
Design, Synthesis and Biological Evaluation of 6-(2,6-Dichloro-3,5-dimethoxyphenyl)-4-substituted-1H-indazoles as Potent Fibroblast Growth Factor Receptor Inhibitors

Abstract
:1. Introduction
2. Results and Discussion
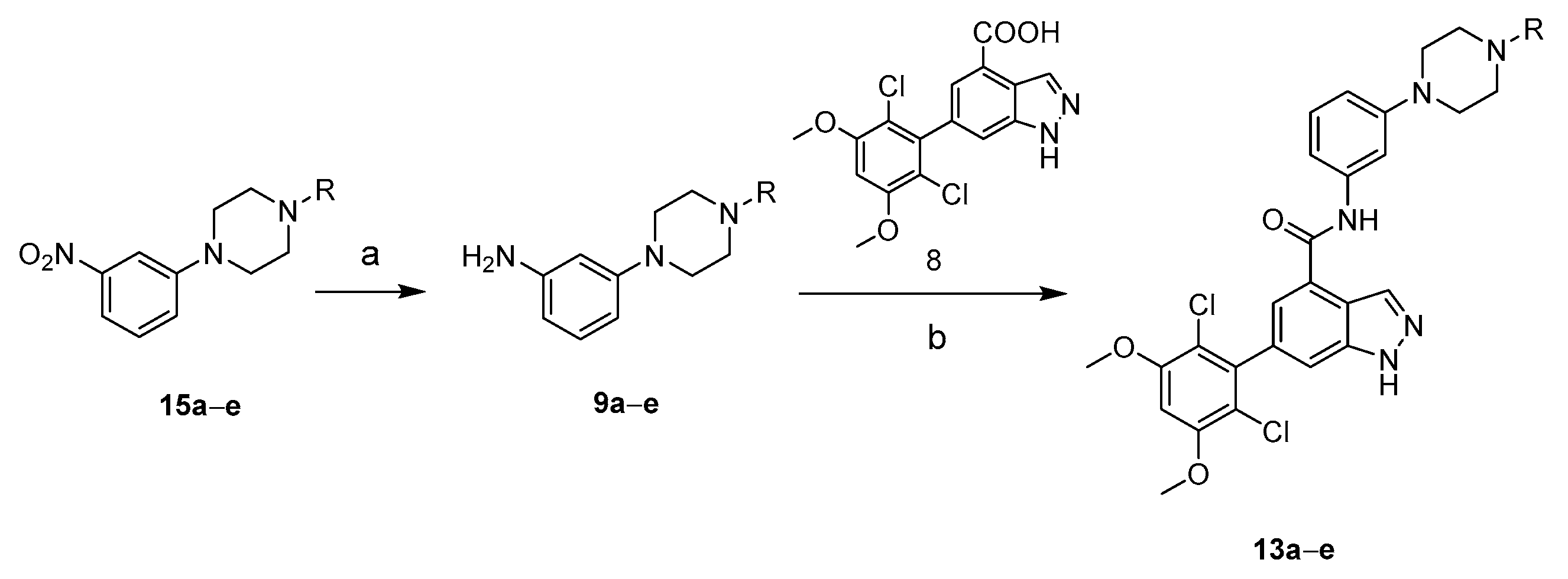
2.1. Chemistry
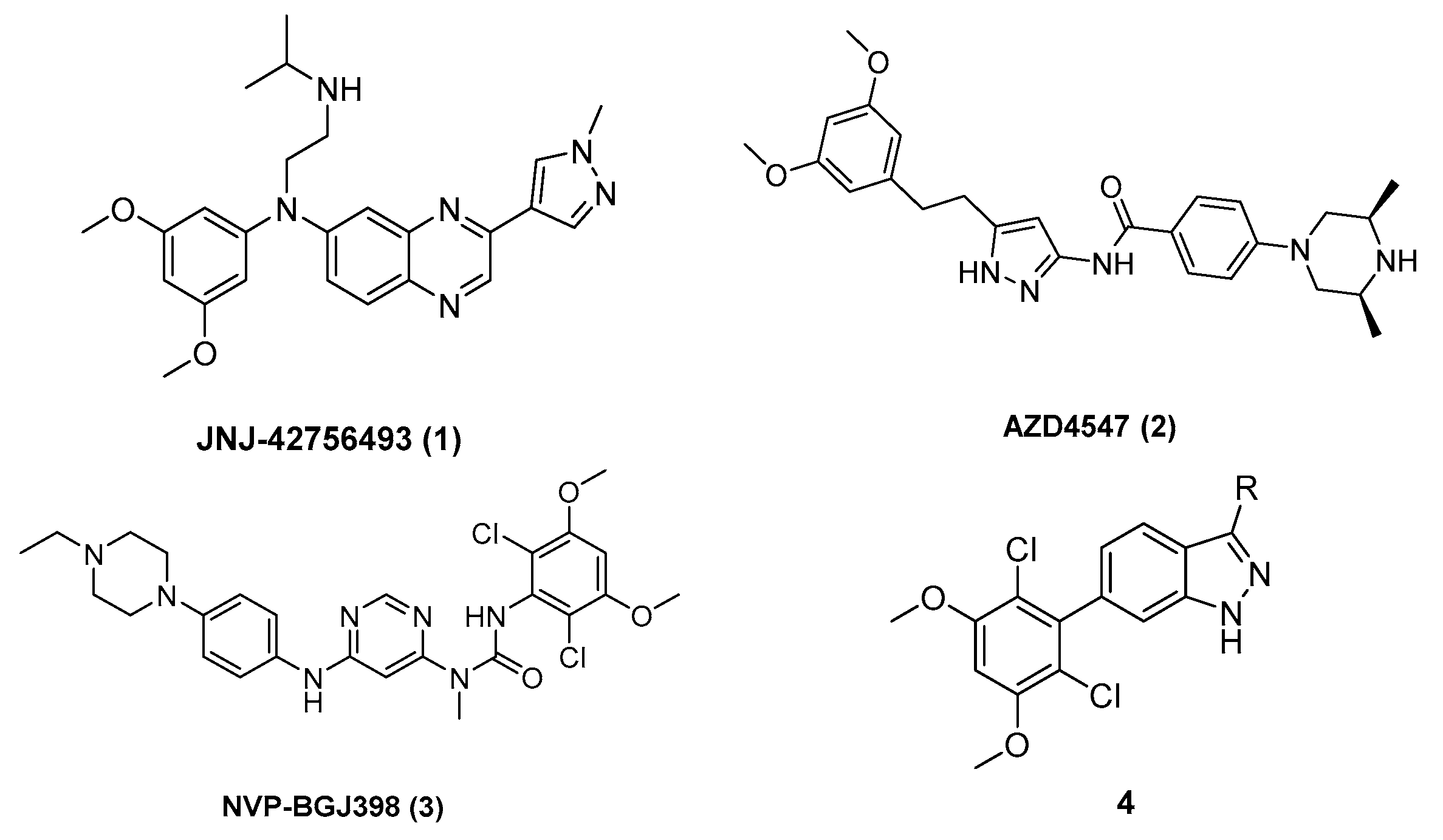
2.2. Inhibitor Design
2.3. The Structure–Activity Relationship of Indazole Derivatives
3. Experimental Section
3.1. General Information
3.1.1. Synthesis of 2-(2,6-Dichloro-3,5-dimethoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (6a)
3.1.2. Synthesis of Methyl 6-(2,6-Dichloro-3,5-dimethoxyphenyl)-1H-indazole-4-carboxylate (7)
3.1.3. Synthesis of 6-(2,6-Dichloro-3,5-dimethoxyphenyl)-1H-indazole-4-carboxylic acid (8)
3.1.4. Synthesis of 6-(2,6-Dichloro-3,5-dimethoxyphenyl)-N-phenyl-1H-indazole-4-carboxamides 10a–e, 11a–e, 12a–i
3.1.5. Synthesis of 6-(2,6-Dichloro-3,5-dimethoxyphenyl)-N-(3-(4-piperazin-1-yl)phenyl-1H-indazole-4-carboxamides 13a–e
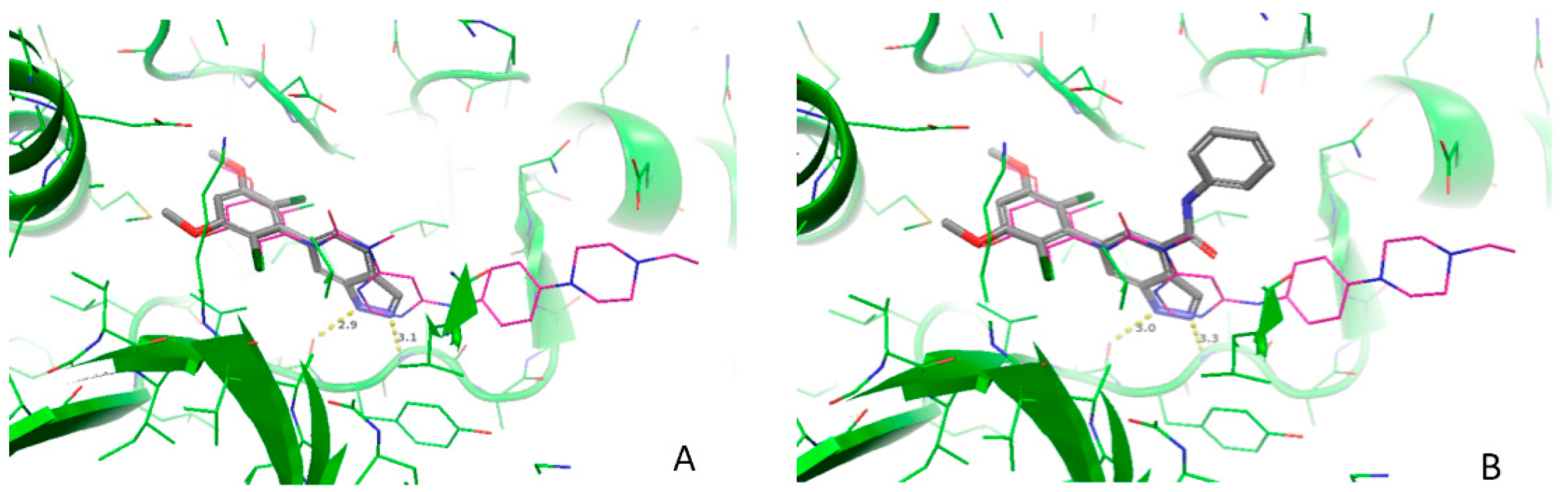
3.2. Molecular Docking
3.3. Elisa Kinase Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Carter, E.P.; Fearon, A.E.; Grose, R.P. Careless talk costs lives: Fibroblast growth factor receptor signalling and the consequences of pathway malfunction. Trends Cell Biol. 2015, 25, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Laestander, C.; Engström, W. Role of fibroblast growth factors in elicitation of cell responses. Cell Prolif. 2014, 47, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.N.; Kilgour, E.; Smith, P.D. Molecular Pathways: Fibroblast Growth Factor Signaling: A New Therapeutic Opportunity in Cancer. Clin. Cancer Res. 2012, 18, 1855–1862. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crinó, L.; Ahn, M.-J.; de Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.C.; Annala, M.J.; Cogdell, D.E.; Granberg, K.J.; Sun, Y.; Ji, P.; Li, X.; Gumin, J.; Zheng, H.; Hu, L.M.; et al. The tumorigenic FGFR3–TACC3 gene fusion escapes miR-99a regulation in glioblastoma. J. Clin. Investig. 2013, 123, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Rorrati, P.; Pellegatta, S.; et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, L.; Li, Y.; Hu, H.; Shen, L.; Shen, X.; Pan, Y.; Ye, T.; Zhang, Y.; Luo, X.; et al. FGFR1/3 tyrosine kinase fusions define a unique molecular subtype of non-small cell lung cancer. Clin. Cancer Res. 2014, 1, 4107–4114. [Google Scholar] [CrossRef] [PubMed]
- Touat, M.; Ileana, E.; Postel-Vinay, S.; André, F.; Soria, J.C. Targeting FGFR signaling in cancer. Clin. Cancer Res. 2015, 21, 2684–2694. [Google Scholar] [CrossRef] [PubMed]
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Dieci, M.V.; Arnedos, M.; Andre, F.; Soria, J.C. Fibroblast growth factor receptor inhibitors as a cancer treatment: From a biologic rationale to medical perspectives. Cancer Discov. 2013, 3, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Hallinan, N.; Finn, S.; Cuffe, S.; Rafee, S.; O'Byrne, K.; Gately, K. Targeting the fibroblast growth factor receptor family in cancer. Cancer Treat. Rev. 2016, 46, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Ronca, R.; Giacomini, A.; Rusnati, M.; Presta, M. The potential of fibroblast growth factor/fibroblast growth factor receptor signaling as a therapeutic target in tumor angiogenesis. Expert Opin. Ther. Targets 2015, 19, 1361–1377. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, F.C.; O'Sullivan, H.; Smyth, E.; McDermott, R.; Viterbo, A. Fibroblast growth factor receptors, developmental corruption and malignant disease. Carcinogenesis 2013, 34, 2198–2205. [Google Scholar] [CrossRef] [PubMed]
- Tanner, Y.; Grose, R.P. Dysregulated FGF signalling in neoplastic disorders. Semin. Cell Dev. Biol. 2016, 53, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, S.; Hadden, M.K. Targeting the fibroblast growth factor receptors for the treatment of cancer. Anti-Cancer Agents Med. Chem. 2013, 13, 748–761. [Google Scholar] [CrossRef]
- Izzedine, H.; Ederhy, S.; Goldwasser, F.; Soria, J.C.; Milano, G.; Cohen, A.; Khayat, D.; Spano, J.P. Management of hypertension in angiogenesis inhibitor-treated patients. Ann. Oncol. 2009, 20, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, S.; Tomao, S.; de Marinis, F. Toxicity of targeted therapy in non-small-cell lung cancer management. Clin. Lung Cancer 2009, 10, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Bahleda, R.; Dienstmann, R.; Adamo, B.; Gazzah, A.; Infante, J.R.; Zhong, B.; Tabernero, J.; Mita, A.; Italiano, A.; Calvo, E.; et al. Phase 1 study of JNJ-42756493, a pan-fibroblast growth factor receptor (FGFR) inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 3401–3408. [Google Scholar]
- Gavine, P.R.; Mooney, L.; Kilgour, E.; Thomas, A.P.; Al-Kadhimi, K.; Beck, S.; Rooney, C.; Coleman, T.; Baker, D.; Mellor, M.J.; et al. AZD4547: An orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. 2012, 72, 2045–2056. [Google Scholar] [CrossRef] [PubMed]
- Guagnano, V.; Furet, P.; Spanka, C.; Bordas, V.; le Douget, M.; Stamm, C.; Brueggen, J.; Jensen, M.R.; Schnell, C.; Schmid, H.; et al. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J. Med. Chem. 2011, 54, 7066–7083. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Peng, X.; Dai, Y.; Zhang, W.; Ren, S.M.; Ai, J.; Geng, M.; Li, Y.X. Design, synthesis and biological evaluation of novel FGFR inhibitors bearing an indazole scaffold. Org. Biomol. Chem. 2015, 13, 7643–7654. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, Y.X.; Xu, P.; Dai, Y.; Luo, C.; Sun, Y.M.; Ai, J.; Geng, M.Y.; Duan, W.H. Discovery of substituted 1H-pyrazolo[3,4-b]pyridine derivatives as potent and selective FGFR kinase inhibitors. ACS Med. Chem. Lett. 2016, 7, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 10a–e, 11a–e, 12a–i, 13a–e are available from the authors.






| Compound No. | R | Enzyme Inhibition (%) a | |
|---|---|---|---|
| 1 μmol/L | 0.1 μmol/L | ||
| 10a | H | 84.1 | 58.8 |
| 10b | m-acetyl | 82.4 | 76.4 |
| 10c | p-acetyl | 52.9 | 32.2 |
| 10d | m-methoxyl | 78.4 | 57.0 |
| 10e | p-methoxyl | 68.5 | 11.9 |
| AZD4547 | / | 94.3 | 93.2 |

| Compound No. | R | Enzyme Inhibition (%) | |
|---|---|---|---|
| 1 μmol/L | 0.1 μmol/L | ||
| 10d |  | 78.4 | 57.0 |
| 11a |  | 79.2 | 47.2 |
| 11b |  | 84.3 | 49.5 |
| 11c |  | 80 | 42.3 |
| 11d |  | 78.5 | 43.1 |
| 11e |  | 57.4 | 31 |

| Compound No. | R | Enzyme Inhibition (%) | IC50 (nM) a | |
|---|---|---|---|---|
| 0.1 μmol/L | 0.01 μmol/L | |||
| 10a | H | 58.8 | 27.3 | 69.1 ± 19.8 |
| 12a |  | 35.7 | 17.1 | / |
| 12b |  | 62.3 | 42.1 | 38.6 ± 0.2 |
| 12c |  | 36.8 | 9.9 | / |
| 12d |  | 31.6 | 17.4 | / |
| 12e |  | 36.7 | 17.2 | / |
| 12f |  | 58.7 | 28.3 | 54.0 ± 8.7 |
| 12g |  | 60.9 | 45.1 | 78.8 ± 14.2 |
| 12h |  | 52.7 | 34.5 | 102.9 ± 0.6 |
| 12i |  | 60.3 | 35.1 | 86.2 ± 17.0 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | R | Enzyme Inhibition (%) | IC50 (nM) a | |
|---|---|---|---|---|
| 0.1 μmol/L | 0.01 μmol/L | |||
| 12g | H | 60.9 | 45.1 | 78.8 ± 14.2 |
| 13a | –CH3 | 72.7 | 48.3 | 30.2 ± 1.9 |
| 13b | –C2H5 | 26.4 | 13.7 | 328.4 ± 65.7 |
| 13c |  | 41.3 | 35.7 | 463.9 ± 99.1 |
| 13d |  | 18.5 | 7.2 | / |
| 13e |  | 64.7 | 27.7 | 117.9 ± 3.9 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Zhao, D.; Dai, Y.; Cheng, M.; Geng, M.; Shen, J.; Ma, Y.; Ai, J.; Xiong, B. Design, Synthesis and Biological Evaluation of 6-(2,6-Dichloro-3,5-dimethoxyphenyl)-4-substituted-1H-indazoles as Potent Fibroblast Growth Factor Receptor Inhibitors. Molecules 2016, 21, 1407. https://doi.org/10.3390/molecules21101407
Zhang Z, Zhao D, Dai Y, Cheng M, Geng M, Shen J, Ma Y, Ai J, Xiong B. Design, Synthesis and Biological Evaluation of 6-(2,6-Dichloro-3,5-dimethoxyphenyl)-4-substituted-1H-indazoles as Potent Fibroblast Growth Factor Receptor Inhibitors. Molecules. 2016; 21(10):1407. https://doi.org/10.3390/molecules21101407
Chicago/Turabian StyleZhang, Zhen, Dongmei Zhao, Yang Dai, Maosheng Cheng, Meiyu Geng, Jingkang Shen, Yuchi Ma, Jing Ai, and Bing Xiong. 2016. "Design, Synthesis and Biological Evaluation of 6-(2,6-Dichloro-3,5-dimethoxyphenyl)-4-substituted-1H-indazoles as Potent Fibroblast Growth Factor Receptor Inhibitors" Molecules 21, no. 10: 1407. https://doi.org/10.3390/molecules21101407
APA StyleZhang, Z., Zhao, D., Dai, Y., Cheng, M., Geng, M., Shen, J., Ma, Y., Ai, J., & Xiong, B. (2016). Design, Synthesis and Biological Evaluation of 6-(2,6-Dichloro-3,5-dimethoxyphenyl)-4-substituted-1H-indazoles as Potent Fibroblast Growth Factor Receptor Inhibitors. Molecules, 21(10), 1407. https://doi.org/10.3390/molecules21101407