2. Results and Discussion
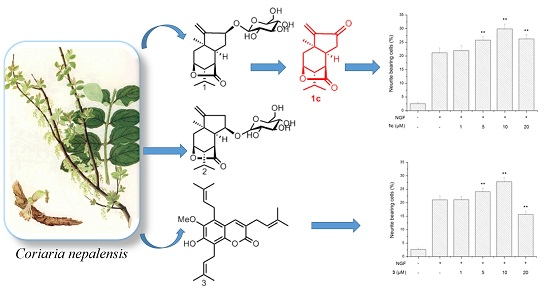
Compound
1 was isolated as a white, amorphous powder. The molecular formula was established as C
21H
32O
8 via HRESIMS (
m/z [M + Na]
+ 435.1991; calcd. 435.1995) in combination with the
13C-NMR spectrum, requiring six degrees of unsaturation. Its IR spectrum indicated the presence of hydroxy (3563 cm
−1) and γ-lactone carbonyl (1765 cm
−1) groups. The
13C and DEPT NMR spectra of
1 (
Table 1) exhibited 21 carbon signals corresponding to three methyl groups, four methylene groups (including one olefinic carbon at δ 111.5 and one oxygenated carbon at δ 61.6), 11 methine groups (seven oxygenated carbons including five carbons of sugar), and three quaternary carbons (one lactone carbonyl at δ 180.6 and one olefinic carbon at δ 158.2). The
1H and
13C-NMR spectra of
1 displayed signals for tertiary methyl (δ 1.25, s, 3H), isopropyl (δ 0.99, 1.01, d, each 3H, and δ 1.83, m, 1H), and exocyclic olefinic bond (δ
H 5.21, s, 1H, 5.42, s, 1H) groups. A series of sugar signals were also detected in the
1H-NMR spectrum (
Table 1) between δ
H 3.22–3.87, with an anomeric proton at δ
H 4.35 (d, 1H,
J= 7.5 Hz, H-1′). The
13C-NMR spectrum indicated the presence of a glucose unit due to a set of carbon signals at δ
C 101.5, 73.5, 76.2, 70.2, 76.6, and 61.6. These data suggested the presence of a glucopyranosyl moiety in
1. The glucose in nepalactone A derivative
1a appeared to be tetraacetylated, based on NMR spectra (
Table 1) and HRESIMS (
m/
z 603.2416 [M + Na]
+). Furthermore, the
1H and
13C-NMR data of
1 were similar to those of the known picrotoxane sesquiterpenoid, corialactone D (
1b), which has been previously identified from the same plant [
8], except for the presence of the glucose unit at C-12 in
1. The attachment of the glucose moiety to C-12 of the aglycone
1b was deduced from the HMBC correlations from H-1′ (δ
H 4.35) to C-12 (δ
C 79.4). These spectroscopic data suggested that
1 is a glucoside of corialactone D. The
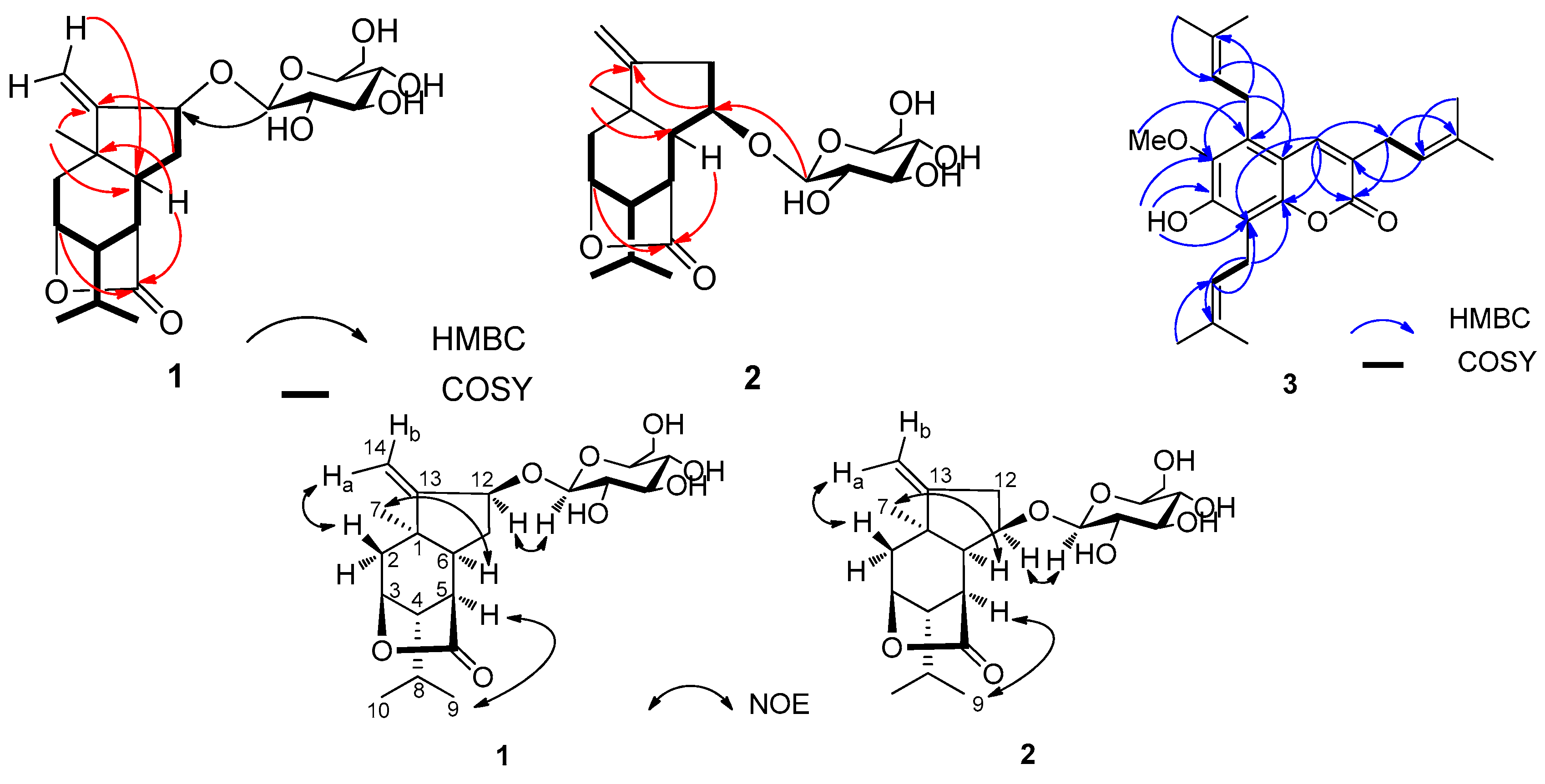
1H-
1H COSY and HSQC correlations indicated the presence of two substructures–CH
2(2)–CH(3)–CH(4)–CH(5)–CH(6)–CH(11)–CH(12)–, and –CH(4)–CH(8)[CH
3(9)]–CH
3(10)–units, whose connectivity was defined by the key HMBC correlations (
Figure 2).
The relative configuration of the aglycone (
1b) of
1 was defined by a NOESY experiment (
Figure 2). The NOE correlations of H
3-7/H-6, H
3-9/H-5, and H-12/H-1′ indicated that CH
3-7, H-5, H-6, H-12, and the isopropyl group were at the same orientation (α-configuration). Additionally, enzymatic hydrolysis of
1 afforded the known intact aglycone
1b, which was identified from
1H NMR, and its absolute configuration (1
R,3
S,4
S,5
R,6
S,12
S) was previously determined from CD spectrum [
8]. In addition, the coupling constant (
J = 7.5 Hz) of the anomeric proton (δ
H 4.35, H-1′) suggested a
β-configuration for the glucose unit. Indeed, compound
1 was hydrolyzable via
β-glucocidase. GC analysis suggested that acid hydrolyzate of
1 contained the glucose moiety with the
d-configuration. Hence, the structure of
1 was determined unambiguously as shown, and named nepalactone A.
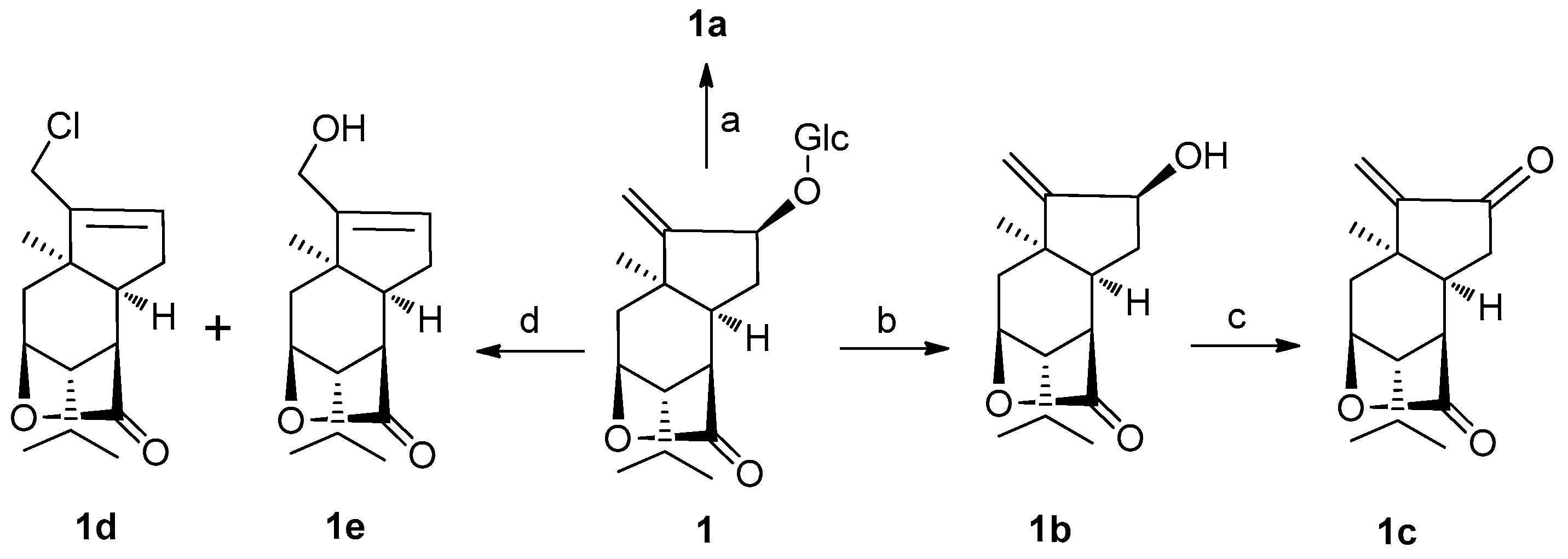
In addition, in order to enhance molecular diversity of sesquiterpene compounds, additional three derivatives of
1. The conversion of the aglycone
1b using Dess-Martin periodinane (DMP) in CH
2Cl
2 at room temperature afforded
1c; and treatment of
1 with 4 N HCl in dioxane at 100 °C yielded two compounds
1d and
1e (
Scheme 1). These three new congeners were characterized by an application of spectroscopic data and HRESIMS.
Compound
2 was obtained as white powder with a molecular formula C
21H
32O
8, as established via HRESIMS (
m/
z [M + Na]
+ 435.1987; calcd. 435.2417). Its molecular formula was identical to that of
1, indicating that
2 was a positional isomer of
1. The similar IR spectrum of
2 compared to
1 indicated the presence of similar functional groups. The
1H and
13C-NMR spectral data (
Table 1) of
2 were highly similar to those of nepalactone A (
1). Compound
1 significantly differed from
2 by the signals for the glucopyranosyl group, attached at C-12 for
1 while at C-11 (δ
C 81.9) for
2. The HMBC correlations from the anomeric proton H-1′ at δ
H 4.33 (d,
J = 7.5 Hz) to C-11 (δ
C 81.9) implied that the glucose group is attached to C-11 (
Figure 2). The stereochemistry of
2 was established from a NOESY experiment (
Figure 2). The most significant NOE correlations observed were between H-6/H
3-7, H-5/H
3-9, and H-1′/H-11, indicating that H-5, H-6, H-6, the methyl group at C-1, and the isopropyl group at C-4 were all on the same α-face of the molecule. The NMR data of
2 were fully assigned by analysis of the 2D-NMR data including its HSQC, HMBC, and
1H-
1H COSY spectra. Thus, the structure of
2 was established as shown for nepalactone B.
Compound
3, yellowish crystalline needles, was assigned the molecular formula C
25H
32O
4 from [M − H]
− at
m/z 395.2226 in HRESIMS, requiring 10 degrees of unsaturation. The IR spectrum displayed the presence of hydroxy (3319 cm
−1), C=O (1691 cm
−1) groups and a benzene ring (1627, 1583, and 1458 cm
−1). The
13C-NMR and DEPT data showed 25 carbon signals for seven methyls, three methylenes, four methines, and 11 quaternary carbons. The
13C-NMR data suggested the presence of three isopentenyl groups (δ
C 119.8, δ
H 5.34, δ
C 135.2; and δ
C 121.1, δ
H 5.33, δ
C 133.1; and δ
C 122.6, δ
H 5.11, δ
C 132.8), one methoxy group (δ
C 62.2). Besides three isopentenyl and one methoxy substituent, the NMR characteristic signals and UV spectra were indicative of a coumarin skeleton [
11]. One downfield singlet (δ
H 7.51, s) in the
1H-NMR spectrum of
3 suggested that
3 was a pentasubstituted coumarin. Furthermore, the
1H and
13C-NMR data of
3 were similar to those of the known coumarin, 7-hydroxy-6-methoxy-3,8-bis(3-methyl-2-butenyl)coumarin, which was previously identified from the same plant [
11], except for the presence of an additional isopentenyl unit [δ
H 5.11 (1H, m, H-2′′), 3.56 (2H, d,
J = 6.7 Hz, H-1′′), 1.86 (s, H-5′′), 1.75 (s, H-4′′); δ
C 132.8 (C-3′′), 122.6 (C-2′′), 25.6 (C-4′′), 24.7 (C-1′′), 18.1 (C-5′′)] at C-5 in 3. The location of the isopentenyl unit at C-5 was supported by the HMBC correlations of H-2′′ to C-5, and H-1′′ to C-6 and C-10. Full analyses of the
1H and
13C-NMR spectral data of
3, supported by the
1H-
1H COSY, DEPT, HSQC, and HMBC experiments (
Figure 2), permitted the assignment of all proton and carbon resonances. Consequently, the structure of
3 was established as 7-hydroxy-6-methoxy-3,5,8-tri(3-methyl-2-butenyl) coumarin and named nepalin C. To our knowledge, this is a rare coumarin with three isopentenyl units in natural source.
3. Experimental Section
3.1. General Procedures
Optical rotations were recorded on an Autopol III automatic polarimeter. UV spectra were obtained using a Thermo Scientific Evolution 300 UV-VIS Spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). 1H and 13C-NMR spectra were recorded on 400 MHz and 500 MHz NMR instruments. Chemical shifts were reported using the solvent residual peak as the internal standard. ESI-MS spectra were performed on a Thermo Fisher LTQ Fleet instrument spectrometer (Thermo Fisher Scientific Inc.). HRESIMS data were obtained on an API QSTAR time-offlight spectrometer. Column chromatography (CC) was performed on silica gel (90–150 μm) (Qingdao Marine Chemical Inc., Qingdao, China), Sephadex LH-20 (40–70 μm), and Lichroprep RP-18 gel (40–63 μm; Merck, Darmstadt, Germany). GF254 plates were used for thin-layer chromatography (TLC). Preparative TLC (PTLC) was carried out on silica gel 60 GF254; semi-preparative HPLC was performed via a Thermo BDS Hypersil column (250 mm× 4.6 mm and 250 mm× 10 mm, 5 μm).
3.2. Plant Materials
The dried root barks of Coriaria nepalensis were collected from the Qinling mountains, in the Shaanxi province of China in November 2013. The plant was authenticated by Professor Wu. A voucher specimen (0023466) was deposited at the Herbarium of College of Life Sciences, Northwest A&F University, P. R. China.
3.3. Extraction, Isolation, and Purification
The dried and powdered root-bark of C. nepalensis (4.2 kg) were extracted by 95% EtOH at room temperature for three times. After removal of the solvent, the residue (1.9 kg) obtained was suspended in water and then extracted successively with petroleum ether (PE), EtOAc, and n-BuOH. The EtOAc portion (911 g) was chromatographed on silica gel over CC eluting with PE/EtOAc mixtures of increasing polarity (9:1, 8:2, 7:3, 6:4, 4:6) to yield five fractions (Frs. 1–5). Fr. 2 was separated by repeated CC over silica gel (EtOAc/MeOH 9:1 to 6:4) to yield five fractions (2-1–2-5). Fraction 2-2 (2 g) was submitted to a RP-18 column (MeOH/H2O, 10:90 to 100:0) to yield two further fractions (2-2-1 and 2-2-2), and Fraction 2-2-1 was separated by Sephadex LH-20 (CHCl3/MeOH 1:1) and silica gel CC (CHCl3/MeOH 8:2) to yield 1 (216 mg). Fraction 2-3 was separated by silica gel CC (PE/acetone 20:1) to afford four fractions (2-3-1–2-3-4). Fraction 2-3-3 was separated by CC on LH-20 (CHCl3/MeOH 1:1), RP-18 CC (70% MeOH), and then silica gel with (PE/acetone, 50:1 to 40:1) to afford 3 (1.23 g). The n-butanol fraction (242 g) was separated by silica gel CC using EtOAc-MeOH (30:1–15:1) as eluent to afford seven fractions (Frs. A–G). Fr. C was separated by CC over silica gel (CHCl3/MeOH, 12:1 to 5:1), Sephadex LH-20 (MeOH), and then RP-18 to yield 1 (500 mg). Purification of Fr. C-3 by LH-20 CC (MeOH) followed by HPLC (40% EtOH, flow rate = 2 mL/min) provided 2 (2 mg, tR = 17.8 min).
Nepalactone A (
1). White, amorphous powder;
−26.3 (
c 0.24, MeOH); IR (KBr)ν
max 3402, 1765, 1076, 1025 cm
−1;
1H and
13C-NMR data, see
Table 1,
Figures S1–S9; ESIMS
m/z 435.17 [M + Na]
+, 846.62 [2M + Na]
+; HRESIMS
m/z 435.1991 [M + Na]
+ (calcd. for C
21H
32O
8Na, 435.1995).
Nepalactone B (
2). White powder;
+ 6.9 (
c 0.24, MeOH); IR (KBr) ν
max 3390, 1769, 1076, 1028 cm
−1;
1H and
13C-NMR data, see
Table 1,
Figures S19–27; ESIMS
m/z 823.15 [2M − H]
−; HRESIMS
m/z 435.1987 [M + Na]
+ (calcd. for C
21H
32O
8Na, 435. 1995).
Nepalarin (3). Pale yellow needles; UV(MeOH): λmax (logε) nm: 221 (4.28), 336 (4.22); IR (KBr) νmax 3355, 1731, cm−1; IR (KBr) νmax 3319, 1692, 1583, 1458, 1394, 1306, 1067, 1041, 1015 cm−1; 1H-NMR (500 MHz, CDCl3) δ 7.51 (1H, s, H-4), 5.34 (1H, m, H-2′), 5.33 (1H, m, H-2′′′), 5.11 (1H, m, H-2′′), 3.84 (3H, s, 6-OCH3), 3.58 (2H , d, J = 7.5 Hz, H-1′′′), 3.56 (2H, d, J = 6.7 Hz, H-1′′), 3.26 (2H, d, J = 7.0 Hz, H-1′), 1.89 (3H, s, H-4′′′), 1.86 (3H, s, H-5′′), 1.83 (3H, s, H-4′), 1.75(3H, s, H-4′′), 1.72 (3H, s, H-5′′′), 1.71 (3H, s, H-5′); 13C-NMR (125 MHz, CDCl3) δ 162.3 (C-2), 149.5 (C-7), 149.3 (C-9), 142.0 (C-6), 135.9 (C-4), 135.2 (C-3′), 133.1 (C-3′′′), 132.8 (C-3′′), 128.2 (C-10), 124.6 (C-3), 122.6 (C-2′′), 121.1 (C-2′′′), 119.8 (C-2′), 114.3 (C-8), 111.8 (C-5), 62.2 (6-OCH3), 28.5 (C-1′), 25.9 (C-4′), 25.8 (C-5′′′), 25.6 (C-4′′), 24.7 (C-1′′), 22.4 (C-1′′′), 18.1 (C-5′′), 18.0 (C-4′′′), 17.8 (C-5′); ESI-MS m/z 419.49 [M + Na]+, 815.18 [2M + Na]+, 341.19 [M − C4H7]+; ESI-MS m/z 395.41 [M − H]−; HRESIMS m/z 395.2226 [M − H]− (calcd. for C25H31O4, 395.2222).
3.4. Nepalactones A Tetraacetate 1a
To a solution of
1 (3.2 mg, 0.03 mmol) in pyridine (1 mL), acetic anhydride (1 mL) was added, and the mixture stood at RT for 24 h. The reaction mixture was concentrated to dryness, and the residue were purified on silica gel CC using EtOAc, affording tetraacetate
1a (ca. 3 mg) as an off-white gum.
−20.9 (
c 0.2, MeOH);
1H and
13C-NMR data, see
Table 1,
Figures S10–18; HRESIMS
m/
z [M + Na]
+ 603.2416 (calcd. for C
29H
40O
12Na, 603.2417).
3.5. Acid Hydrolysis of 1 and theDetermination of Sugar
Compound 1 (2 mg) was hydrolyzed with 2N HCl-dioxane (1:1, 1 mL) at 100 °C for 2 h. The reaction mixture was partitioned between chloroform and H2O three times. The aqueous layer was neutralized with 2 M Ag2CO3 and evaporated in vacuo. The residue was dissolved in pyridine (0.5 mL), to which l-cysteine methyl ester hydrochloride in pyridine (0.1 M, 0.5 mL) was added. After reacting at 60 °C for 1 h, trimethylsilylimidazole (0.05 mL) was added to the reaction mixture and kept at 4 °C for another 8 h. The mixture was analyzed via GC–MS. By comparison of the retention time of the authentic sample, the monosaccharide of 1 was determined to be d-glucose (tR 11.85 min) (tR 12.15 min of l-glucose).
3.6 Enzymatic Hydrolysis of 1
To a solution of 1 (20 mg) in a mixture of a citrate buffer (pH = 5, 1 mL) and water (1 mL), β-gluosidase (1.3 mg, Sigma-Aldrich, St. Louis, MO, USA) was added. After the reaction mixture was incubated at 30 °C for 5 days, it was extracted with EtOAc, dried over anhydrous sodium sulfate, concentrated, and purified via column chromatography using MeOH/CHCl3 (1:15) to yield aglycon 1b as a white solid (8.2 mg, 68%), which was identified as a sesquiterpenoid, corialactone D, via comparison with the 1H-NMR spectrum. 1H-NMR (500 MHz, CDCl3) δ 1.73 (d, J = 15.0 Hz, H-2α), 2.46 (dd, J = 3.5, 15.0 Hz, H-2β), 4.68 (m, H-3), 2.02 (m, H-4), 2.36 (m, H-5), 2.36 (m, H-6), 1.30 (s, 3H, H-7), 1.83 (m, 1H, H-8), 1.02 (d, J = 6.9 Hz, 3H, H-9), 1.00 (d, J = 6.9 Hz, 3H, H-10), 2.37 (m, 1H, H-11α), 2.10 (m, 1H, H-11β), 4.86 (m, 1H, H-12), 5.33 (s, 1H, H-14a), 5.18 (s, 1H, H-14b).
3.7 Preparation of 1c
To a solution of 1b (8.2 mg, 0.03 mmol) in CH2Cl2 (1 mL) at room temperature, NaHCO3 (13.8 mg, 0.16 mmol, 5 equiv.) and Dess-Martin periodinane (DMP) (20.9 mg, 0.05 mmol, 1.5 equiv.) were added. After 40 min of stirring, the reaction mixture was poured into a mixture of a saturated aqueous solution of Na2S2O3 (5 mL), a saturated aqueous solution of NaHCO3 (5 mL), and water (5 mL). The layers were separated, and the aqueous phase was extracted with CH2Cl2. The combined organic extracts were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified via flash chromatography on silica gel (PE/EtOAc 5/1 to 2/1), affording 1c (7.8 mg, yield 98%) as a slightly yellow oil; 1H-NMR (500 MHz, CDCl3) δ 6.06 (s, 1H, H-14a), 5.25 (s, 1H, H-14b), 4.66 (t, J = 4.0 Hz, 1H, H-3), 2.65 (dd, J = 19.3, 8.8 Hz, 1H, H-11α), 2.46 (d, J = 19.3 Hz, 1H, H-11β), 2.38 (m, 1H, H-2β), 2.36 (m, 1H, H-5), 2.34 (m, 1H, H-6), 1.99 (m, 1H, H-4), 1.78 (m, 1H, H-8), 1.77 (m, 1H, H-2α), 1.15 (s, 3H, H-7), 0.96 (d, J = 7.0 Hz, 3H, H-10), 0.94 (d, J = 7.0 Hz, 3H, H-9); 13C-NMR (125 MHz, CDCl3) δ 203.7 (C-12), 176.7 (C-15), 151.9 (C-13), 117.5 (C-14), 79.0 (C-3), 51.2 (C-4), 45.0 (C-5), 40.1 (C-1), 39.8 (C-11), 35.5 (C-6), 33.5 (C-2), 32.2 (C-7), 25.3 (C-8), 20.6 (C-10), 19.73 (C-9); ESIMS m/z 249.08 [M + H]+, 271.15 [M + Na]+; HRESIMS m/z 271.1307 [M + Na]+ (calcd. for C15H20O3Na, 271.1305).
3.8 Preparation of 1d and 1e
Compound 1 (30 mg) was hydrolyzed with 4 N HCl–dioxane (1:1, 15 mL) at 100 °C for 2 h. The reaction mixture was partitioned between chloroform and H2O three times. The organic layer was neutralized with 20 M Ag2CO3 and filtered. The resulting solution was washed with brine (50 mL), dried over Na2SO4, and then filtered and evaporated. The residue was subjected to column chromatography on silica gel using petroleum ether and ethyl acetate as eluent to yield 1d (11 mg, 56%) and 1e (7 mg, 38%) as a yellowish oil.
1d: amorphous powder; IR (KBr)νmax 3402, 1765, 1076, 1025 cm−1; 1H-NMR (500 MHz, CDCl3) δ 5.74 (s, 1H, H-12), 4.64 (s, 1H, H-3), 4.23 (d, J = 13.0 Hz, 1H, H-14a), 4.12 (d, J = 13.0 Hz, 1H, H-14b), 1.79 (m, 1H, H-8), 1.25 (s, 3H, H-7), 0.99 (d, J = 6.5 Hz, 3H, H-10), 0.96 (d, J = 6.5 Hz, 3H, H-9); 13C-NMR (125 MHz, CDCl3) δ 178.1 (C-15), 144.9 (C-13), 130.2 (C-12), 79.6 (C-3), 50.9 (C-4), 46.4 (C-1), 44.7 (C-5), 43.8 (C-6), 40.8 (C-14), 34.0 (C-11), 30.8 (C-2), 29.1 (C-7), 25.8 (C-8), 20.6 (C-10), 19.8 (C-9); HRESIMS m/z 291.1128 [M + Na]+ (calcd. for C15H21O2ClNa, 291.1127).
1e: amorphous powder; IR (KBr)νmax 3402, 1765, 1076, 1025 cm−1; 1H-NMR (500 MHz, CDCl3) δ 5.59 (s, 1H, H-12), 4.65 (s, 1H, H-3), 4.18 (s, 2H, H-14), 1.79 (m, 1H, H-8), 1.15 (s, 3H, H-7), 1.00 (d, J = 6.5 Hz, 3H, H-10), 0.97 (d, J = 6.5 Hz, 3H, H-9); 13C-NMR (125 MHz, CDCl3) δ 178.3 (C-15), 149.1 (C-13), 126.4 (C-12), 79.9 (C-3), 59.8 (C-14), 51.0 (C-4), 46.1 (C-1), 44.8 (C-5), 42.7 (C-6), 34.2 (C-11), 31.4 (C-2), 28.4 (C-7), 25.2 (C-8), 20.7 (C-10), 19.8 (C-9); HRESIMS m/z 273.1470 [M + Na]+ (calcd. for C15H22O3Na, 273.1471).
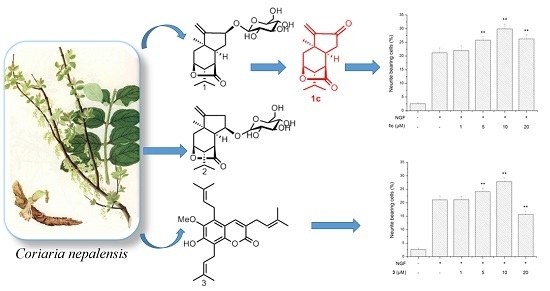
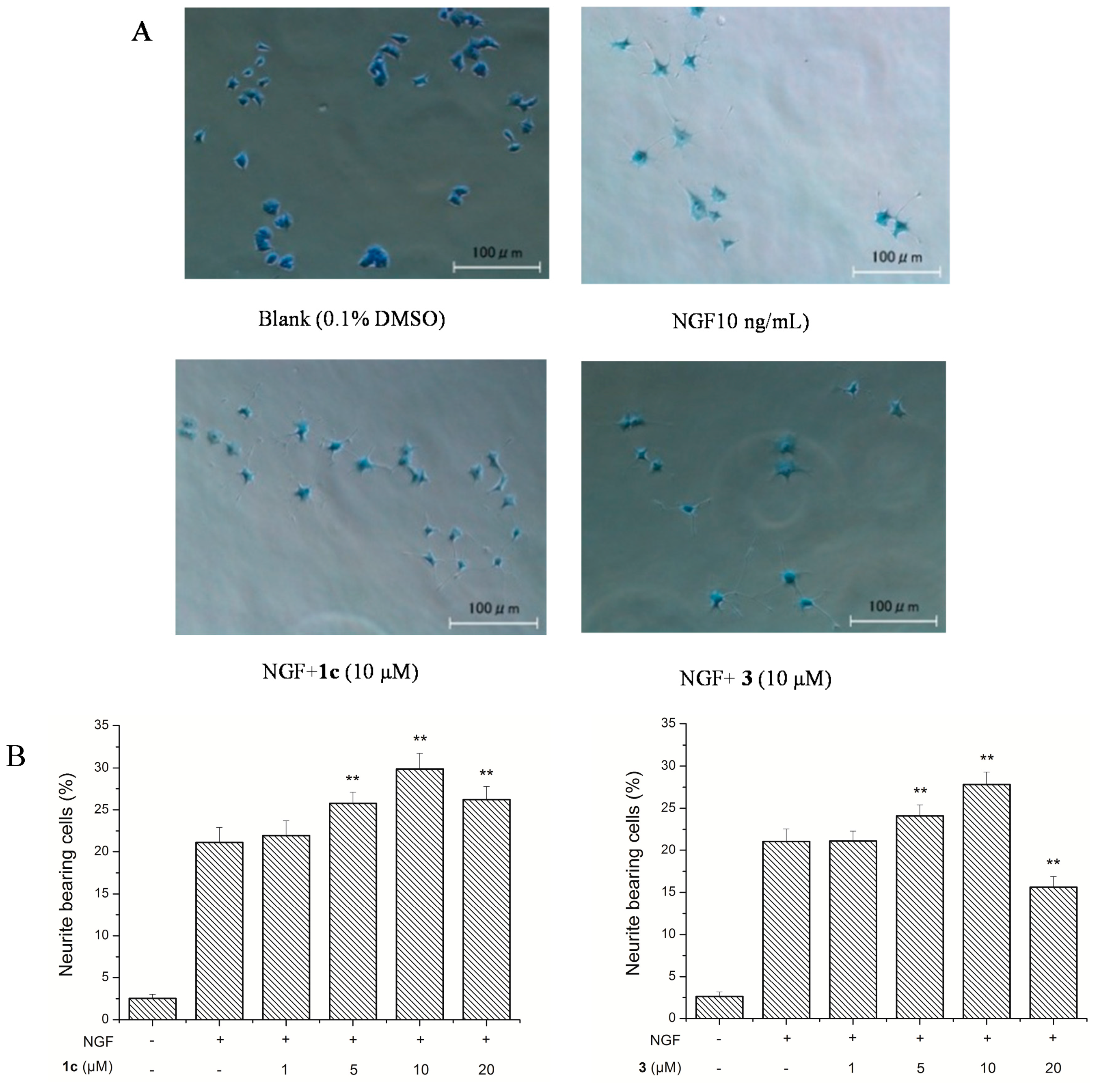
3.9. Neurite Outgrowth-Promoting Assay
Neurite outgrowth-promoting activity of these isolated compounds was assessed as described previously by our group [
17,
18]. Experiments were repeated at least three times, and data are expressed as mean ± SD (**
p < 0.01). Statistical analyses were performed using Dunnett’s
t-test.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}