R.; Majumdar, S. Isocyanide-Based Multicomponent Reactions for the Synthesis of Heterocycles. Molecules 2016, 21, 19. https://doi.org/10.3390/molecules21010019
Váradi A, Palmer TC, Notis Dardashti R, Majumdar S. Isocyanide-Based Multicomponent Reactions for the Synthesis of Heterocycles. Molecules. 2016; 21(1):19. https://doi.org/10.3390/molecules21010019
Chicago/Turabian StyleVáradi, András, Travis C. Palmer, Rebecca Notis Dardashti, and Susruta Majumdar. 2016. "Isocyanide-Based Multicomponent Reactions for the Synthesis of Heterocycles" Molecules 21, no. 1: 19. https://doi.org/10.3390/molecules21010019
APA StyleVáradi, A., Palmer, T. C., Notis Dardashti, R., & Majumdar, S. (2016). Isocyanide-Based Multicomponent Reactions for the Synthesis of Heterocycles. Molecules, 21(1), 19. https://doi.org/10.3390/molecules21010019