
Diphenylcarbene Protected by Four ortho-Iodine Groups: An Unusually Persistent Triplet Carbene

Abstract
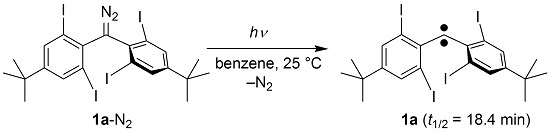
:
1. Introduction
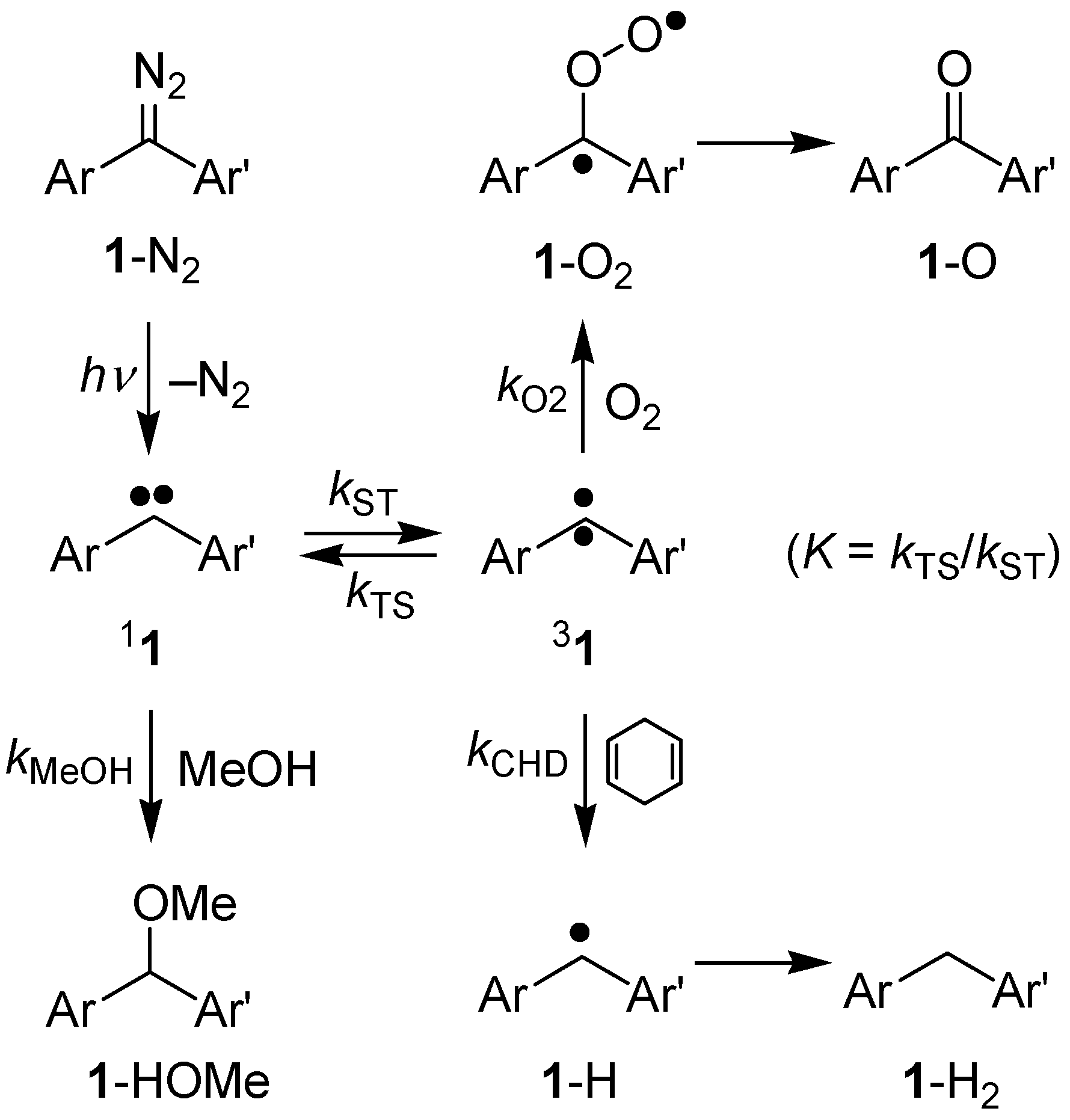
2. Results and Discussion
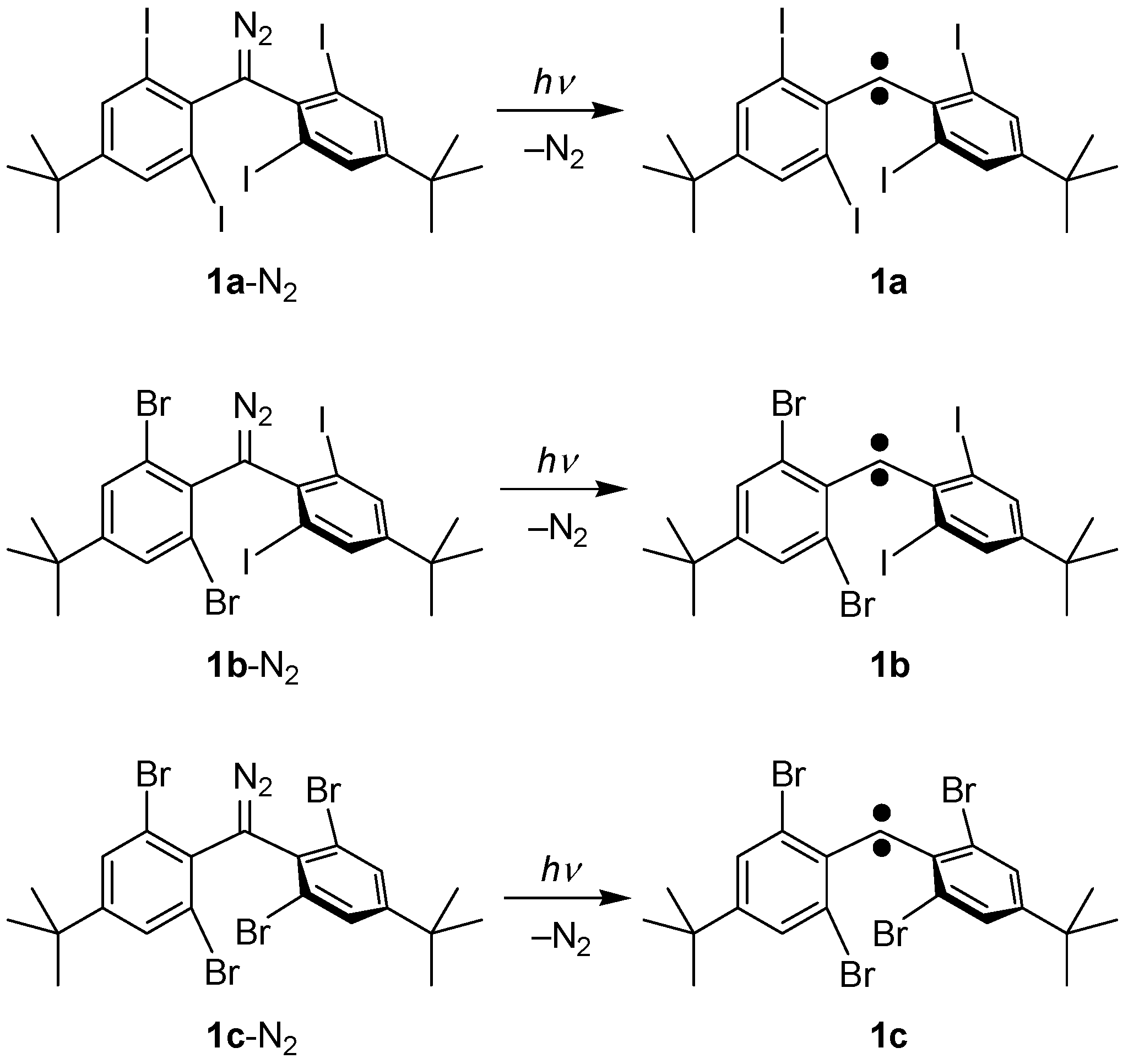
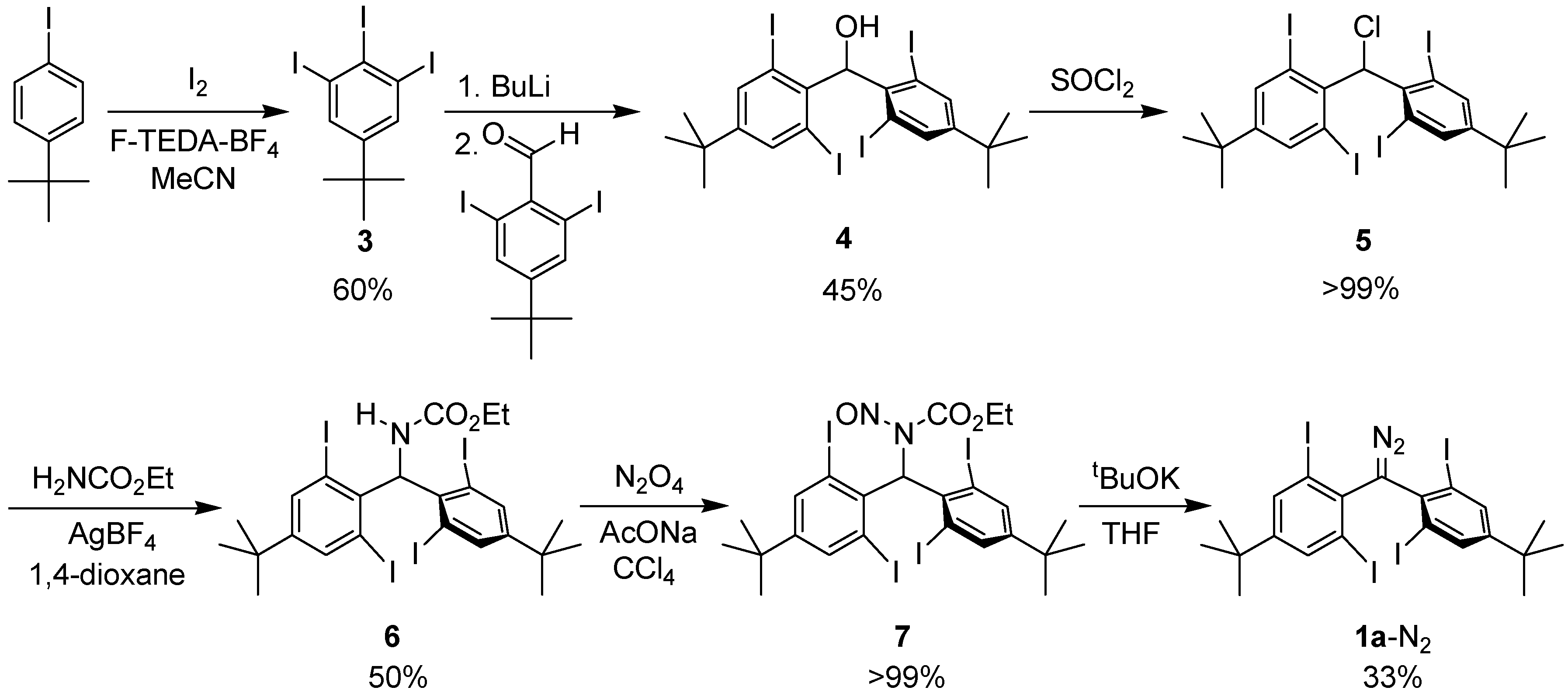
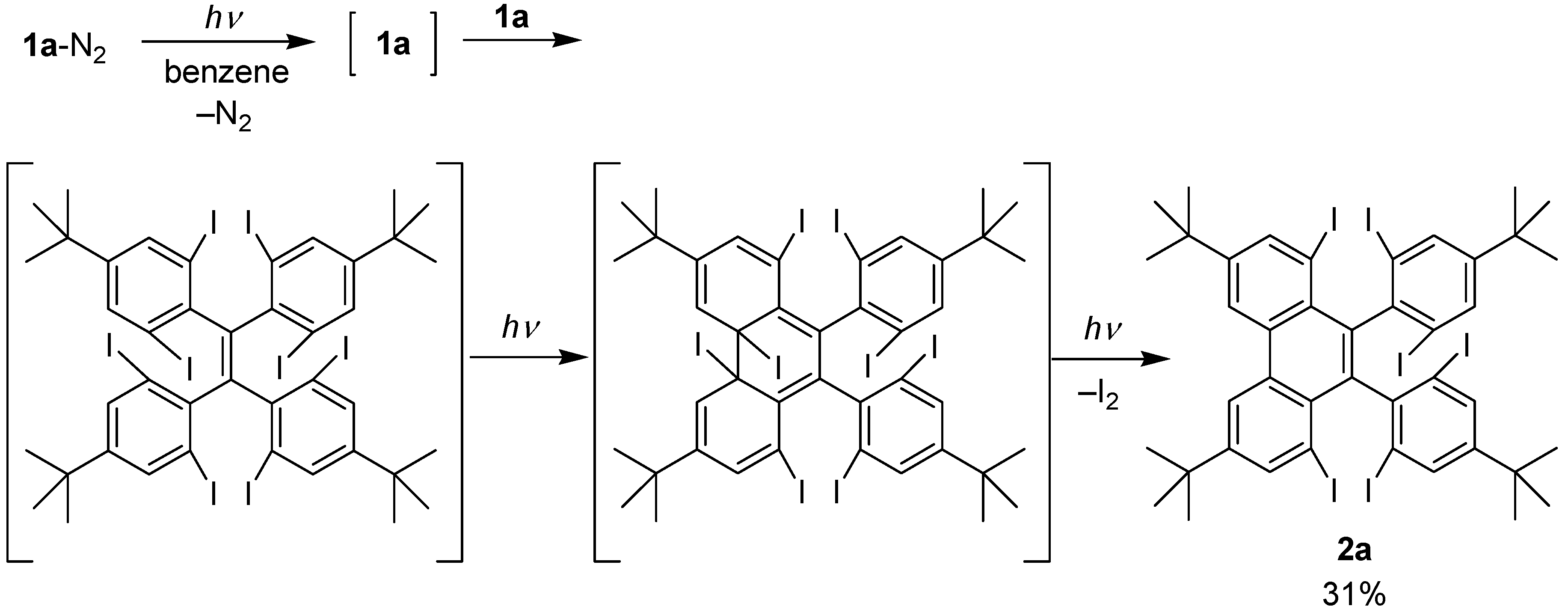
2.1. Preparation of Precursor Diazomethane
2.2. Spectroscopic Studies
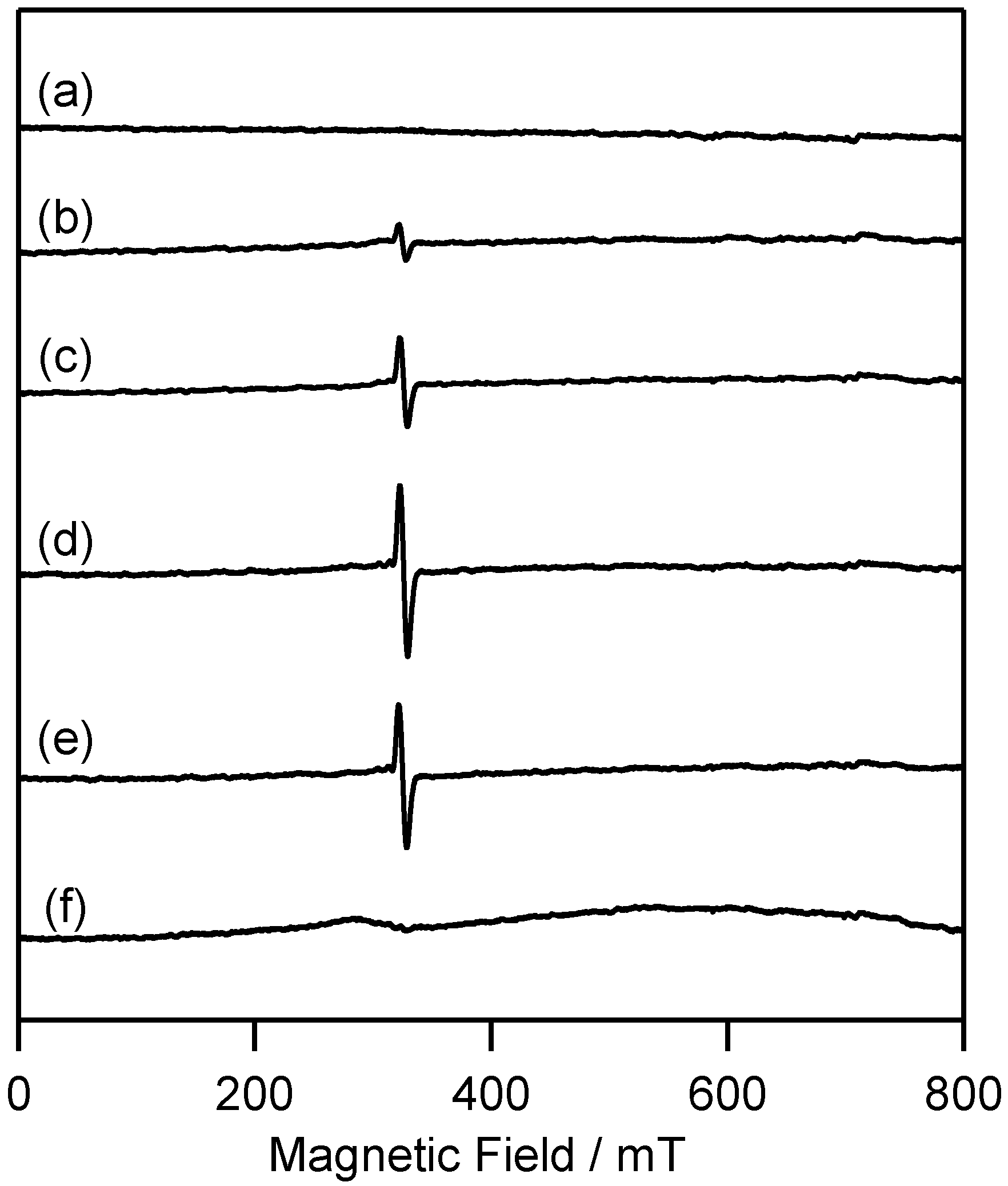
2.2.1. ESR Studies in Rigid Matrix at Low Temperature
2.2.2. Magnetic-Susceptibility Measurements
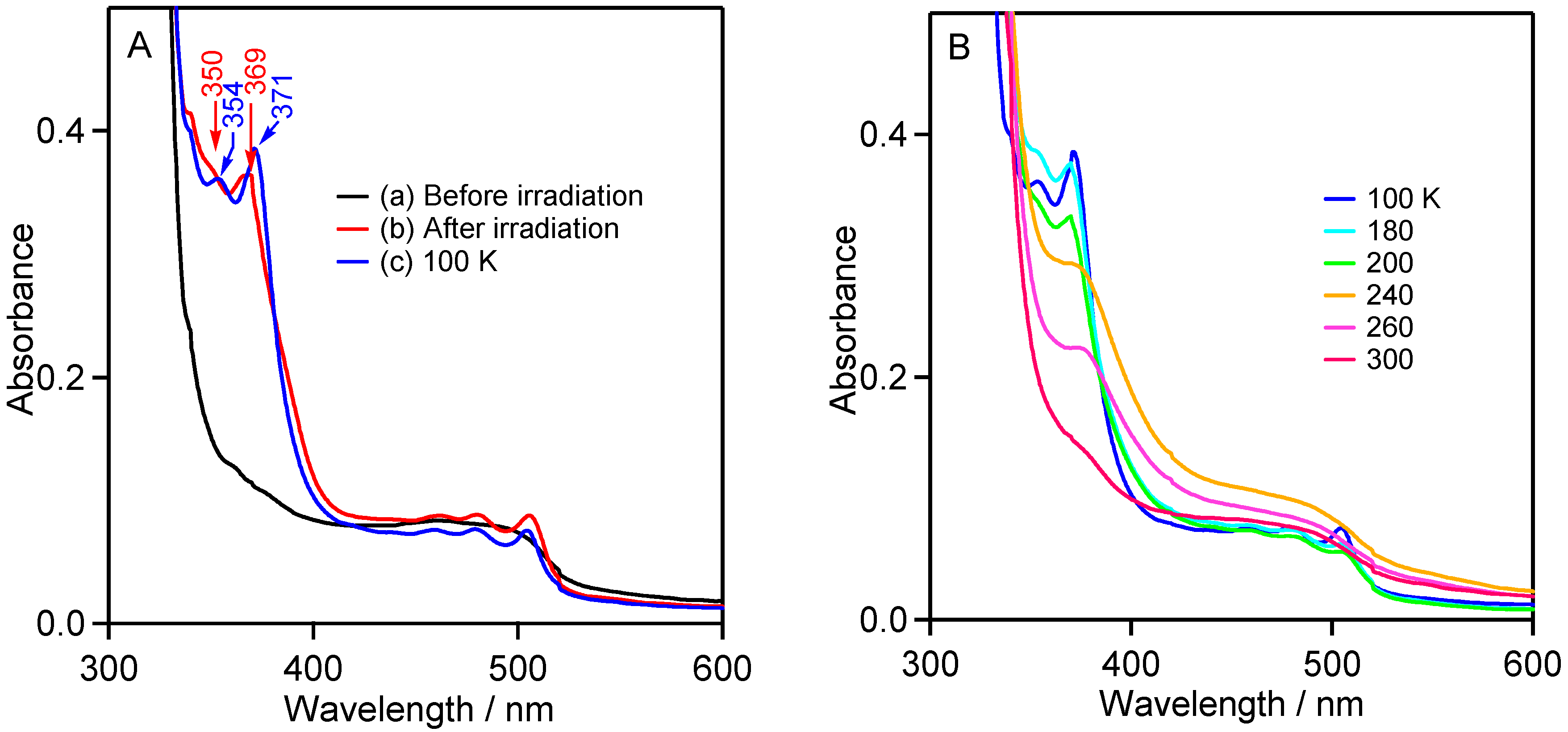
2.2.3. UV-Vis Studies in Rigid Matrix at Low Temperature
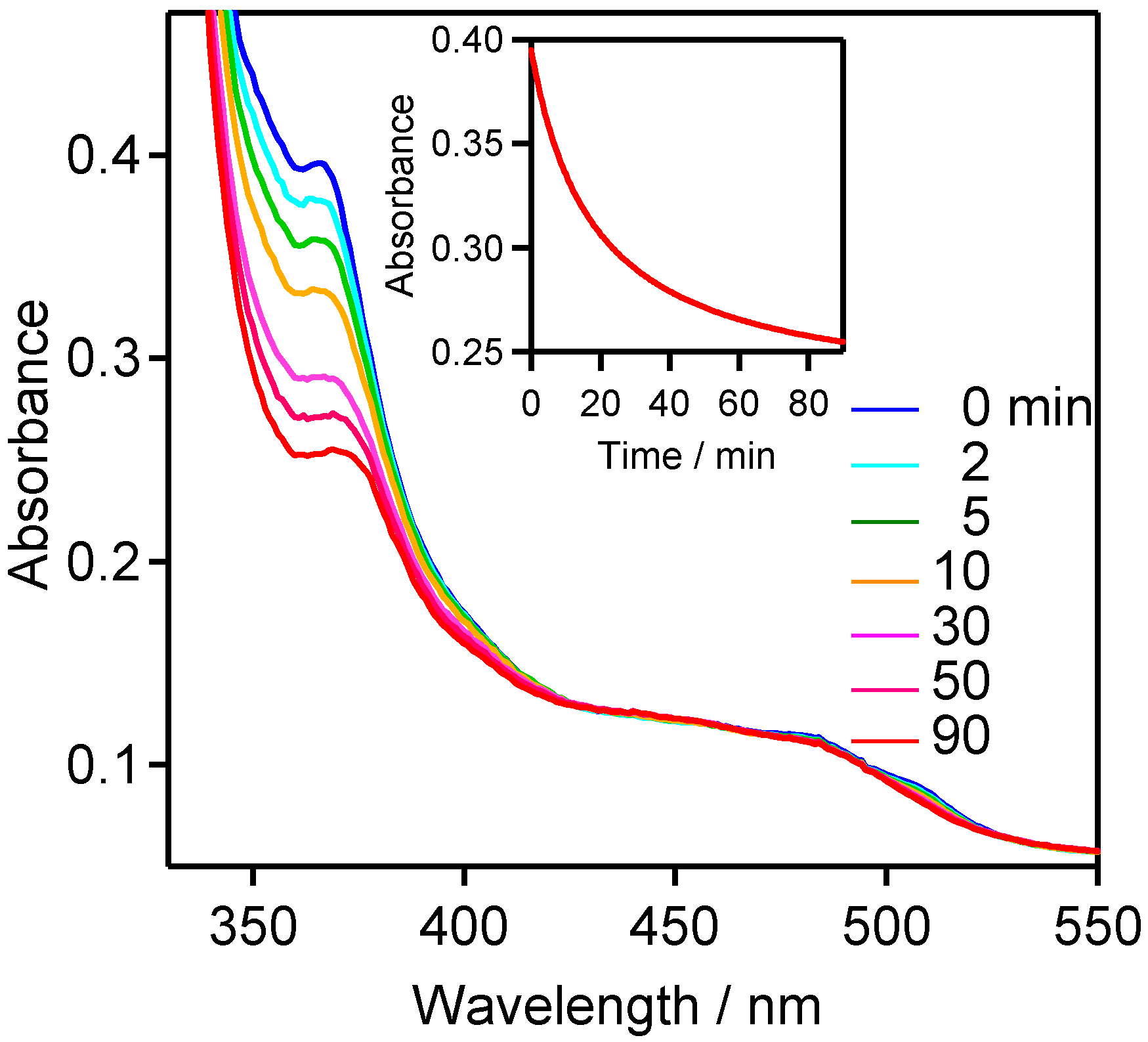
2.2.4. Laser Flash Photolysis Studies in Solution at Room Temperature
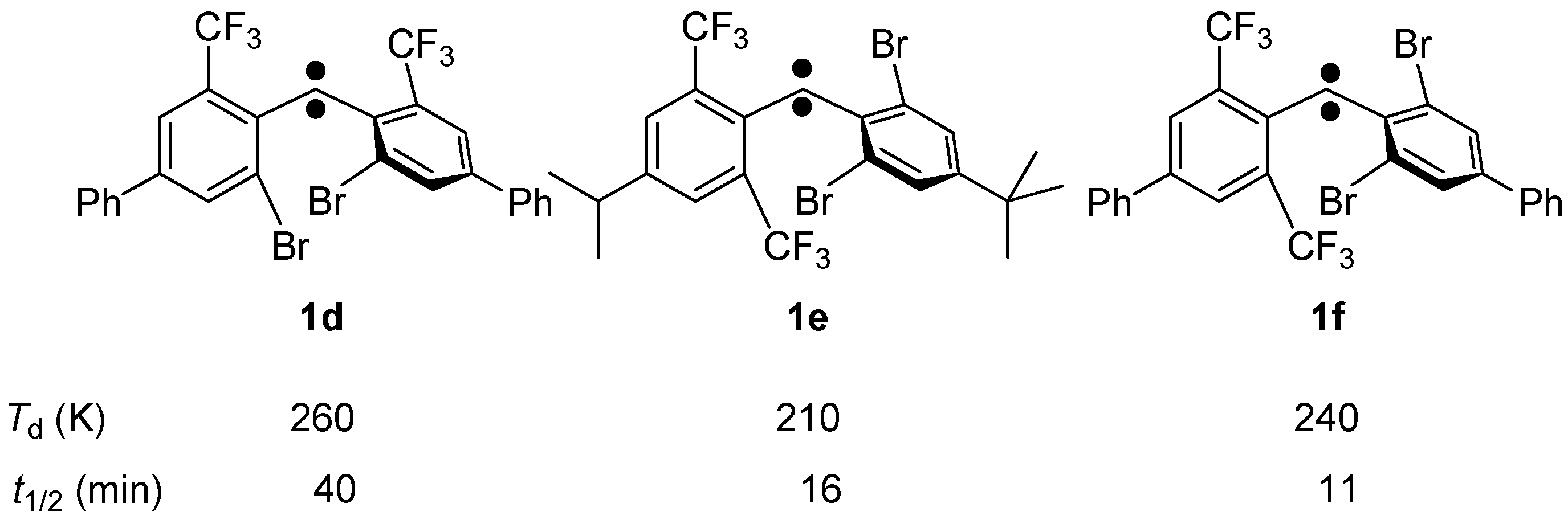
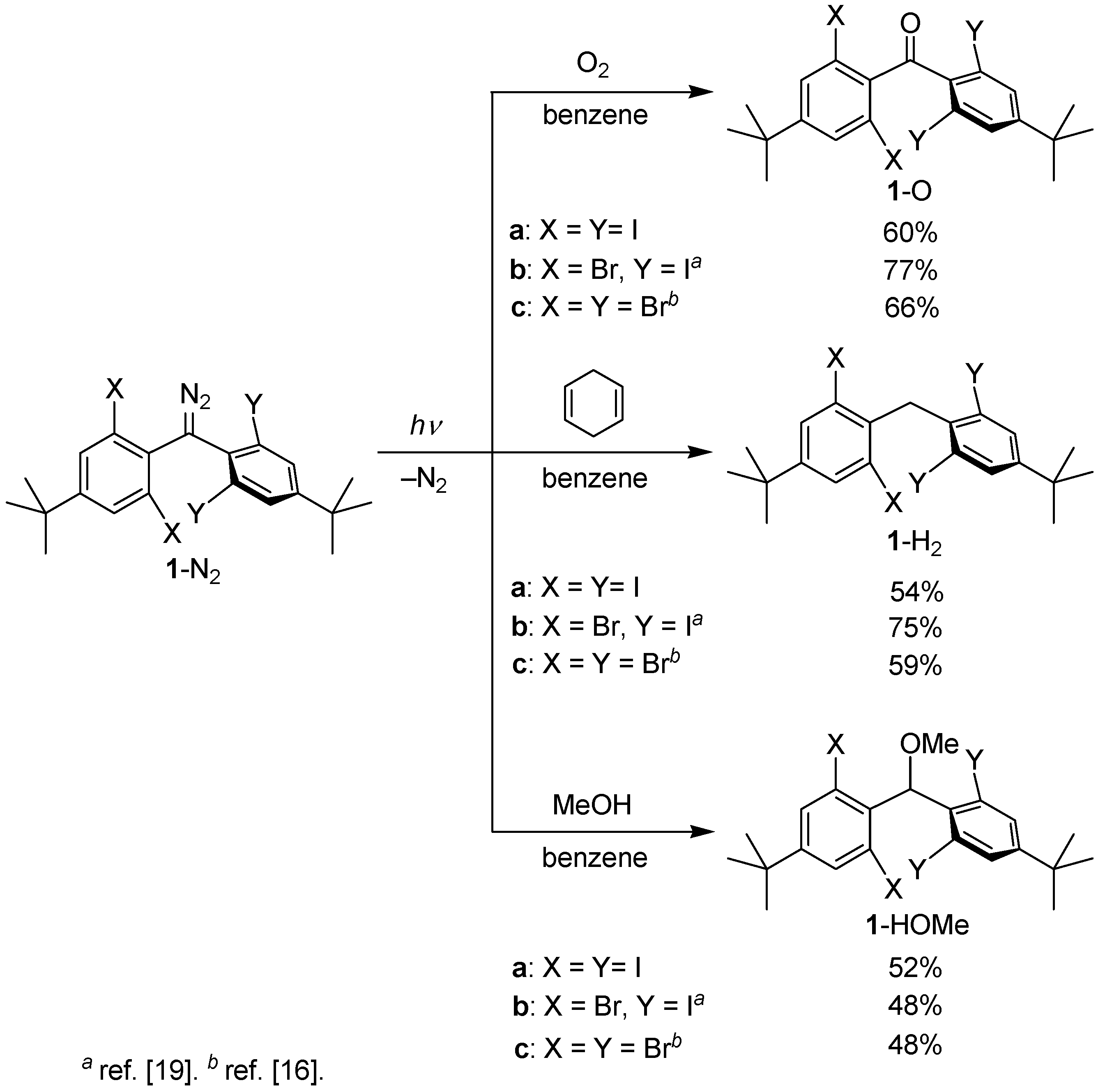
2.3. Product Analysis Studies
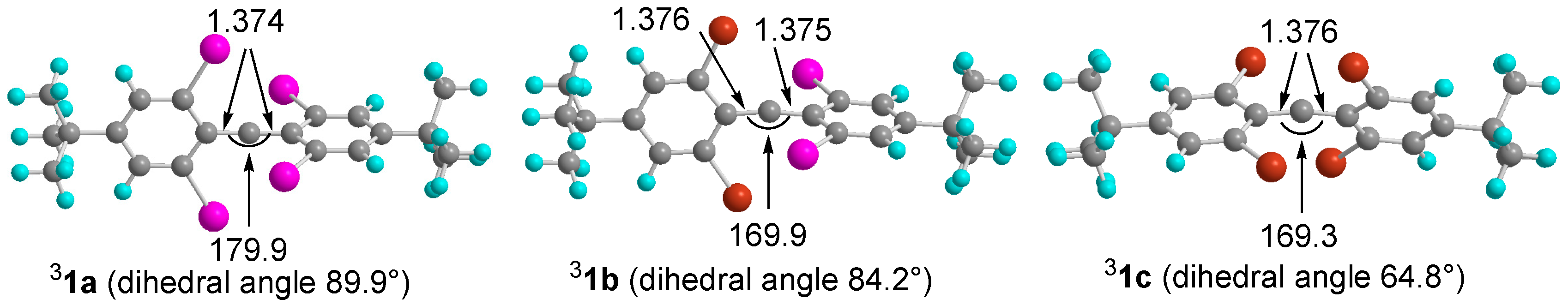
2.4. DFT Calculations
3. Materials and Methods
3.1. General
3.2. Synthesis of Bis(4-tert-butyl-2,6-diiodophenyl)diazomethane (1a-N2)
3.3. Product Analysis
3.4. ESR Measurements
3.5. SQUID Measurements
3.6. Low-Temperature UV-Vis Measurements
3.7. Laser Flash Photolysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Glorius, F.; Rogers, M.M.; Stahl, S.S.; Diez-Gonzalez, S.; Nolan, S.P.; Peris, E.; Gade, L.H.; Bellemin-Laponnaz, S.; Tekavec, T.; Louie, J.; et al. N-Heterocyclic Carbenes in Transition Metal Catalysis: Topics in Organometallic Chemistry; Glorius, F., Ed.; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2007; Volume 21. [Google Scholar]
- Beligny, S.; Blechert, S.; Burling, S.; Paine, B.M.; Whittlesey, M.M.; Scott, N.M.; Nolan, S.P.; Nielsen, D.J.; Cavell, K.J.; Schultz, M.J.; et al. N-Heterocyclic Carbenes in Synthesis; Nolan, S.P., Ed.; Wiley-VCH: New York, NY, USA, 2006. [Google Scholar]
- Rajca, A. Organic Diradicals and Polyradicals: From Spin Coupling to Magnetism? Chem. Rev. 1994, 94, 871–893. [Google Scholar] [CrossRef]
- Iwamura, H. High-spin Organic Molecules and Spin Alignment in Organic Molecular Assemblies. Adv. Phys. Org. Chem. 1990, 26, 179–253. [Google Scholar]
- McConnell, H.M.; Berson, J.A.; Breslow, R.; Klein, D.J.; Borden, W.T.; Shultz, D.A.; Nimura, S.; Yabe, A.; Baumgarten, M.; Blackstock, S.C.; et al. Magnetic Properties of Organic Materials; Lahti, P.M., Ed.; Marcel Dekker: New York, NY, USA, 1999. [Google Scholar]
- Tomioka, H. Persistent Triplet Carbenes. Acc. Chem. Res. 1997, 30, 315–321. [Google Scholar] [CrossRef]
- Tomioka, H. Advances in Carbene Chemistry; Brinker, U.H., Ed.; JAI Press: Greenwich, CT, USA, 1998; Volume 2, pp. 175–214. [Google Scholar]
- Tomioka, H. Carbene Chemistry; Bertrand, G., Ed.; Fontis Media, S.A.: Lausanne, Switzerland, 2002; pp. 103–152. [Google Scholar]
- Hirai, K.; Itoh, T.; Tomioka, H. Persistent Triplet Carbenes. Chem. Rev. 2009, 109, 3275–3332. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, H.; Okada, H.; Watanabe, T.; Banno, K.; Komatsu, K.; Hirai, K. Polymethylated and Poly(tert)butylated Diphenylcarbenes. Generation, Reactions, Kinetics, and Deuterium Isotope Effects of Sterically Congested Triplet Carbenes. J. Am. Chem. Soc. 1997, 119, 1582–1593. [Google Scholar] [CrossRef]
- Rowland, R.S.; Taylor, R. Intermolecular Nonbonded Contact Distances in Organic Crystal Structures: Comparison with Distances Expected from van der Waals Radii. J. Phys. Chem. 1996, 100, 7384–7391. [Google Scholar] [CrossRef]
- Griffin, G.W.; Bertoniere, N.R.; Baron, W.J.; Moss, R.A. Carbenes; Moss, R.A., Jones, M., Jr., Eds.; Wiley: New York, NY, USA, 1973; Volume 1. [Google Scholar]
- Mackay, C.; Hartzler, H.D.; Seyferth, D.; Closs, G.L.; Trozzolo, A.M.; Wasserman, E.; Gasper, P.P.; Hammond, G.S. Carbenes; Moss, R.A., Jones, M., Jr., Eds.; Wiley: New York, NY, USA, 1975; Volume 2. [Google Scholar]
- Kirmse, W. Carbene Chemistry, 2nd ed.; Academic Press: New York, NY, USA, 1971. [Google Scholar]
- Tomioka, H.; Hattori, M.; Hirai, K.; Murata, S. Anomalous Effects of Para Substituents on the Stability of Sterically Congested Triplet Diarylcarbenes. The First Triplet Carbene Surviving over Min under Normal Conditions. J. Am. Chem. Soc. 1996, 118, 8723–8724. [Google Scholar] [CrossRef]
- Tomioka, H.; Watanabe, T.; Hattori, M.; Nomura, N.; Hirai, K. Generation, Reactions, and Kinetics of Sterically Congested Triplet Diphenylcarbenes. Effects of Bromine Groups. J. Am. Chem. Soc. 2002, 124, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Bsssho, K.; Hirai, K.; Kitagawa, T. Steric Protection of Triplet Diphenylcarbenes by Iodo Groups. Mie University: Tsu, Japan, Unpublished work. 2010. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2003. [Google Scholar]
- Hirai, K.; Bessho, K.; Kitagawa, T.; Tomioka, H. Triplet diphenylcarbene protected by iodine and bromine groups. J. Phys. Org. Chem. 2010, 23, 347–356. [Google Scholar] [CrossRef]
- Hirai, K.; Tomioka, H. A Triplet Carbene That Can Almost Be Bottled. J. Am. Chem. Soc. 1999, 121, 10213–10214. [Google Scholar] [CrossRef]
- Itoh, T.; Nakata, Y.; Hirai, K.; Tomioka, H. Triplet Diphenylcarbenes Protected by Trifluoromethyl and Bromine Groups. A Triplet Carbene Surviving a Day in Solution at Room Temperature. J. Am. Chem. Soc. 2006, 128, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, H.; Hirai, K.; Nakayama, T. Chemistry, Kinetics, and Spectroscopy of Highly Hindered Diarylcarbenes. The case of Decachlorodiphenylcarbene. J. Am. Chem. Soc. 1993, 115, 1285–1289. [Google Scholar] [CrossRef]
- Hu, Y.; Ishikawa, Y.; Hirai, K.; Tomioka, H. Spectroscopic and Product Studies of the Effect of Para Substituents on the Reactivity of Triplet Bis(2,6-dimethylphenyl)carbenes. Bull. Chem. Soc. Jpn. 2001, 74, 2207–2218. [Google Scholar] [CrossRef]
- Itoh, T.; Jinbo, Y.; Hirai, K.; Tomioka, H. Triplet Diphenylcarbenes Protected by (Trimethylsilyl)ethynyl Groups. J. Org. Chem. 2004, 69, 4238–4244. [Google Scholar] [CrossRef] [PubMed]
- Sander, W.; Grote, D.; Kossmann, S.; Neese, F. 2,3,5,6-Tetrafluorophenylnitren-4-yl: Electron Paramagnetic Resonance Spectroscopic Characterization of a Quartet-Ground-State Nitreno Radical. J. Am. Chem. Soc. 2008, 130, 4396–4403. [Google Scholar] [CrossRef] [PubMed]
- Hattori, M.; Hirai, K.; Tomioka, H. Synthesis of Stable Triplet Carbenes. Mie University: Tsu, Japan, Unpublished work. 1997. [Google Scholar]
- Platz, M.S. Diradicals; Borden, W.T., Ed.; Wiley: New York, NY, USA, 1982; pp. 195–258. [Google Scholar]
- Wasserman, E.; Hutton, R.S. Electron Paramagnetic Resonance of Triplet States: Cyclic 4π-Electron Systems, Methylene and Environmental Effects. Acc. Chem. Res. 1977, 10, 27–32. [Google Scholar] [CrossRef]
- Breslow, R.; Chang, H.W.; Wasserman, E. Stable Triplet States of Some Cyclopentadienyl Cations. J. Am. Chem. Soc. 1967, 89, 1112–1119. [Google Scholar] [CrossRef]
- Nakamura, N.; Inoue, K.; Iwamura, H.; Fujioka, T.; Sawaki, Y. Synthesis and characterization of a branched-chain hexacarbene in a tridecet ground state. An approach to superparamagnetic polycarbenes. J. Am. Chem. Soc. 1992, 114, 1484–1485. [Google Scholar] [CrossRef]
- Nakamura, N.; Inoue, K.; Iwamura, H. A branched-chain nonacarbene with a nonadecet ground state: A step nearer to superparamagnetic polycarbenes. Angew. Chem. Int. Ed. Engl. 1993, 32, 872–874. [Google Scholar] [CrossRef]
- Matsuda, K.; Nakamura, N.; Takahashi, K.; Inoue, K.; Koga, N.; Iwamura, H. Design, synthesis, and characterization of three kinds of π-cross-conjugated hexacarbenes with high-spin (S = 6) ground states. J. Am Chem. Soc. 1995, 117, 5550–5560. [Google Scholar] [CrossRef]
- Matsuda, K.; Nakamura, N.; Inoue, K.; Koga, N.; Iwamura, H. Design and synthesis of a “starburst”-type nonadiazo compound and magnetic characterization of its photoproduct. Chem. Eur. J. 1996, 2, 259–264. [Google Scholar] [CrossRef]
- Matsuda, K.; Nakamura, N.; Inoue, K.; Koga, N.; Iwamura, H. Toward Dendritic Two-Dimensional Polycarbenes: Syntheses of ‘Starburst’-Type Nona- and Dodecadiazo Compounds and Magnetic Study of Their Photoproducts. Bull. Chem. Soc. Jpn. 1996, 69, 1483–1494. [Google Scholar] [CrossRef]
- Carlin, R.L. Magnetochemistry; Springer: Berlin, Germany, 1986. [Google Scholar]
- Tomioka, H. Advances in Strained and Interesting Organic Molecules; Halton, B., Ed.; JAI Press: Greenwich, CT, USA, 2000; Volume 8, pp. 83–112. [Google Scholar]
- Tomioka, H.; Iwamoto, E.; Itakura, H.; Hirai, K. Generation and characterization of a fairly stable triplet carbene. Nature 2001, 412, 626–628. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, E.; Hirai, K.; Tomioka, H. A Triplet Carbene Surviving a Week in Solution at Room Temperature. J. Am. Chem. Soc. 2003, 125, 14664–14665. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, H. Reactive Intermediate Chemistry; Moss, R.A., Platz, M.S., Jones, M., Jr., Eds.; Wiley: New York, NY, USA, 2004; pp. 375–461. [Google Scholar]
- Sander, W. Carbonyl Oxides: Zwitterions or Diradicals? Angew. Chem. Int. Ed. Engl. 1990, 29, 344–354. [Google Scholar] [CrossRef]
- Bunnelle, W. Preparation, Properties, and Reactions of Carbonyl Oxides. Chem. Rev. 1991, 91, 335–362. [Google Scholar] [CrossRef]
- Sugawara, T.; Iwamura, H.; Hayashi, H.; Sekiguchi, A.; Ando, W.; Liu, M.T.H. Time-resolved absorption spectroscopic detection of 10, 10-dimethyl-10-silaanthracen-9 (10H)-one oxide. Chem. Lett. 1983, 1261–1262. [Google Scholar] [CrossRef]
- Casal, H.L.; Sugamori, S.E.; Scaiano, J.C. Study of Carbonyl Ooxide Formation in the Reaction of Singlet Oxygen with Diphenyldiazomethane. J. Am. Chem. Soc. 1984, 106, 7623–7624. [Google Scholar] [CrossRef]
- Casal, H.L.; Tanner, M.; Werstiuk, N.H.; Scaiano, J.C. Fluorenone oxide: Transient Spectroscopy and Kinetics of Its Formation and Reactions. J. Am. Chem. Soc. 1985, 107, 4616–4620. [Google Scholar] [CrossRef]
- Barcus, R.L.; Hadel, L.M.; Johnston, L.J.; Platz, M.S.; Savino, T.G.; Scaiano, J.C. 1-Naphthylcarbene: Spectroscopy, Kinetics, and Mechanisms. J. Am. Chem. Soc. 1986, 108, 3928–3937. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Tanimoto, Y.; Itoh, M.; Hirai, K.; Tomioka, H. Laser Flash Photolysis Studies of Methoxycarbonyl Phenyl Carbene and Its Derived Carbonyl Oxide at Room Temperature. J. Am. Chem. Soc. 1987, 109, 1942–1946. [Google Scholar] [CrossRef]
- Scaiano, J.C.; McGrimpsey, W.G.; Casal, H.L. Generation and Transient Spectroscopy of Substituted Diaryl Carbonyl Oxides. J. Org. Chem. 1989, 54, 1612–1616. [Google Scholar] [CrossRef]
- Itakura, H.; Mizuno, H.; Hirai, K.; Tomioka, H. Generation, Characterization, and Kinetics of Triplet Di[1,2,3,4,5,6,7,8-octahydro-1,4:5,8-di(ethano)anthryl]carbene. J. Org. Chem. 2000, 65, 8797–8806. [Google Scholar] [CrossRef] [PubMed]
- Stavber, S.; Kralj, P.; Zupan, M. Progressive Direct Iodination of Sterically Hindered Alkyl Substituted Benzenes. Synthesis 2002, 1513–1518. [Google Scholar] [CrossRef]
- Sample Availability: Not available.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carbenes | 2k/εl (s−1) | t1/2 (s) | kO2 (M−1·s−1) | kCHD (M−1·s−1) | KkMeOH 1 (M−2·s−1) |
|---|---|---|---|---|---|
| 1a | 0.0055 0.33 min−1 | 1.1 × 103 18.4 min | 6.5 × 105 | 1.5 | 0.033 |
| 1b | 0.17 | 2.4 × 10 | 7.3 × 106 | 3.9 × 10 | 0.30 |
| 1c | 0.35 | 1.6 × 10 | 2.1 × 107 | 5.3 × 102 | 0.42 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirai, K.; Bessho, K.; Tsujita, K.; Kitagawa, T. Diphenylcarbene Protected by Four ortho-Iodine Groups: An Unusually Persistent Triplet Carbene. Molecules 2016, 21, 1545. https://doi.org/10.3390/molecules21111545
Hirai K, Bessho K, Tsujita K, Kitagawa T. Diphenylcarbene Protected by Four ortho-Iodine Groups: An Unusually Persistent Triplet Carbene. Molecules. 2016; 21(11):1545. https://doi.org/10.3390/molecules21111545
Chicago/Turabian StyleHirai, Katsuyuki, Kana Bessho, Kosaku Tsujita, and Toshikazu Kitagawa. 2016. "Diphenylcarbene Protected by Four ortho-Iodine Groups: An Unusually Persistent Triplet Carbene" Molecules 21, no. 11: 1545. https://doi.org/10.3390/molecules21111545
APA StyleHirai, K., Bessho, K., Tsujita, K., & Kitagawa, T. (2016). Diphenylcarbene Protected by Four ortho-Iodine Groups: An Unusually Persistent Triplet Carbene. Molecules, 21(11), 1545. https://doi.org/10.3390/molecules21111545