
DeBouganin Diabody Fusion Protein Overcomes Drug Resistance to ADCs Comprised of Anti-Microtubule Agents

Abstract
:1. Introduction
2. Results
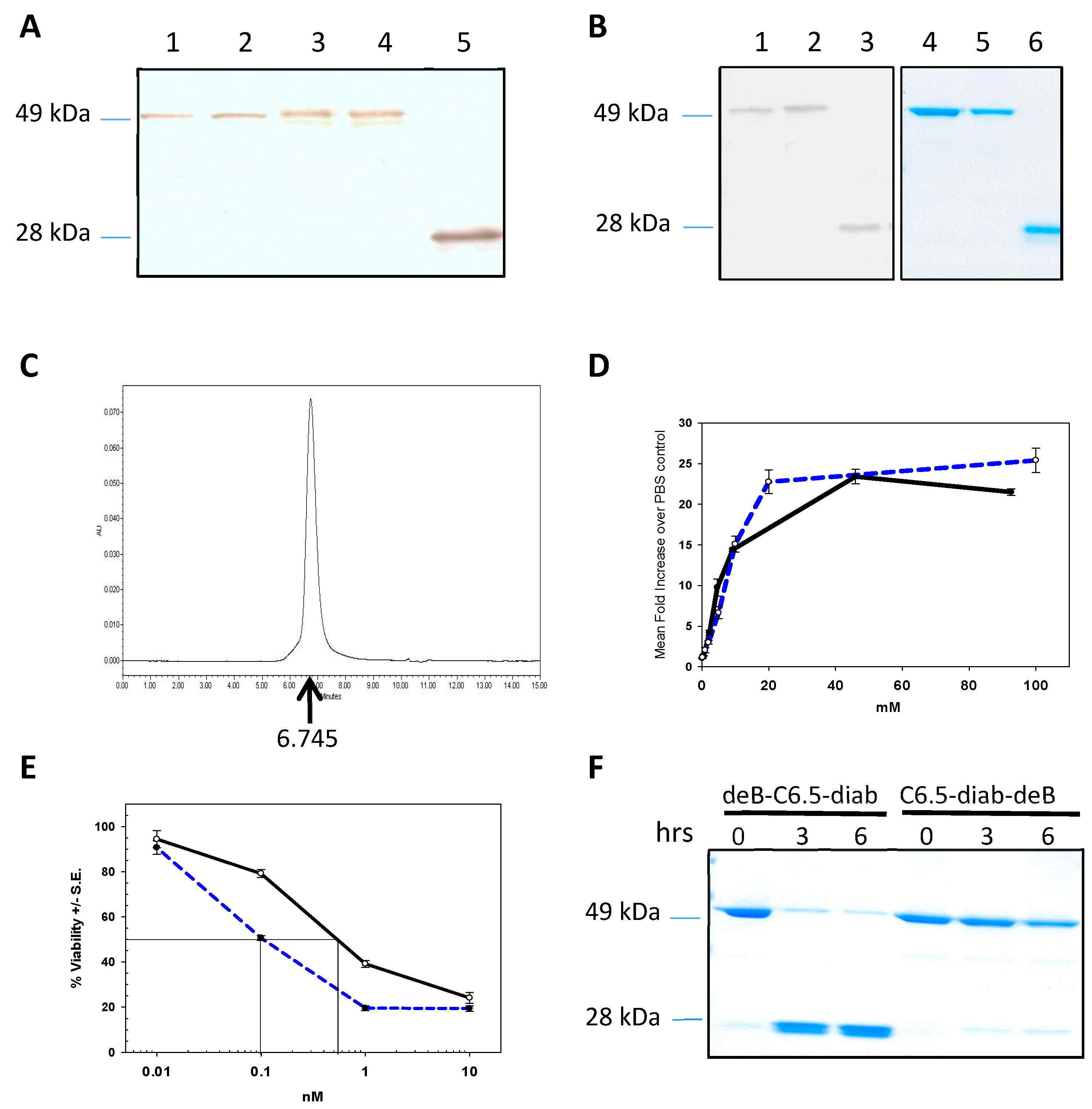
2.1. Engineering and Selection of deB-C6.5 Diabody
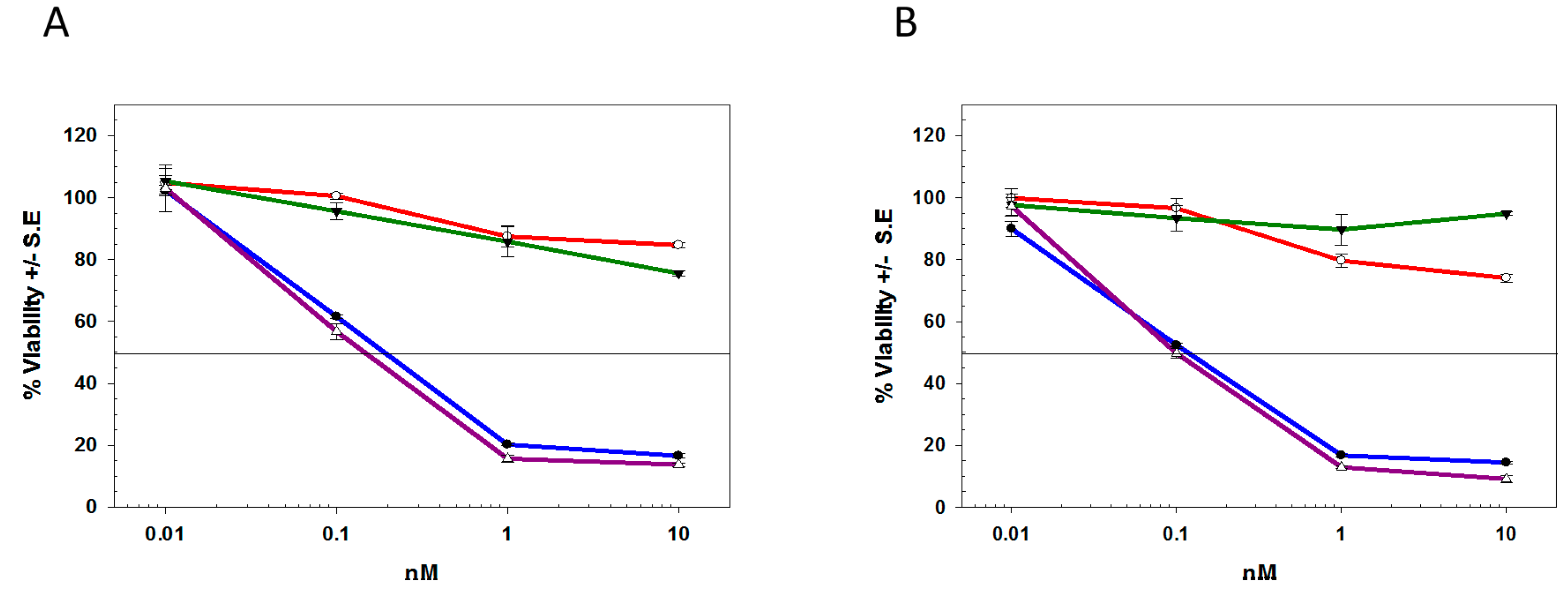
2.2. Biological Characterization of deB-C6.5-diab
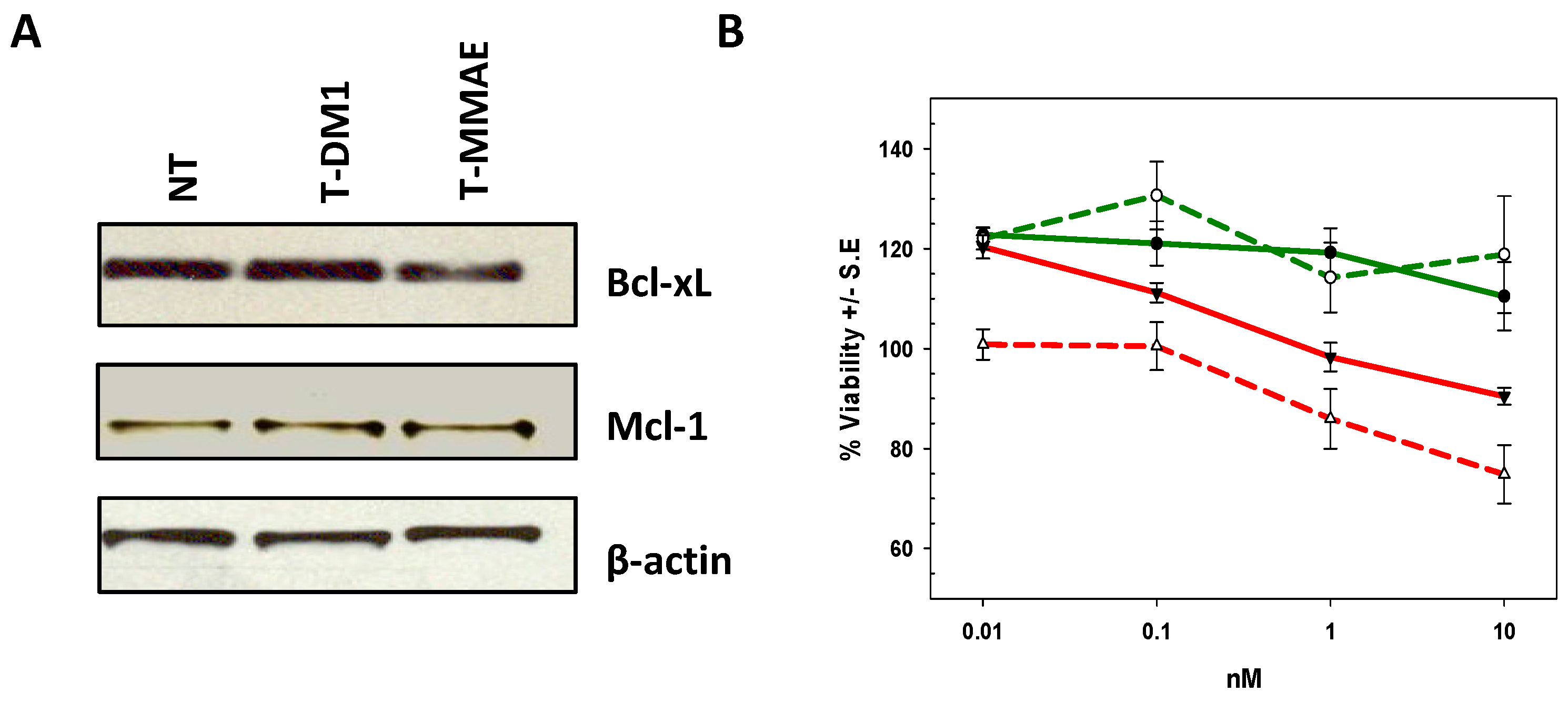
2.3. Differentiating deBouganin MOA from Anti-Microtubule Agents
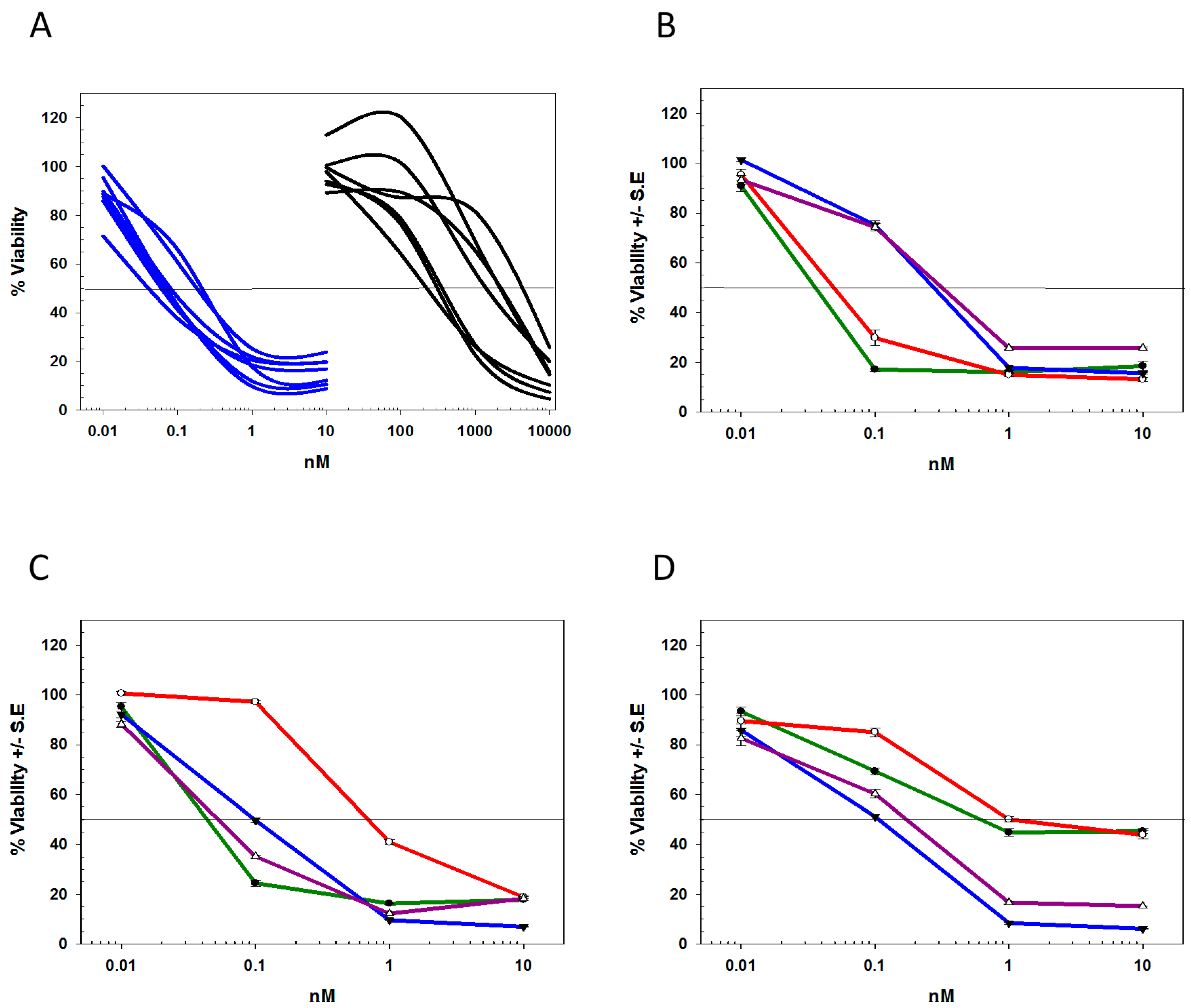
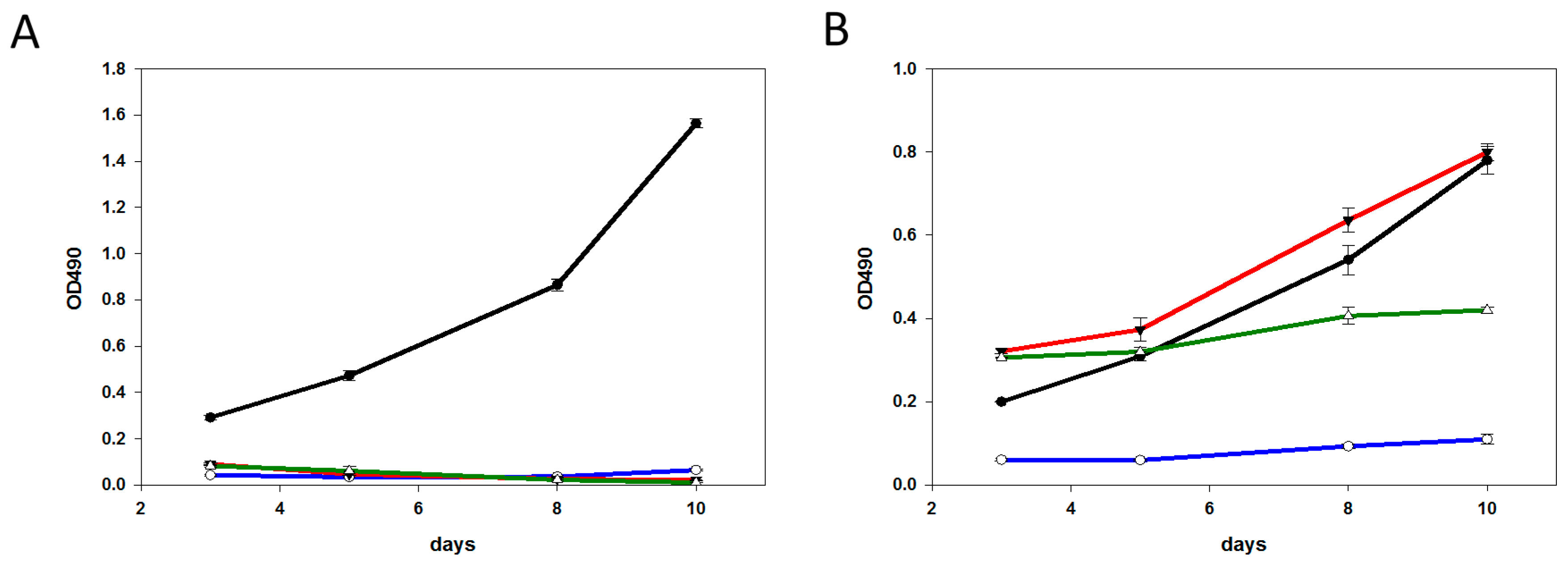
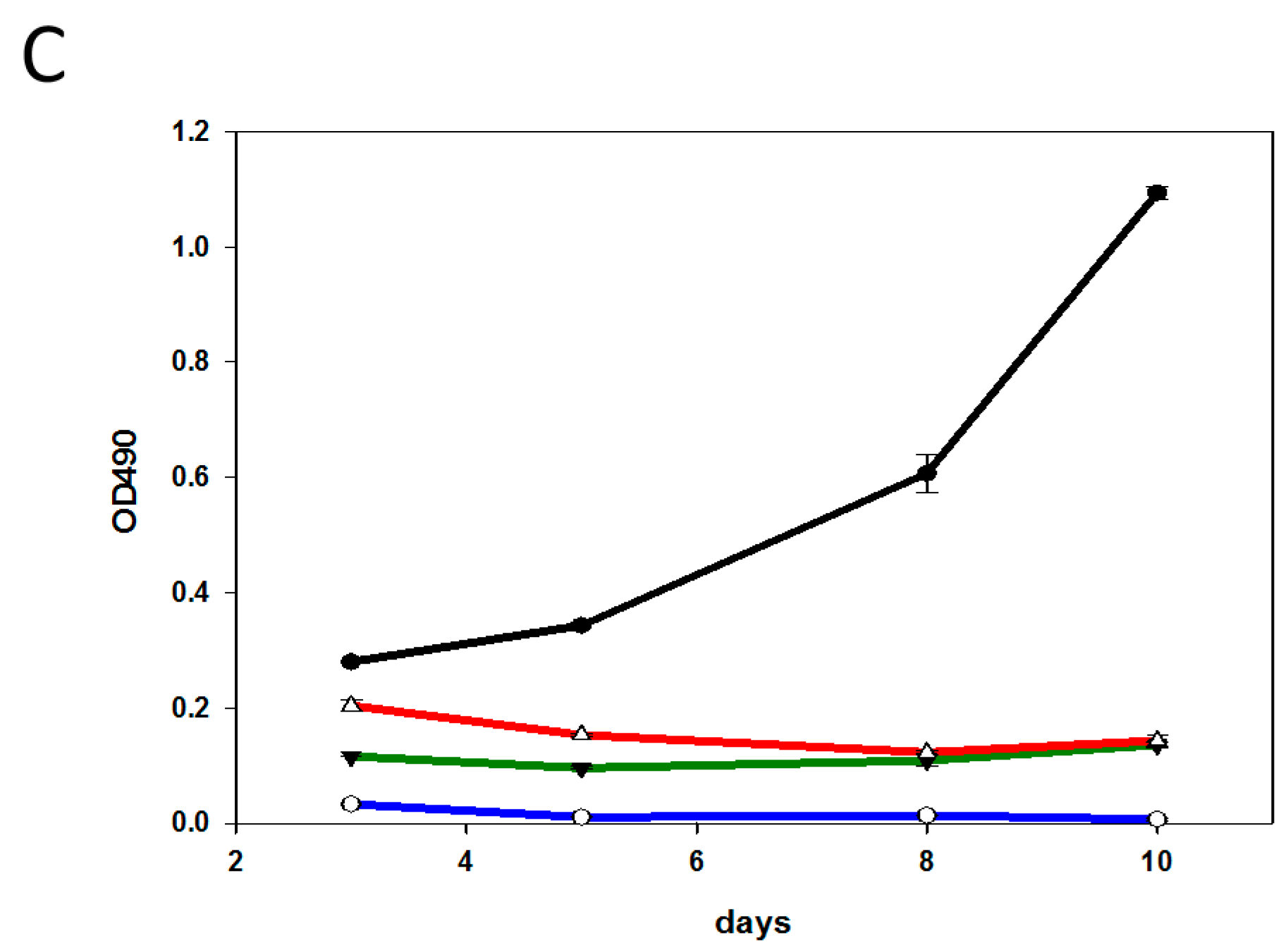
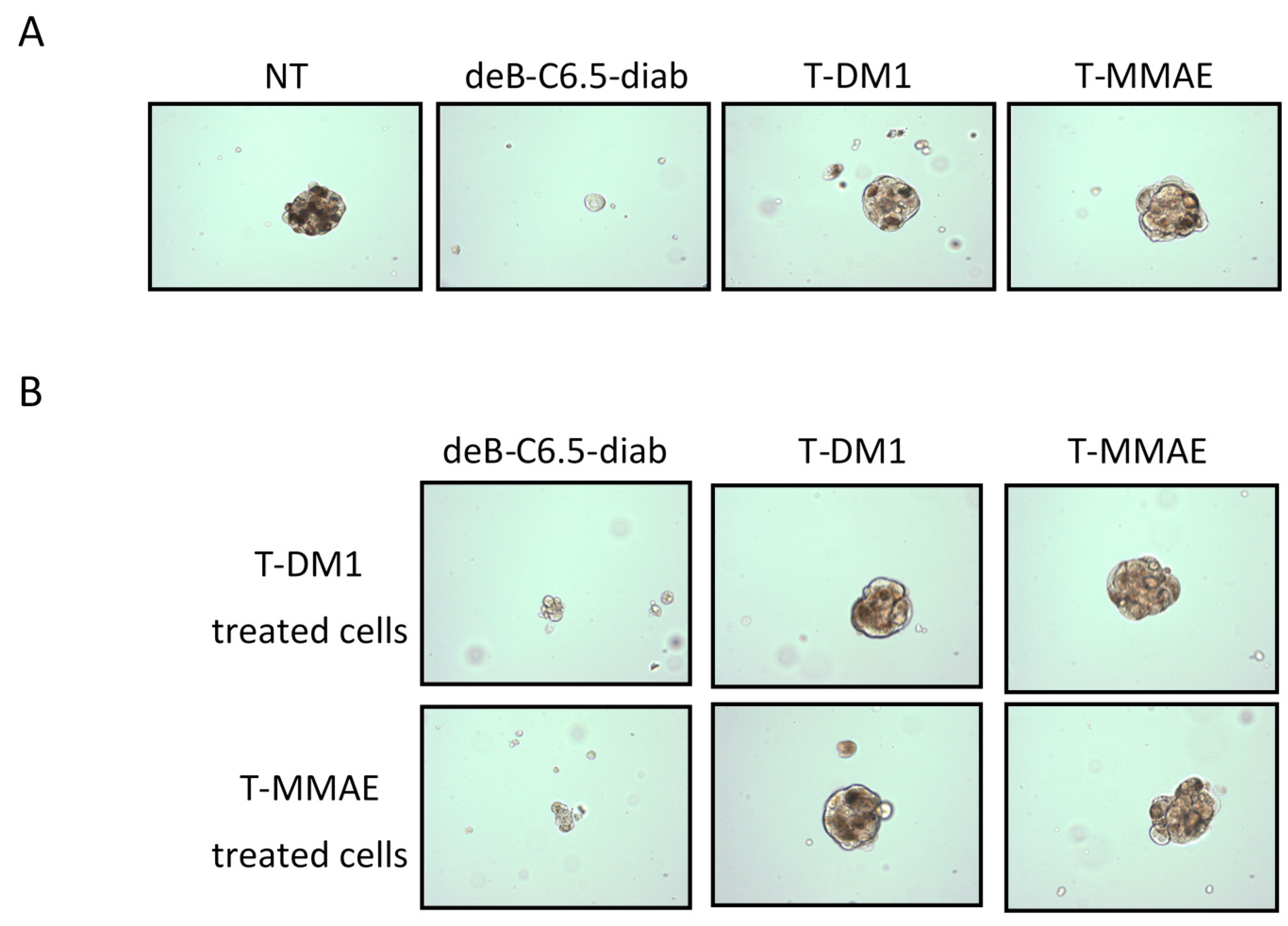
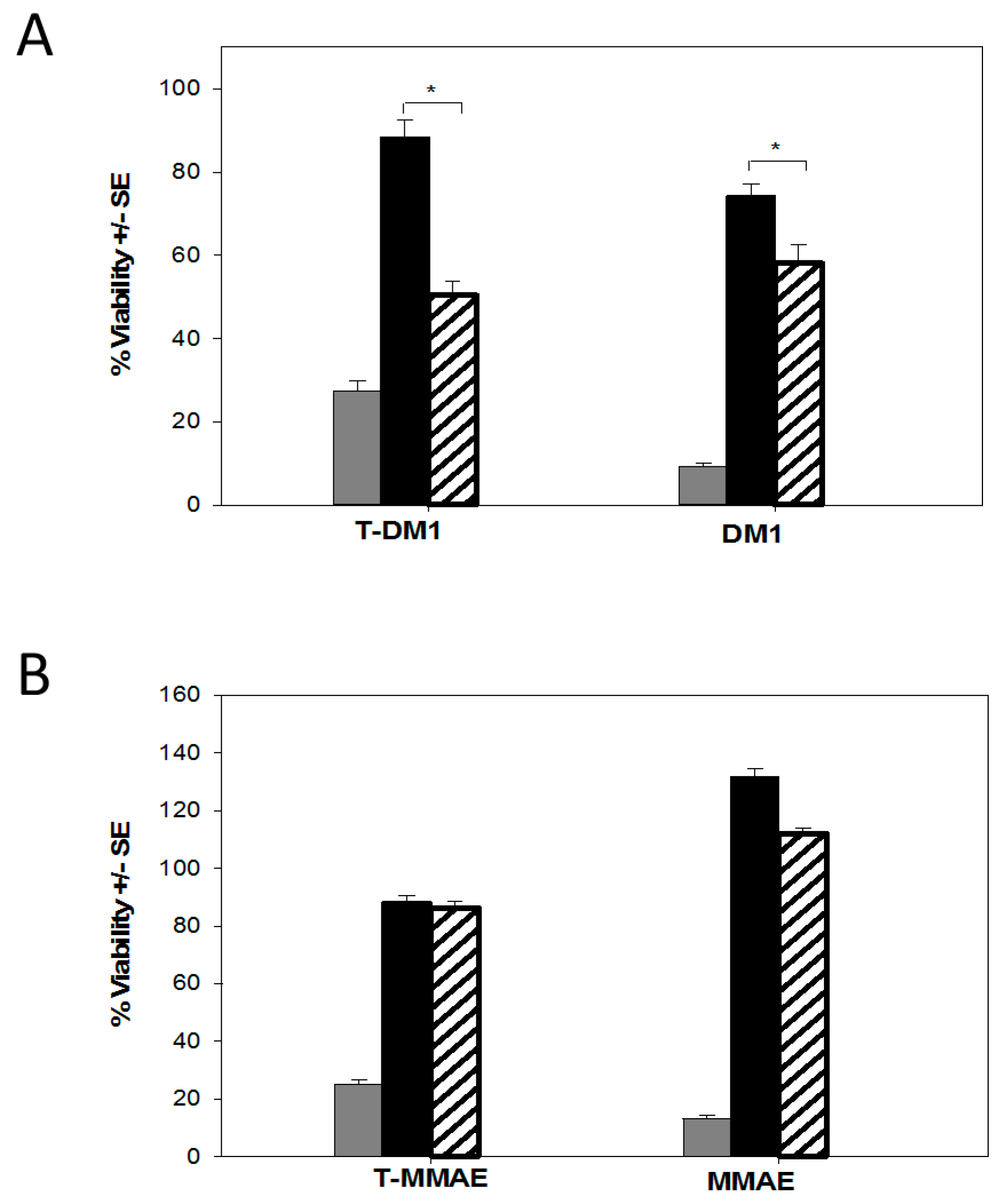
2.4. Characterization of the T-DM1 and T-MMAE Surviving Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Antibodies, Toxins, and Conjugates
4.3. Plasmid Construction
4.4. Research Scale Expression Study
4.5. Large-Scale Expression and Purification of Fusion Proteins
4.6. Western Blot Analysis
4.7. Cell Surface Reactivity
4.8. In Vitro Furin Cleavage Assay
4.9. Potency
4.10. Tumorosphere Assays
4.11. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Carter, P.J.; Senter, P.D. Antibody-drug conjugates for cancer therapy. Cancer J. 2008, 14, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Sievers, E.L.; Senter, P.D. Antibody-drug conjugates in cancer therapy. Annu. Rev. Med. 2013, 64, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Fang, L. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Bartlett, N.L.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.L.; Forero-Torres, A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N. Engl. J. Med. 2010, 363, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 209. [Google Scholar] [CrossRef] [PubMed]
- Gebleux, R.; Casi, G. Antibody-drug conjugates: Current status and future perspectives. Pharmacol. Ther. 2016, 167, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Garraway, L.A.; Jänne, P.A. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012, 2, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Loganzo, F.; Sung, M.; Gerber, H.P. Mechanisms of Resistance to Antibody-Drug Conjugates. Mol. Cancer Ther. 2016, 15, 2825–2834. [Google Scholar] [CrossRef] [PubMed]
- Stirpe, F.; Battelli, M.G. Ribosome-inactivating proteins: Progress and problems. Cell. Mol. Life Sci. 2006, 63, 1850–1866. [Google Scholar] [CrossRef] [PubMed]
- Stirpe, F. Ribosome-inactivating proteins. Toxicon 2004, 44, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Akkouh, O.; Ng, T.B.; Cheung, R.C.; Wong, J.H.; Pan, W.; Ng, C.C.; Chan, W.Y. Biological activities of ribosome-inactivating proteins and their possible applications as antimicrobial, anticancer, and anti-pest agents and in neuroscience research. Appl. Microbiol. Biotechnol. 2015, 99, 9847–9863. [Google Scholar] [CrossRef] [PubMed]
- Gilabert-Oriol, R.; Weng, A.; Mallinckrodt, B.; Melzig, M.F.; Fuchs, H.; Thakur, M. Immunotoxins constructed with ribosome-inactivating proteins and their enhancers: A lethal cocktail with tumor specific efficacy. Curr. Pharm. Des. 2014, 20, 6584–6643. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Tyagi, M.; Pachauri, M.; Ghosh, P.C. Potential therapeutic applications of plant toxin-ricin in cancer: Challenges and advances. Tumour Biol. 2015, 36, 8239–8246. [Google Scholar] [CrossRef] [PubMed]
- Stirpe, F.; Lappi, D.A. Ribosome-Inactivating Proteins: Ricin and Related Proteins; Willey Blackwell: Ames, IA, USA, 2014. [Google Scholar]
- Walsh, M.J.; Dodd, J.E.; Hautbergue, G.M. Ribosome-inactivating proteins: Potent poisons and molecular tools. Virulence 2013, 4, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Sestili, P.; Alfieri, R.; Carnicelli, D.; Martinelli, C.; Barbieri, L.; Stirpe, F.; Brigotti, M. Shiga toxin 1 and ricin inhibit the repair of H2O2-induced DNA single strand breaks in cultured mammalian cells. DNA Repair 2005, 4, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Brigotti, M.; Alfieri, R.; Sestili, P.; Bonelli, M.; Petronini, P.G.; Guidarelli, A.; Sperti, S. Damage to nuclear DNA induced by Shiga toxin 1 and ricin in human endothelial cells. FASEB J. 2002, 16, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Polito, L.; Bortolotti, M.; Pedrazzi, M.; Mercatelli, D.; Battelli, M.G.; Bolognesi, A. Apoptosis and necroptosis induced by stenodactylin in neuroblastoma cells can be completely prevented through caspase inhibition plus catalase or necrostatin-1. Phytomedicine 2016, 23, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, L.; Battelli, M.G.; Stirpe, F. Ribosome-inactivating proteins from plants. Biochim. Biophys. Acta 1993, 1154, 237–282. [Google Scholar] [CrossRef]
- Stirpe, F. Ribosome-inactivating proteins: From toxins to useful proteins. Toxicon 2013, 67, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Pastan, I.; Hassan, R.; FitzGerald, D.J.; Kreitman, R.J. Immunotoxin treatment of cancer. Annu. Rev. Med. 2007, 58, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, M.; Guindon, J.; Brazas, L.; Moore, C.; Entwistle, J.; Cizeau, J.; MacDonald, G.C. A phase II study of oportuzumab monatox: An immunotoxin therapy for patients with noninvasive urothelial carcinoma in situ previously treated with bacillus Calmette-Guérin. J. Urol. 2012, 188, 1712–1718. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, G.C.; Rasamoelisolo, M.; Entwistle, J.; Cuthbert, W.; Kowalski, M.; Spearman, M.; Glover, N. A phase I clinical study of intratumorally administered VB4–845, an anti-epithelial cell adhesion molecule recombinant fusion protein, in patients with squamous cell carcinoma of the head and neck. Med. Oncol. 2009, 26, 257–264. [Google Scholar] [CrossRef] [PubMed]
- den Hartog, M.T.; Lubelli, C.; Boon, L.; Heerkens, S.; Ortiz Buijsse, A.P.; de Boer, M.; Stirpe, F. Cloning and expression of cDNA coding for bouganin. Eur. J. Biochem. 2002, 269, 1772–1779. [Google Scholar] [CrossRef] [PubMed]
- Cizeau, J.; Grenkow, D.; Brown, J.; Entwistle, J.; MacDonald, G. Engineering and biological characterization of VB6–845, an anti-EpCAM immunotoxin containing a T-cell epitope-depleted variant of the plant toxin bouganin. J. Immunother. 2009, 32, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, A.; Polito, L.; Olivieri, F.; Valbonesi, P.; Barbieri, L.; Battelli, M.G.; Parente, A. New ribosome-inactivating proteins with polynucleotide:adenosine glycosidase and antiviral activities from Basella rubra L. and Bougainvillea spectabilis Willd. Planta 1997, 203, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, A.; Polito, L.; Tazzari, P.L.; Lemoli, R.M.; Lubelli, C.; Fogli, M.; Stirpe, F. In vitro anti-tumour activity of anti-CD80 and anti-CD86 immunotoxins containing type 1 ribosome-inactivating proteins. Br. J. Haematol. 2000, 110, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Dillon, R.L.; Chooniedass, S.; Premsukh, A.; Adams, G.P.; Entwistle, J.; MacDonald, G.C.; Cizeau, J. Trastuzumab-deBouganin Conjugate Overcomes Multiple Mechanisms of T-DM1 Drug Resistance. J. Immunother. 2016, 39, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Luque-Cabal, M.; Garcia-Teijido, P.; Fernandez-Perez, Y.; Sanchez-Lorenzo, L.; Palacio-Vazquez, I. Mechanisms behind the Resistance to Trastuzumab in HER2-Amplified Breast Cancer and Strategies to Overcome It. Clin. Med. Insights Oncol. 2016, 10 (Suppl. S1), 21–30. [Google Scholar] [CrossRef] [PubMed]
- Maximiano, S.; Magalhaes, P.; Guerreiro, M.P.; Morgado, M. Trastuzumab in the Treatment of Breast Cancer. BioDrugs 2016, 30, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F.; Kurvari, V.; Virmani, A.; Gollahon, L.; Sakaguchi, M.; Westerfield, M.; Tomlinson, G. Characterization of paired tumor and non-tumor cell lines established from patients with breast cancer. Int. J. Cancer 1998, 78, 766–774. [Google Scholar] [CrossRef]
- Austin, C.D.; De Maziere, A.M.; Pisacane, P.I.; van Dijk, S.M.; Eigenbrot, C.; Sliwkowski, M.X.; Scheller, R.H. Endocytosis and Sorting of ErbB2 and the Site of Action of Cancer Therapeutics Trastuzumab and Geldanamycin. Mol. Biol. Cell 2004, 15, 5268–5282. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.E.; Hall, P.D.; McLain, C.; Safa, A.R.; Tagge, E.P.; Kreitman, R.J. Cell-specific modulation of drug resistance in acute myeloid leukemic blasts by diphtheria fusion toxin, DT388-GMCSF. Bioconjug. Chem. 1998, 9, 490–496. [Google Scholar] [CrossRef] [PubMed]
- McGrath, M.S.; Rosenblum, M.G.; Philips, M.R.; Scheinberg, D.A. Immunotoxin resistance in multidrug resistant cells. Cancer Res. 2003, 63, 72–79. [Google Scholar] [PubMed]
- Dinota, A.; Tazzari, P.L.; Michieli, M.; Visani, G.; Gobbi, M.; Bontadini, A.; Pileri, S. In vitro bone marrow purging of multidrug-resistant cells with a mouse monoclonal antibody directed against Mr 170,000 glycoprotein and a saporin-conjugated anti-mouse antibody. Cancer Res. 1990, 50, 4291–4294. [Google Scholar] [PubMed]
- Kreitman, R.J.; Pastan, I. Immunoconjugates in the management of hairy cell leukemia. Best Pract. Res. Clin. Haematol. 2015, 28, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Sharon, E.; Thomas, A.; Zhang, J.; Ling, A.; Miettinen, M.; Pastan, I. Phase 1 study of the antimesothelin immunotoxin SS1P in combination with pemetrexed and cisplatin for front-line therapy of pleural mesothelioma and correlation of tumor response with serum mesothelin, megakaryocyte potentiating factor, and cancer antigen 125. Cancer 2014, 120, 3311–3319. [Google Scholar] [PubMed]
- Hassan, R.; Bullock, S.; Premkumar, A.; Kreitman, R.J.; Kindler, H.; Willingham, M.C.; Pastan, I. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin. Cancer Res. 2007, 13, 5144–5149. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xia, X.; Ke, Y.; Nie, H.; Smith, M.A.; Zhu, X. Trichosanthin induced apoptosis in HL-60 cells via mitochondrial and endoplasmic reticulum stress signaling pathways. Biochim. Biophys. Acta 2007, 1770, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Gong, Y.; Ma, H.; An, C.; Chen, D.; Chen, Z.L. Reactive oxygen species involved in trichosanthin-induced apoptosis of human choriocarcinoma cells. Biochem. J. 2001, 355 Pt 3, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.L.; Wong, J.H.; Fang, E.F.; Chan, Y.S.; Ye, X.J.; Ng, T.B. Differential inhibitory potencies and mechanisms of the type I ribosome inactivating protein marmorin on estrogen receptor (ER)-positive and ER-negative breast cancer cells. Biochim. Biophys. Acta 2013, 1833, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Shaw, F.L.; Harrison, H.; Spence, K.; Ablett, M.P.; Simoes, B.M.; Farnie, G.; Clarke, R.B. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J. Mammary Gland Biol. Neoplasia 2012, 17, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not Available.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | HER2 Expression | IC50 (nM) | |||
|---|---|---|---|---|---|
| deB-C6.5-diab | T-deB * | T-DM1 * | T-MMAE | ||
| Breast | |||||
| BT-474 | 3+ | 0.047 (0.005) | 0.082 (0.019) | 0.715 (0.025) | 0.040 (0.002) |
| HCC1419 | 3+ | 0.155 (0.015) | 0.086 (0.025) | 1.900 (0.900) | 5.705 (5.095) |
| HCC1569 | 3+ | 0.195 (0.025) | 0.210 (0.080) | 10.200 (0.200) | 1.045 (0.155) |
| HCC1954 | 3+ | 0.043 (0.005) | 0.045 (0.009) | 0.320 (0.000) | 0.076 (0.005) |
| HCC2218 | 3+ | 0.265 (0.065) | 0.245 (0.095) | 0.290 (0.042) | 0.250 (0.050) |
| HCC202 | 3+ | 0.022 (0.010) | 0.055 (0.015) | 0.100 (0.049) | 0.165 (0.055) |
| SK-BR-3 | 3+ | 0.330 (0.020) | 0.275 (0.005) | 0.047 (0.004) | 0.037 (0.003) |
| MDA-MB-361 | 2+ | 0.685 (0.035) | 1.795 (0.524) | 0.320 (0.000) | 0.051 (0.001) |
| MDA-MB-453 | 2+ | 0.225 (0.085) | 0.335 (0.105) | 0.440 (0.060) | 0.255 (0.035) |
| MCF-7 | 1+ | >10 | >10 | >10 | >10 |
| T47D | 1+ | >10 | >10 | 8.000 (2.000) | >10 |
| MDA-MB-231 | 0 | >10 | >10 | >10 | >10 |
| Lung | |||||
| Calu-3 | 3+ | 0.041 (0.000) | 0.105 (0.005) | 1.400 (0.400) | 0.086 (0.006) |
| Gastric | |||||
| NCI-N87 | 3+ | 0.032 (0.016) | 0.09 (0.012) | 0.265 (0.177) | 0.091 (0.055) |
| OE-19 | 3+ | 0.043 (0.008) | 0.050 (0.009) | 0.037 (0.008) | 0.0425 (0.007) |
| HCC1419 | HCC1419-T-DM1 | HCC1419-T-MMAE | BT-474 | BT-474-T-DM1 | BT-474-T-MMAE | |
|---|---|---|---|---|---|---|
| deB-C6.5-diab | 0.15 (0.02) | 0.19 (0.01) | 0.17 (0.05) | 0.07 (0.02) | 0.33 (0.27) | 0.11 (0.01) |
| T-deB | 0.3 (0.05) | 0.19 (0.038) | 0.27 (0.03) | 0.18 (0.005) | 0.87 (0.02) | 0.42 (0.17) |
| T-DM1 | 1.9 (0.9) | >10 | >10 | 0.85 (0.25) | >10 | >10 |
| T-MMAE | 5.7 (5.1) | >10 | >10 | 0.04 (0.01) | >10 | >10 |
| T-Duo | 0.28 (0.03) | 4.65 (1.85) | >10 | 0.4 (0.1) | >10 | 9.95 (5.05) |
| DM1 | 135 (65) | >100 | >100 | 17 (8) | >100 | >100 |
| MMAE | 7 (3) | >100 | >100 | 0.57 (0.17) | >100 | >100 |
| Taxol | >1000 | >1000 | >1000 | 18 (3) | >1000 | >1000 |
| Duocarmycin | 0.65 (0.05) | 3.55 (0.45) | 4.95 (0.05) | 0.31 (0.08) | 5.45 (1.45) | 3.05 (0.45) |
| BT-474 | BT-474-T-DM1 | BT-474-T-MMAE | HCC1419 | HCC1419-T-DM1 | HCC1419-T-MMAE | |
|---|---|---|---|---|---|---|
| deB-C6.5-diab | 0 | 0.14 (0.14) | 0.6 (0.6) | 2.9 (2.9) | 0.19 (0.01) | 1.8 (0.5) |
| T-DM1 | 91.7 | 42.8 (29.4) | 72.6 (6.9) | 91.3 (11.7) | 110.6 (2.1) | 83.6 (10.1) |
| T-MMAE | 87.8 | 60.1 (28) | 70.6 (6.1) | 90.4 (13.6) | 114 (6.1) | 90.9 (12.9) |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chooniedass, S.; Dillon, R.L.; Premsukh, A.; Hudson, P.J.; Adams, G.P.; MacDonald, G.C.; Cizeau, J. DeBouganin Diabody Fusion Protein Overcomes Drug Resistance to ADCs Comprised of Anti-Microtubule Agents. Molecules 2016, 21, 1741. https://doi.org/10.3390/molecules21121741
Chooniedass S, Dillon RL, Premsukh A, Hudson PJ, Adams GP, MacDonald GC, Cizeau J. DeBouganin Diabody Fusion Protein Overcomes Drug Resistance to ADCs Comprised of Anti-Microtubule Agents. Molecules. 2016; 21(12):1741. https://doi.org/10.3390/molecules21121741
Chicago/Turabian StyleChooniedass, Shilpa, Rachelle L. Dillon, Arjune Premsukh, Peter J. Hudson, Gregory P. Adams, Glen C. MacDonald, and Jeannick Cizeau. 2016. "DeBouganin Diabody Fusion Protein Overcomes Drug Resistance to ADCs Comprised of Anti-Microtubule Agents" Molecules 21, no. 12: 1741. https://doi.org/10.3390/molecules21121741
APA StyleChooniedass, S., Dillon, R. L., Premsukh, A., Hudson, P. J., Adams, G. P., MacDonald, G. C., & Cizeau, J. (2016). DeBouganin Diabody Fusion Protein Overcomes Drug Resistance to ADCs Comprised of Anti-Microtubule Agents. Molecules, 21(12), 1741. https://doi.org/10.3390/molecules21121741