
An Update on the Synthesis of Pyrrolo[1,4]benzodiazepines

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

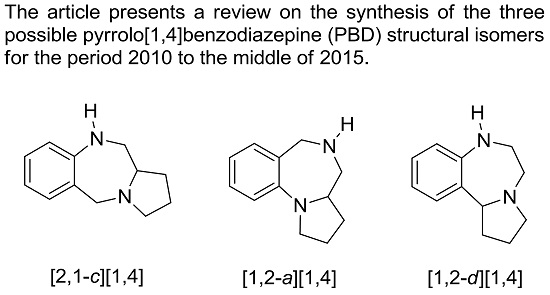
1.1. Pyrrolo[2,1-c][1,4]benzodiazepines

1.2. Pyrrolo[1,2-a][1,4]benzodiazepines
1.3. Pyrrolo[1,2-d][1,4]benzodiazepines

1.4. General Information
2. Synthesis of Pyrrolo[1,4]benzodiazepines
2.1. Pyrrolo[2,1-c][1,4]benzodiazepines
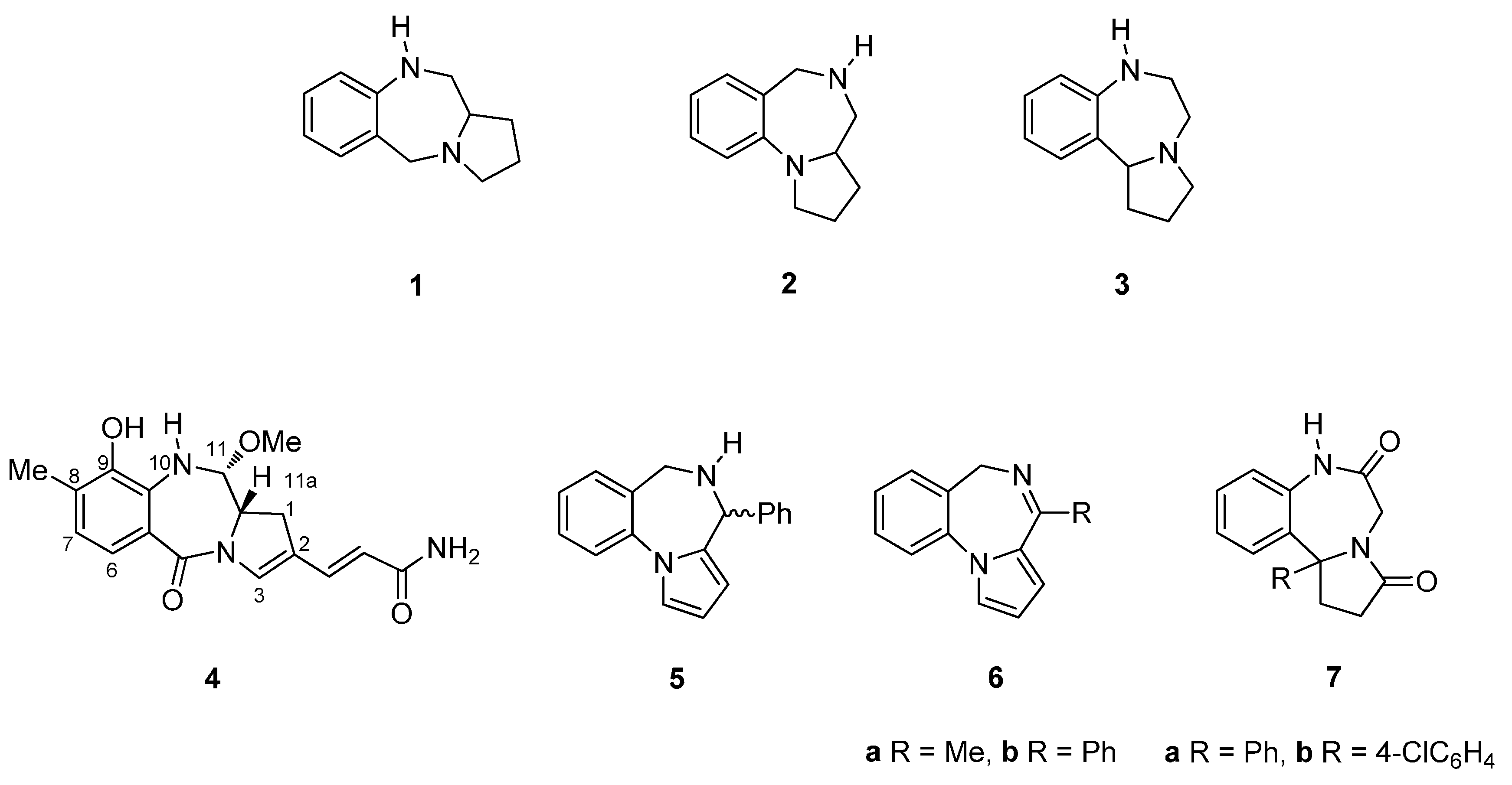
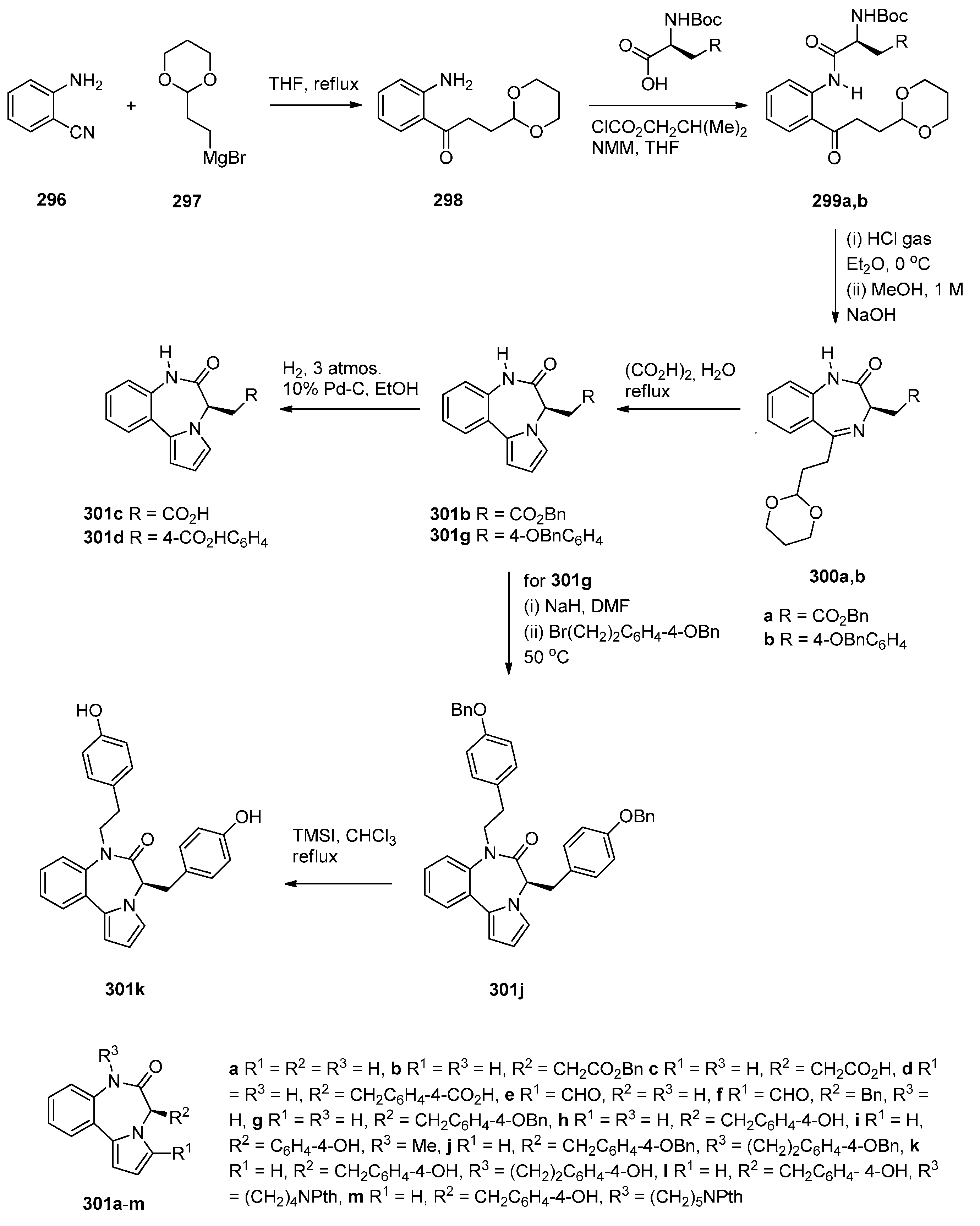
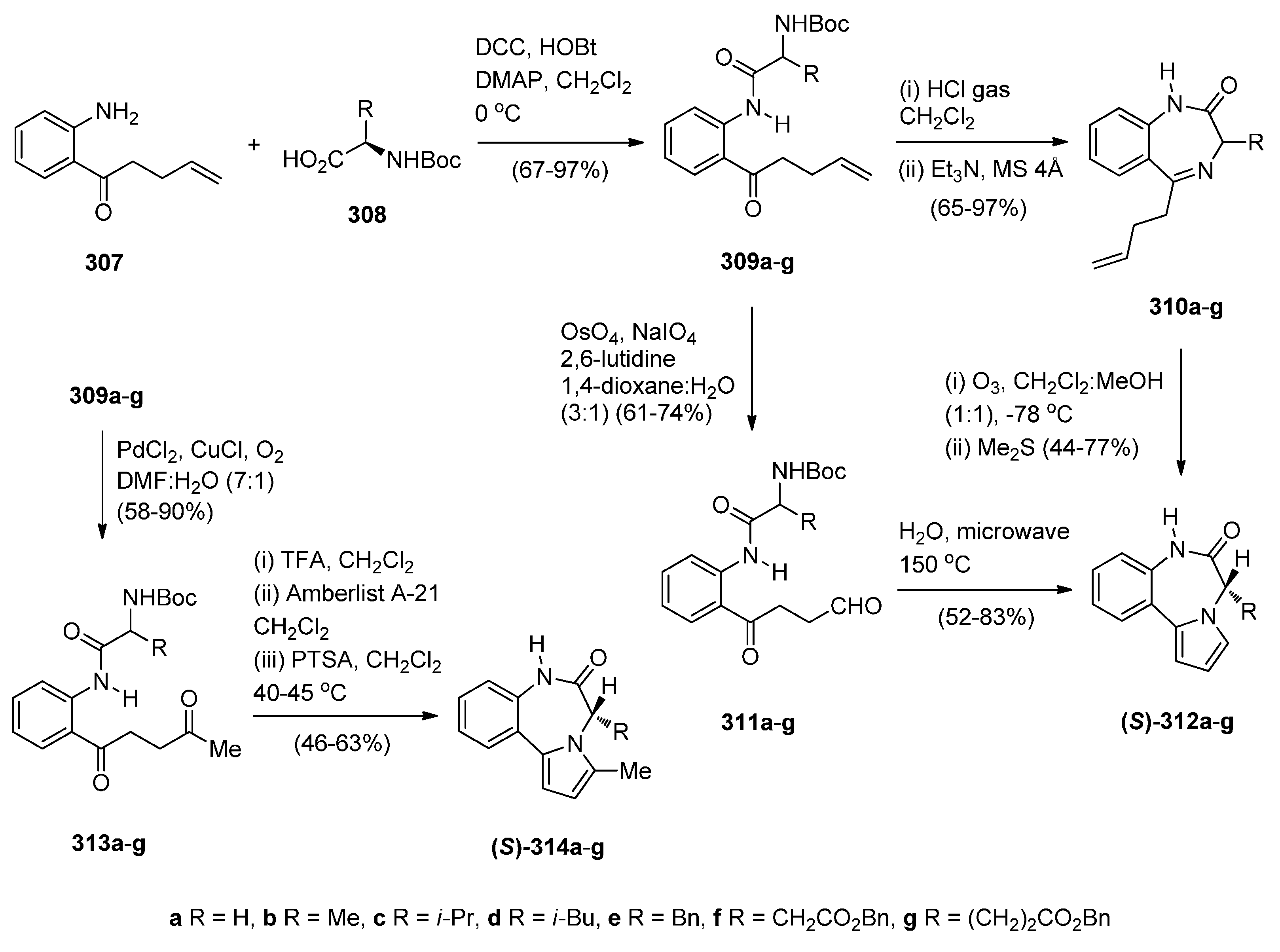
2.1.1. Pyrrolo[2,1-c][1,4]benzodiazepine-5-one Monomers or Dimers with a Non-Aromatic Pyrrole Ring
Cyclocondensation of N-(2-Aminobenzoyl)pyrrolidine-2-Carboxaldehyde Diethyl Thioacetals

















Oxidative Cyclisation of N-(2-(Alloc or Troc)aminobenzoyl)pyrrolidine-2-Methanol Derivatives









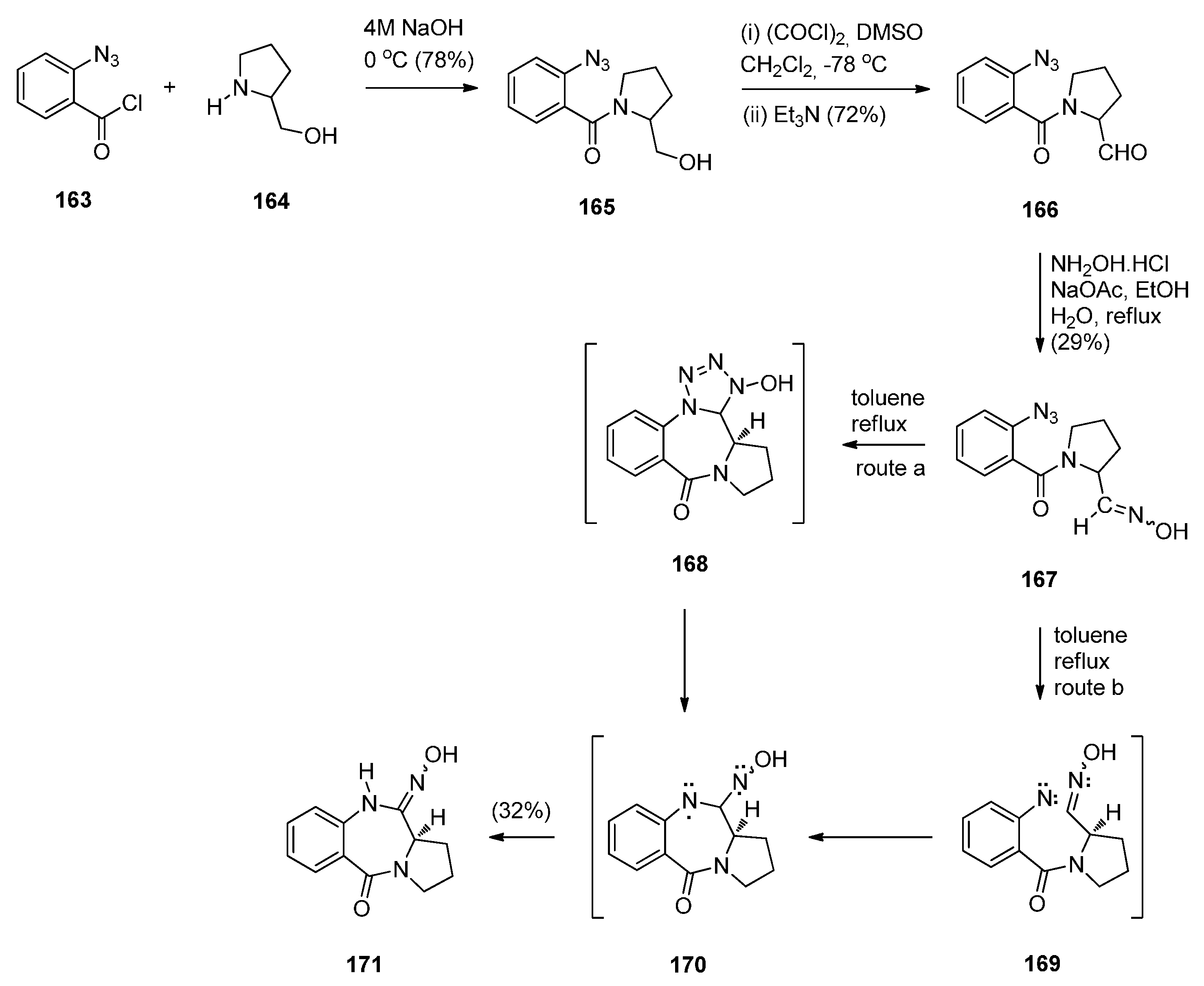
Reductive Cyclisation of N-(2-Azidobenzoyl)pyrrolidine-2-Carboxaldehydes
Cyclisation of N-(2-Azidobenzoyl)pyrrolidine-2-carboxaldehydes



2.1.2. Pyrrolo[2,1-c][1,4]benzodiazepine-5,11-Diones (Dilactams) with a Non-Aromatic Pyrrole Ring
Reductive Cyclisation of Methyl N-(2-Azidobenzoyl)pyrrolidine-2-Carboxylates

Reaction of Isatoic Anhydrides with d-Prolines

2.1.3. Pyrrolo[2,1-c][1,4]benzodiazepine-11-Ones with a Non-Aromatic Pyrrole Ring
Cyclodehydration of N-[2-(Aryl or Heteroaryl)hydroxymethyl]-l-Prolinamides

2.1.4. Pyrrolo[2,1-c][1,4]benzodiazepines with an Aromatic Pyrrole Ring
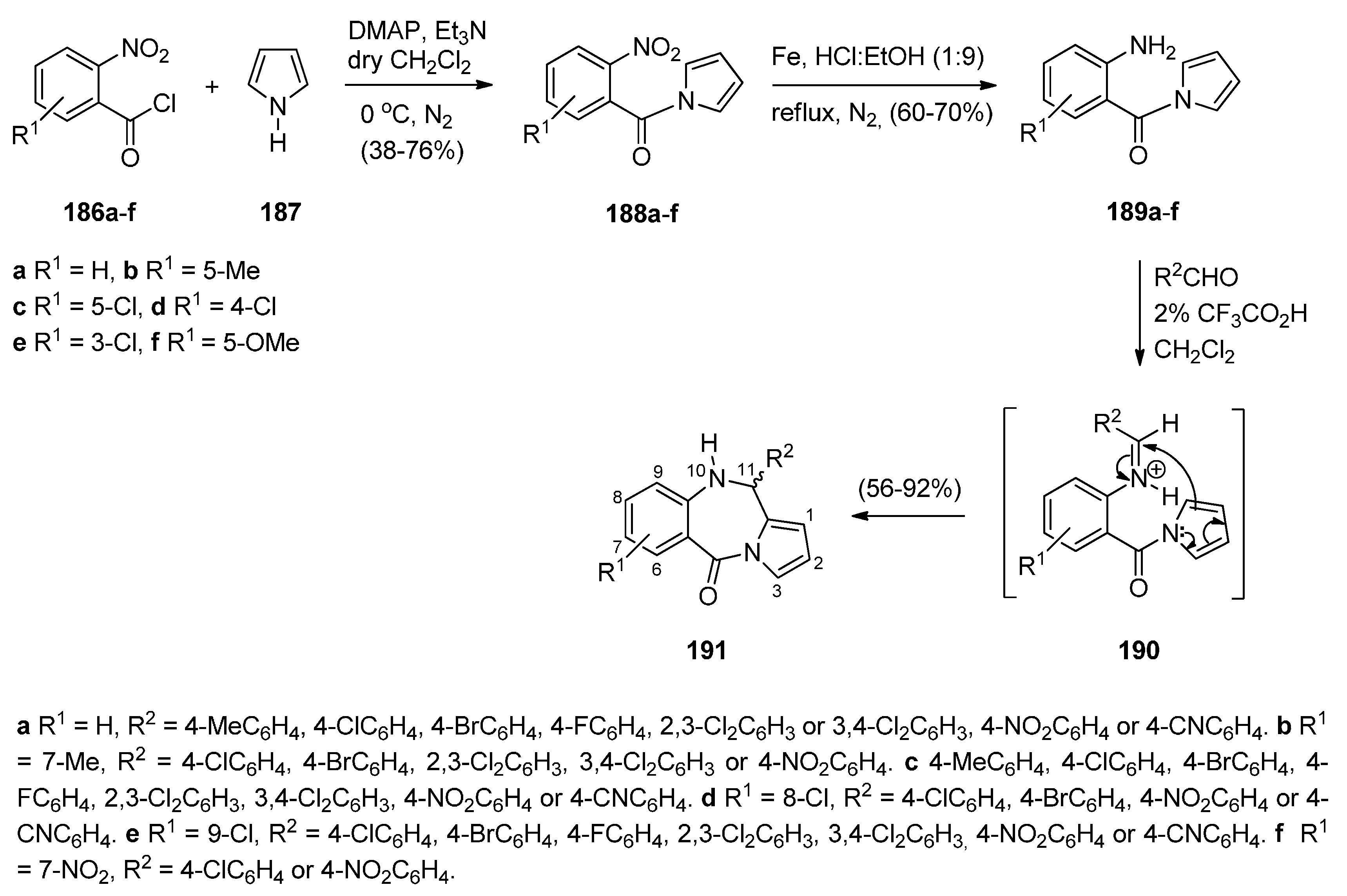
Cyclisation of N-(2-Aminobenzoyl)-1H-Pyrroles with Aldehydes.


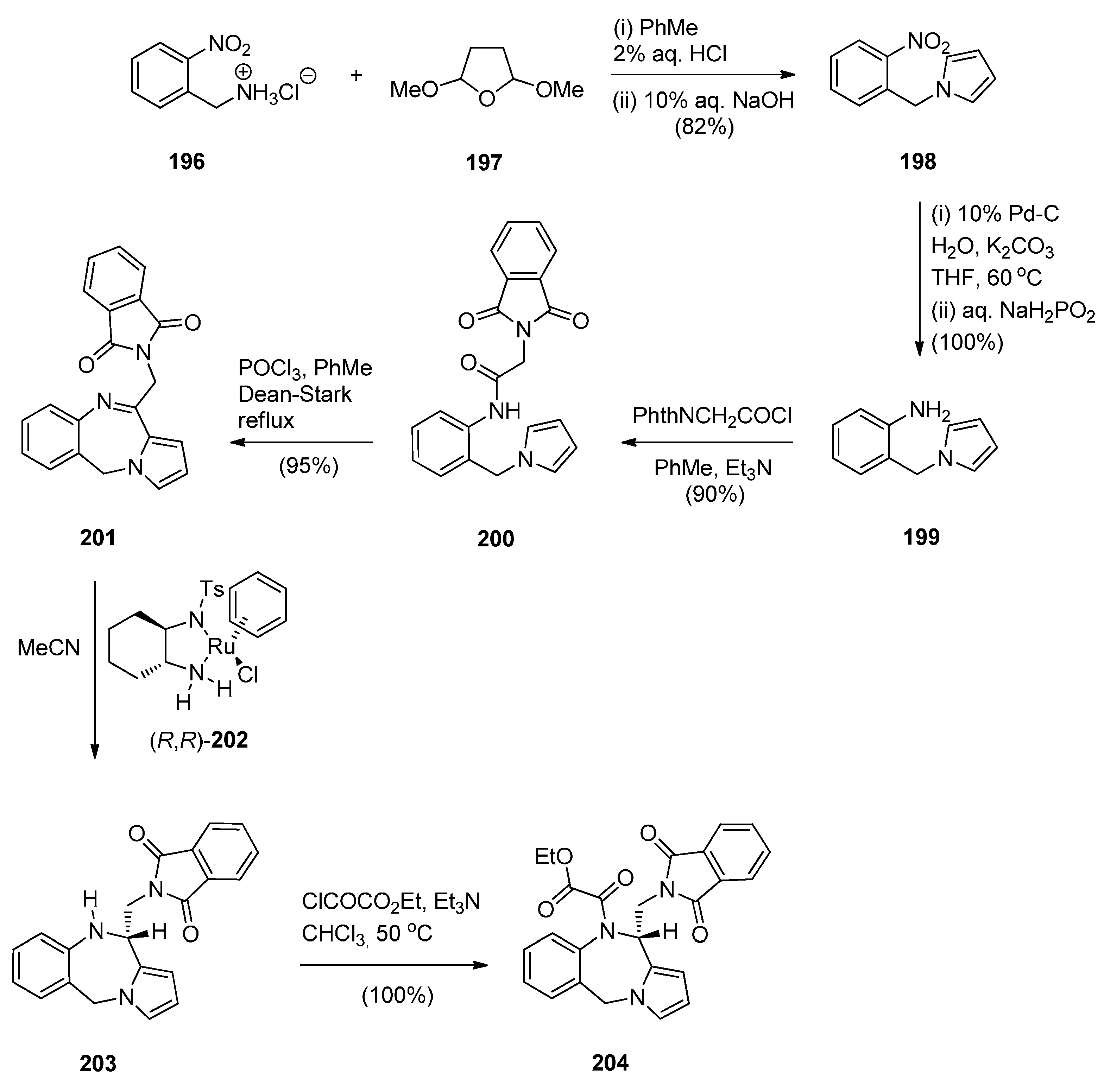
Cyclisation of 2-(N-Phthalimido)-N-[2-(1H-pyrrol-1-yl-methyl)phenyl]-Acetamide

2.1.5. Pyrrolo[2,1-c][1,4]benzodiazepine-3-ones with a Non-Aromatic Pyrrole Ring
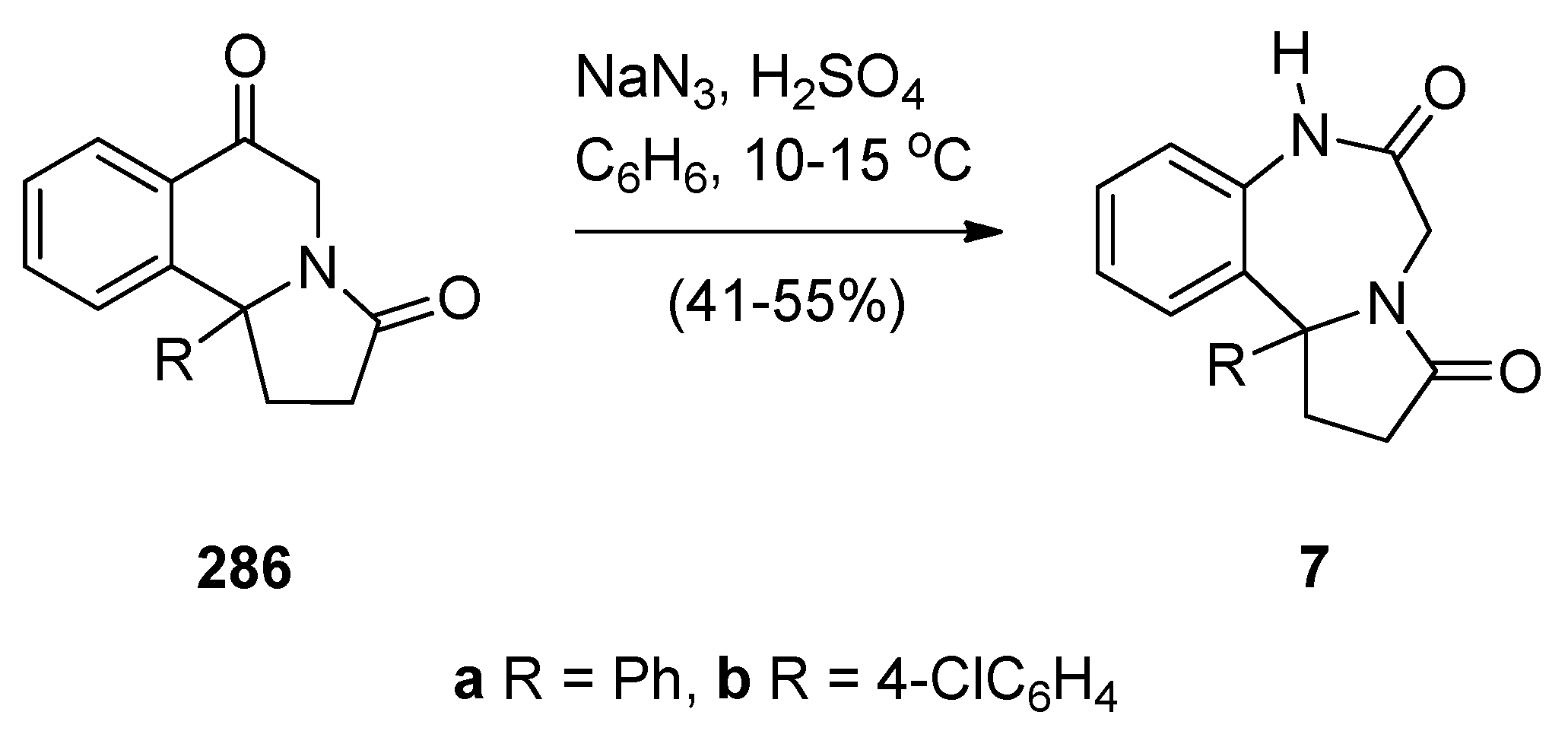
Reductive Cyclisation of in Situ Generated 1-(2-aminobenzyl)-5-aroylpyrrolidin-2-ones


2.2. Pyrrolo[1,2-a][1,4]benzodiazepines
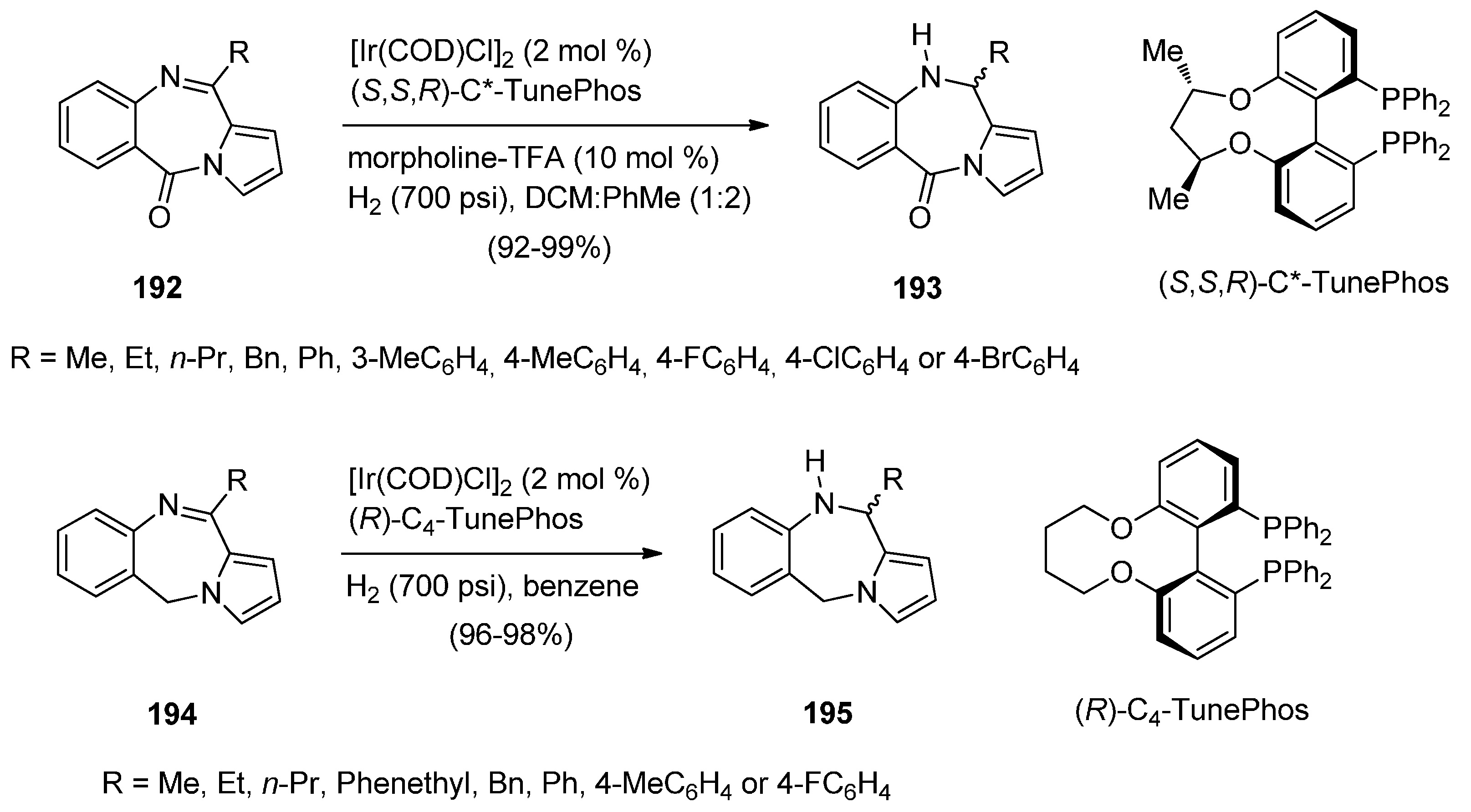
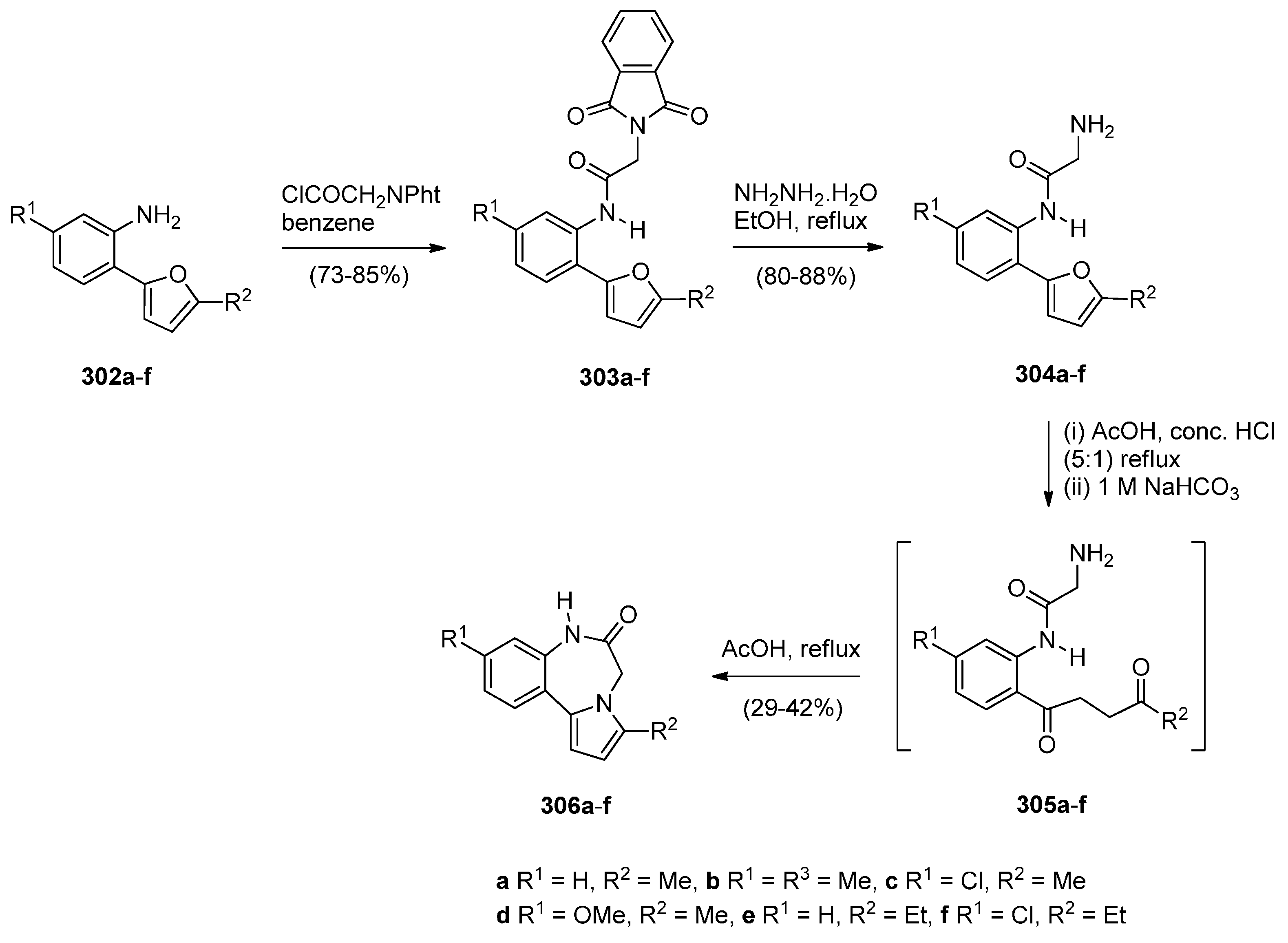
2.2.1. Pyrrolo[1,2-a][1,4]benzodiazepines with an Aromatic Pyrrole Ring
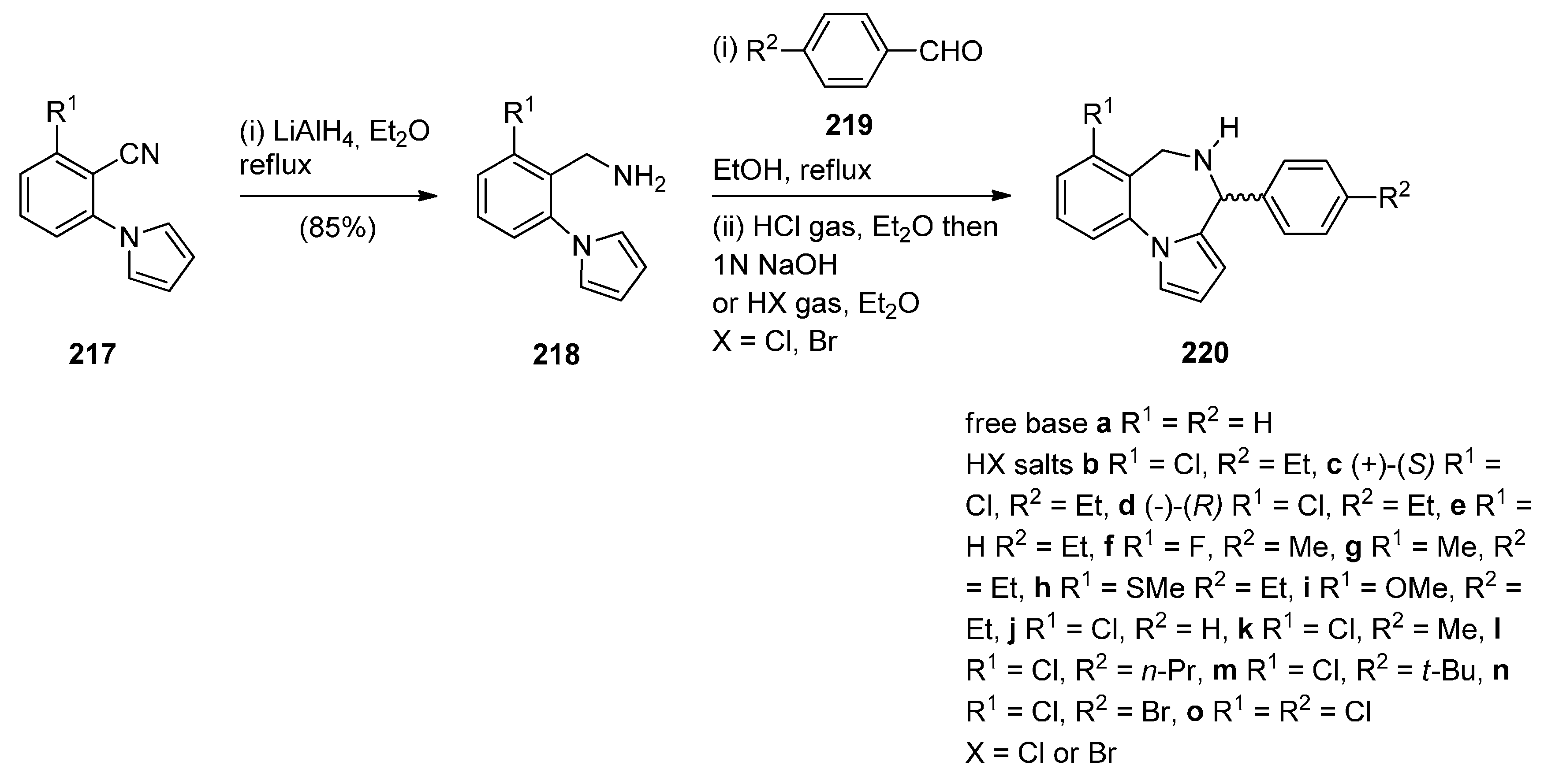
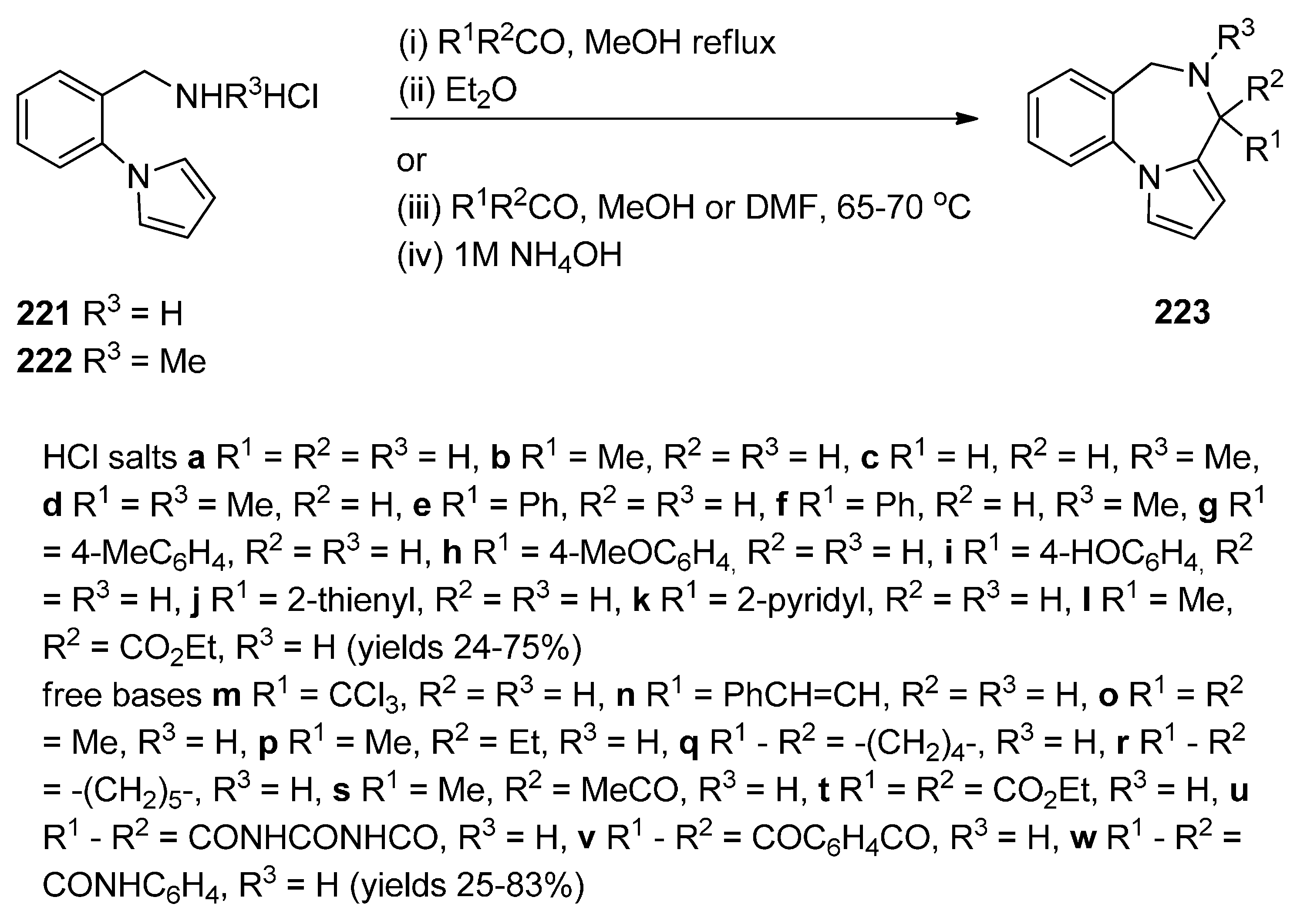
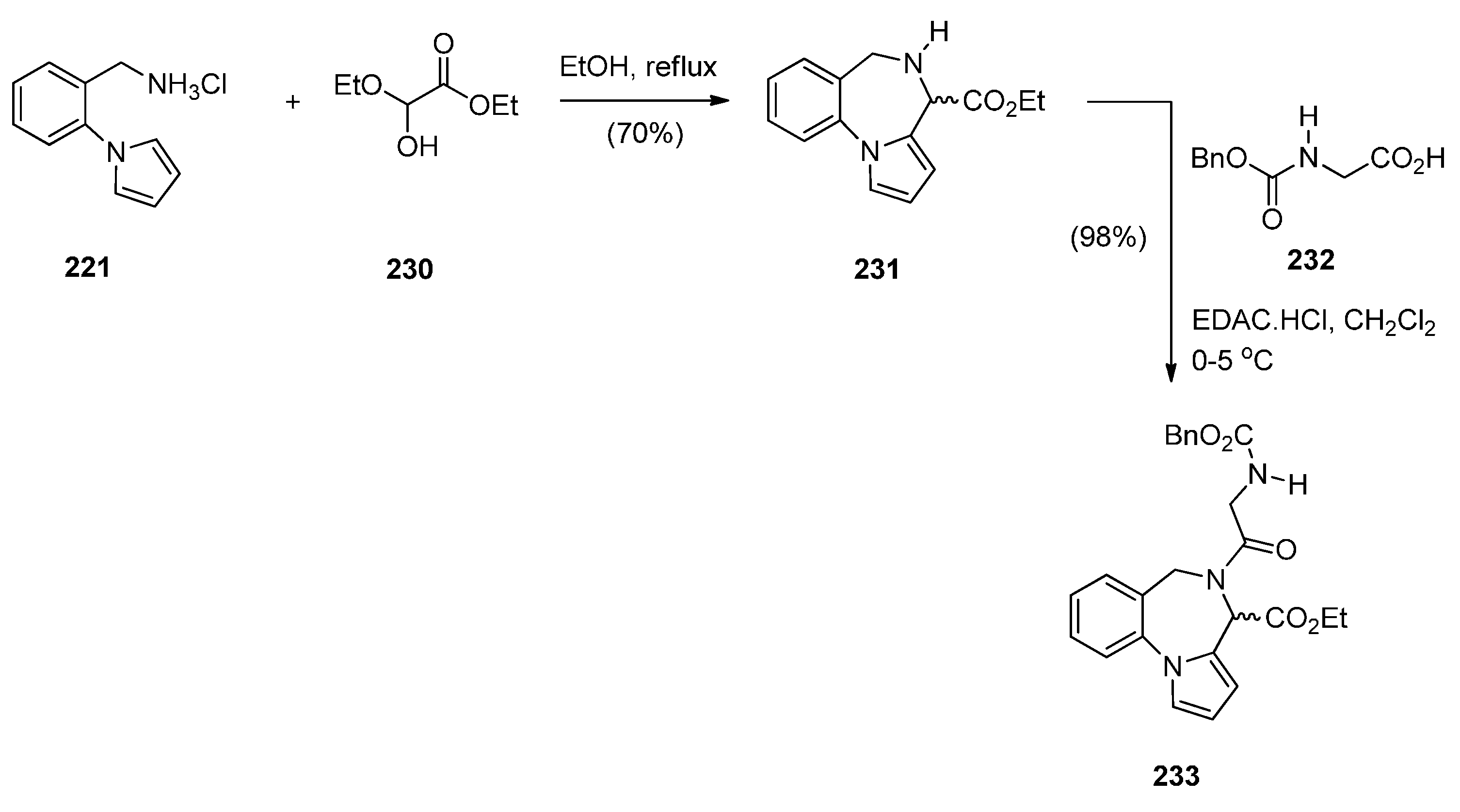
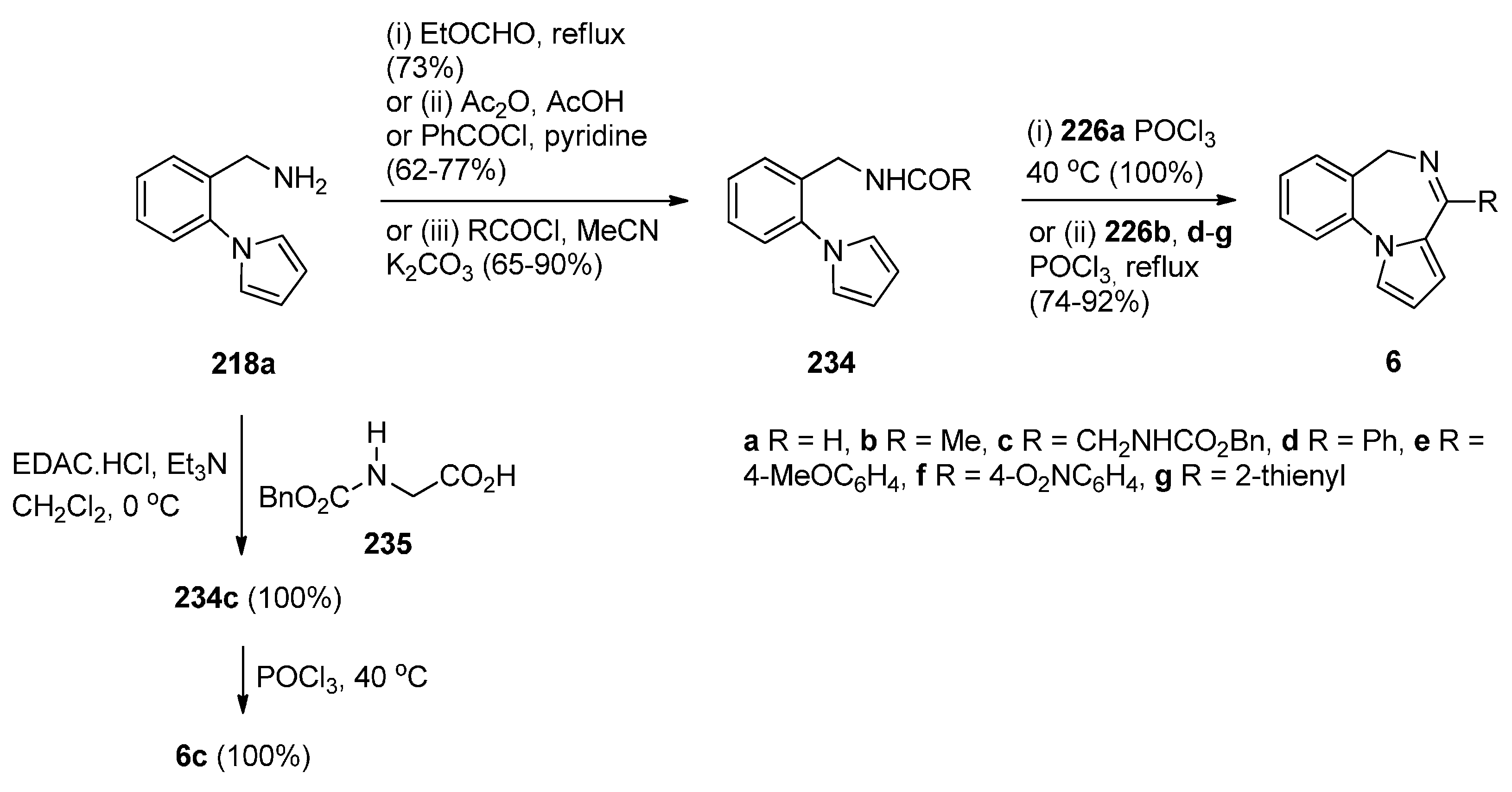
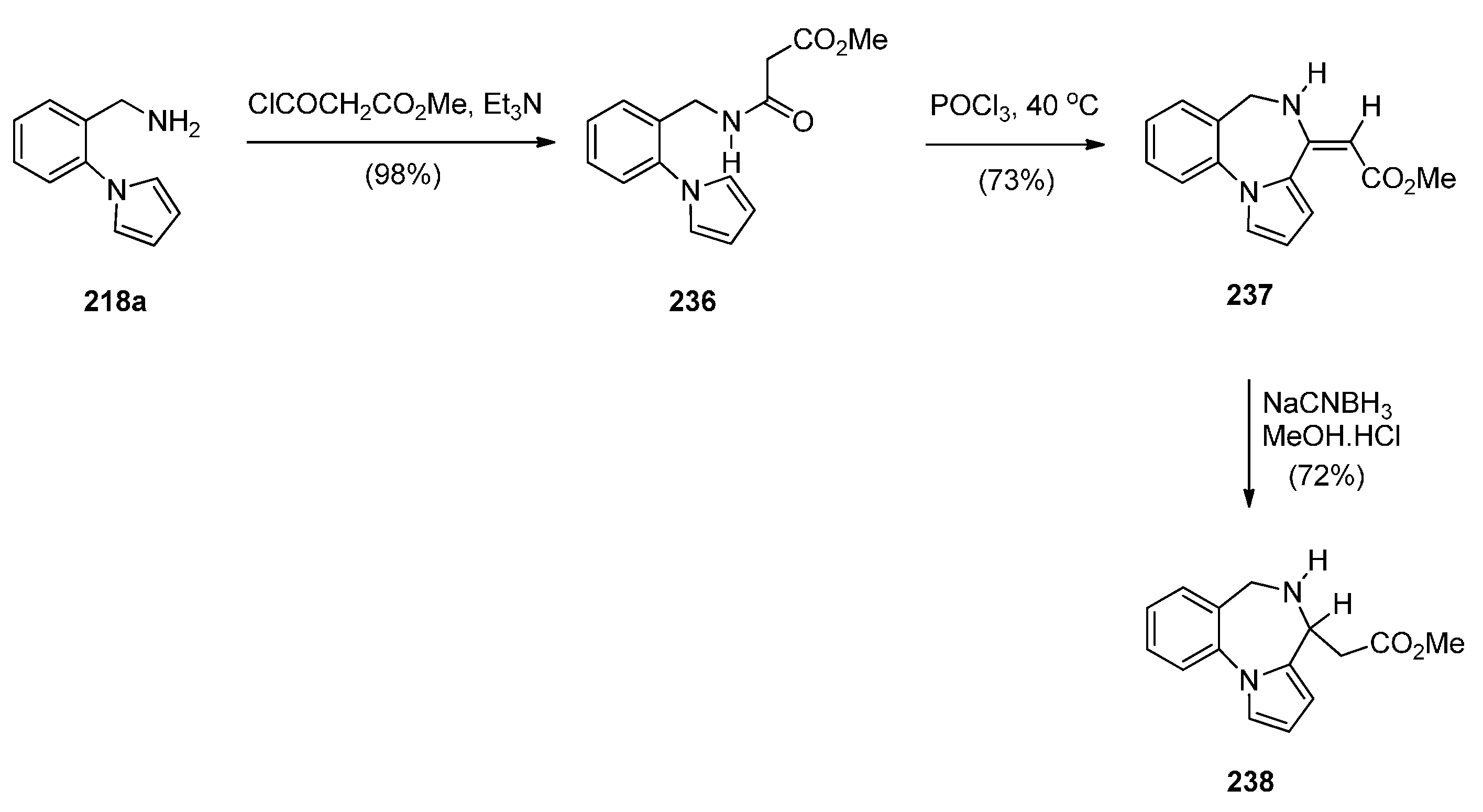
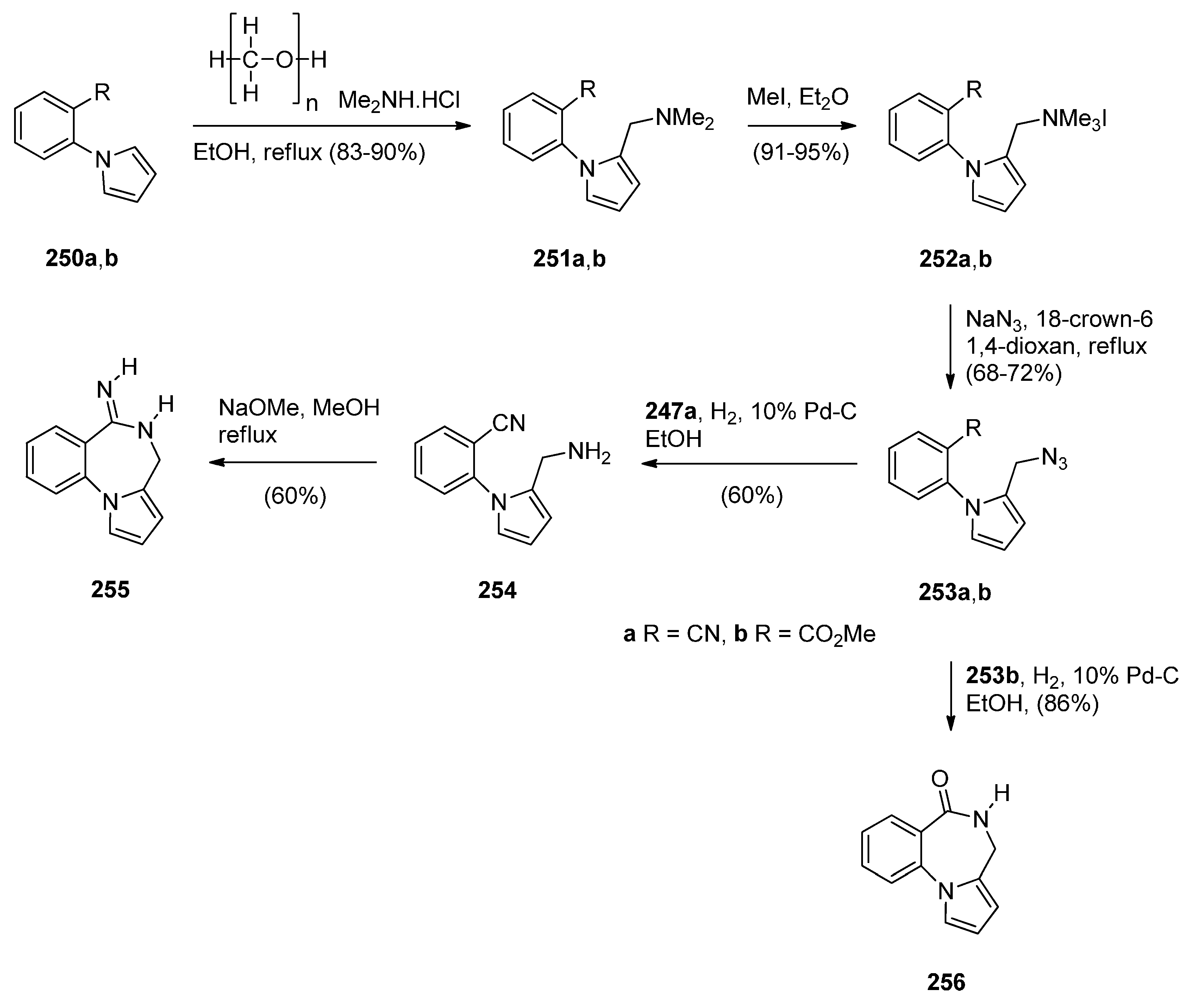
Cyclisation of 1-(2-Aminomethylphenyl)pyrrole with Carbonyl Compounds or a Hemiacetal




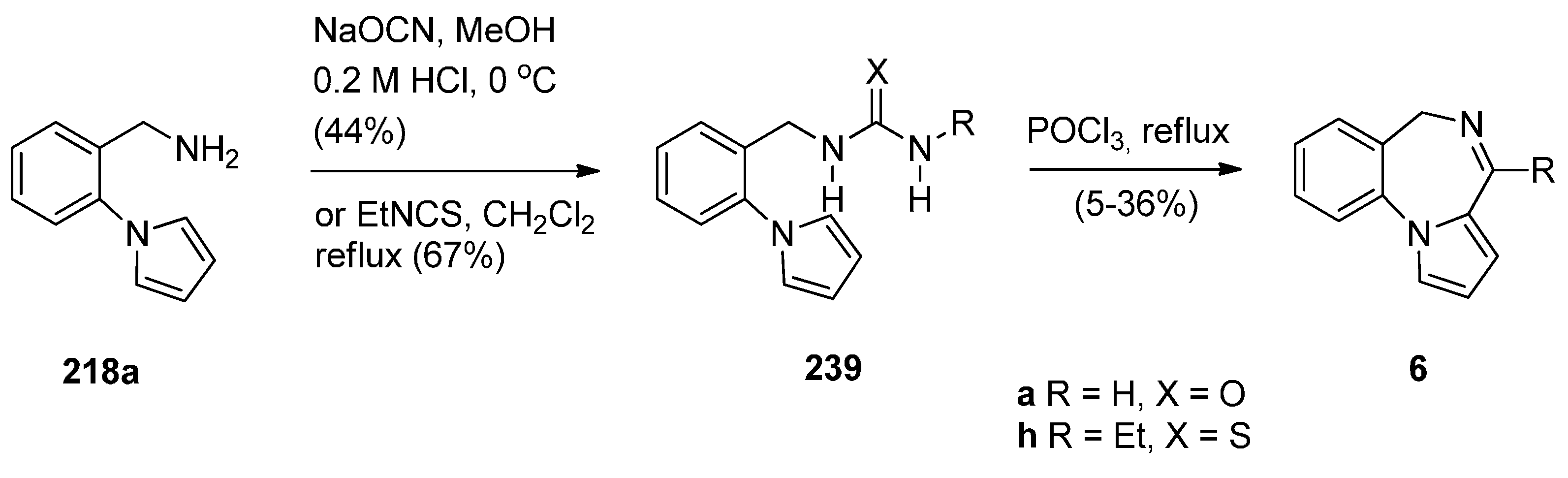
Cyclisation of Amide, Urea And Thiourea Derivatives of 1-(2-Aminomethylphenyl)pyrrole



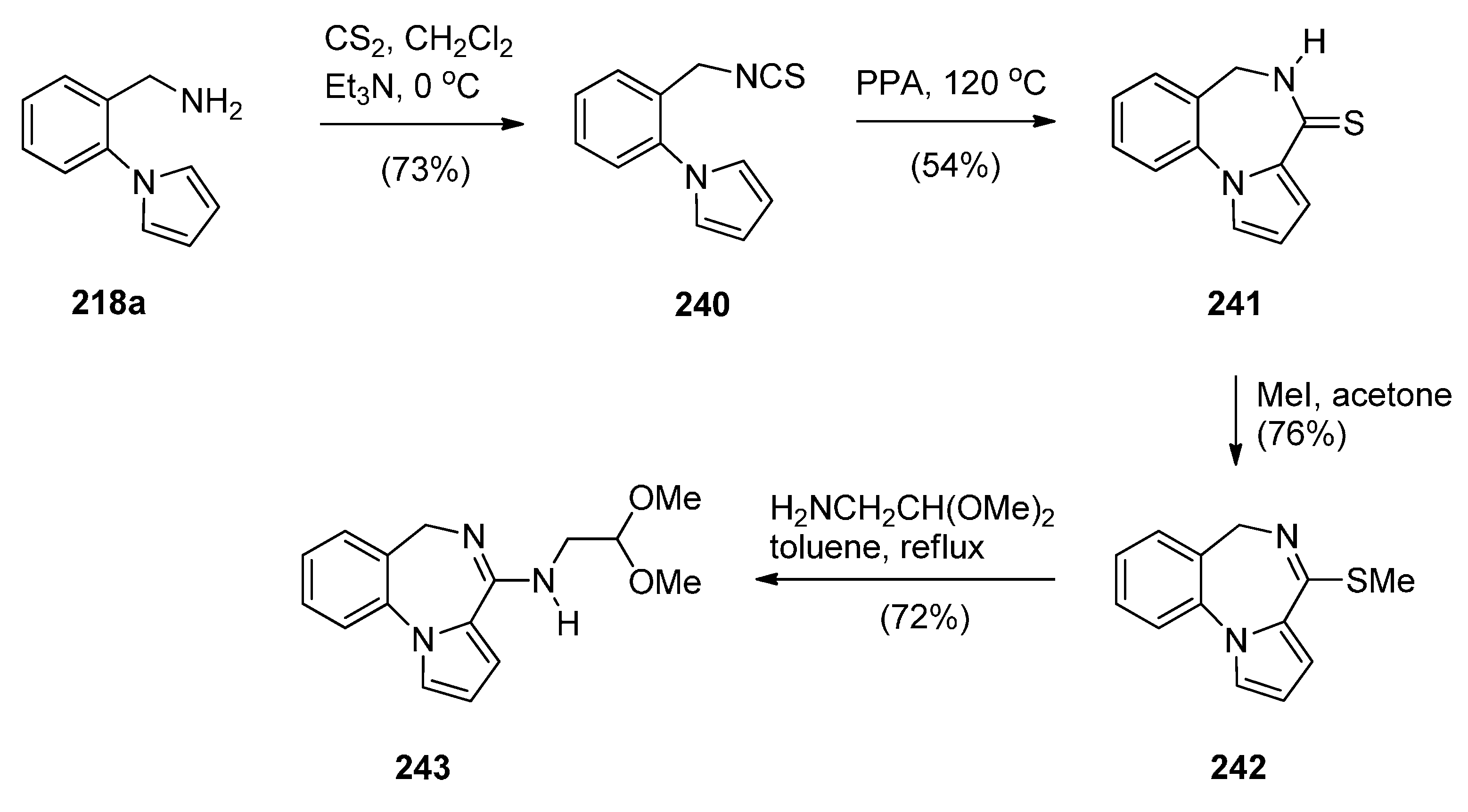
Cyclisation of the Isothiocyanate Derivative of 1-(2-Aminomethyl-phenyl)pyrrole

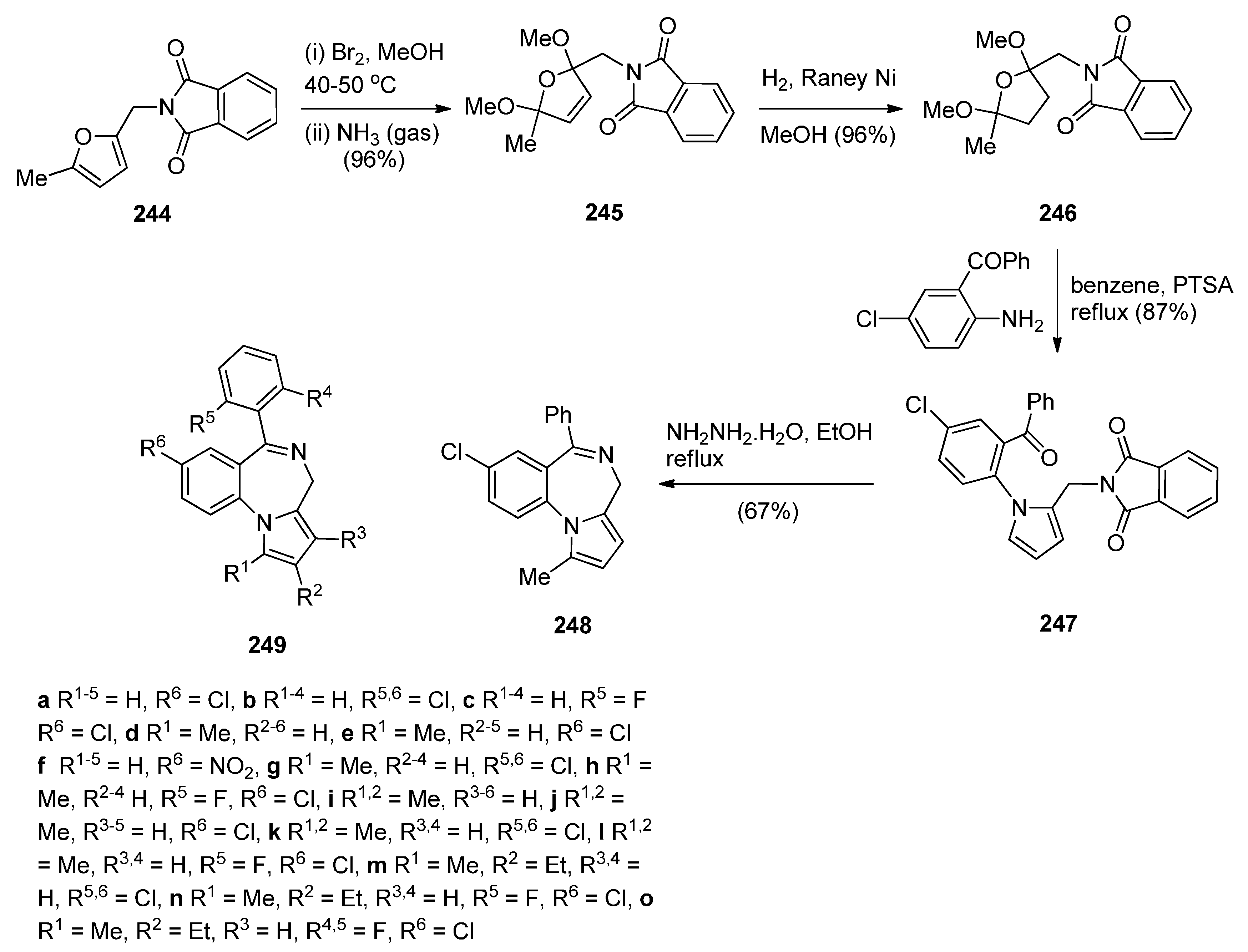
Cyclocondensation of 2-Aminomethyl-1-(2-aroyl)pyrrole Derivatives


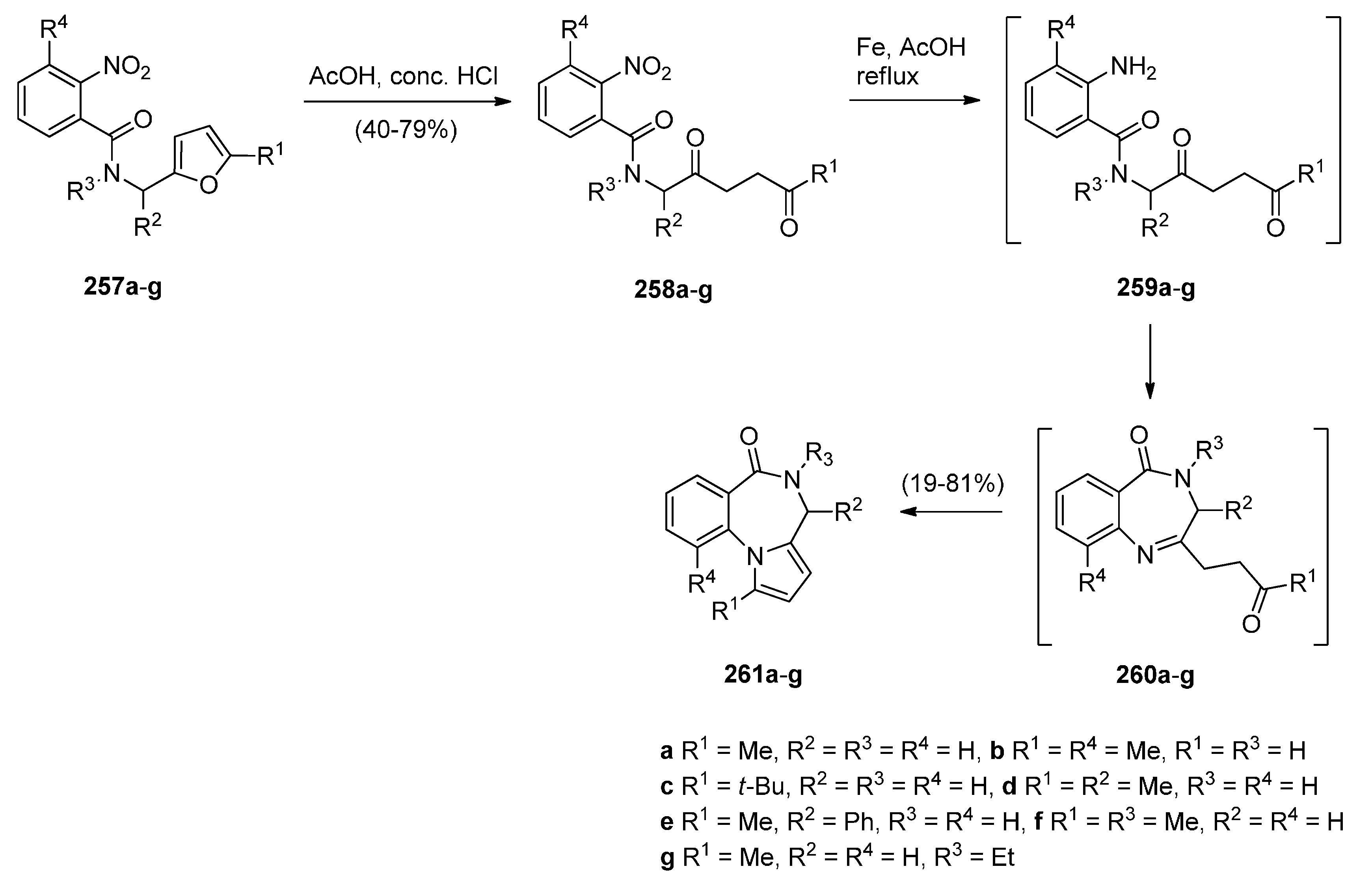
Cyclocondensation of N-(2,5-Dicarbonylalkyl)-2-nitrobenzamide Precursors Then Formation of Pyrrole Ring
2.2.2. Pyrrolo[1,2-a][1,4]benzodiazepines with a Non-Aromatic Pyrrole Ring

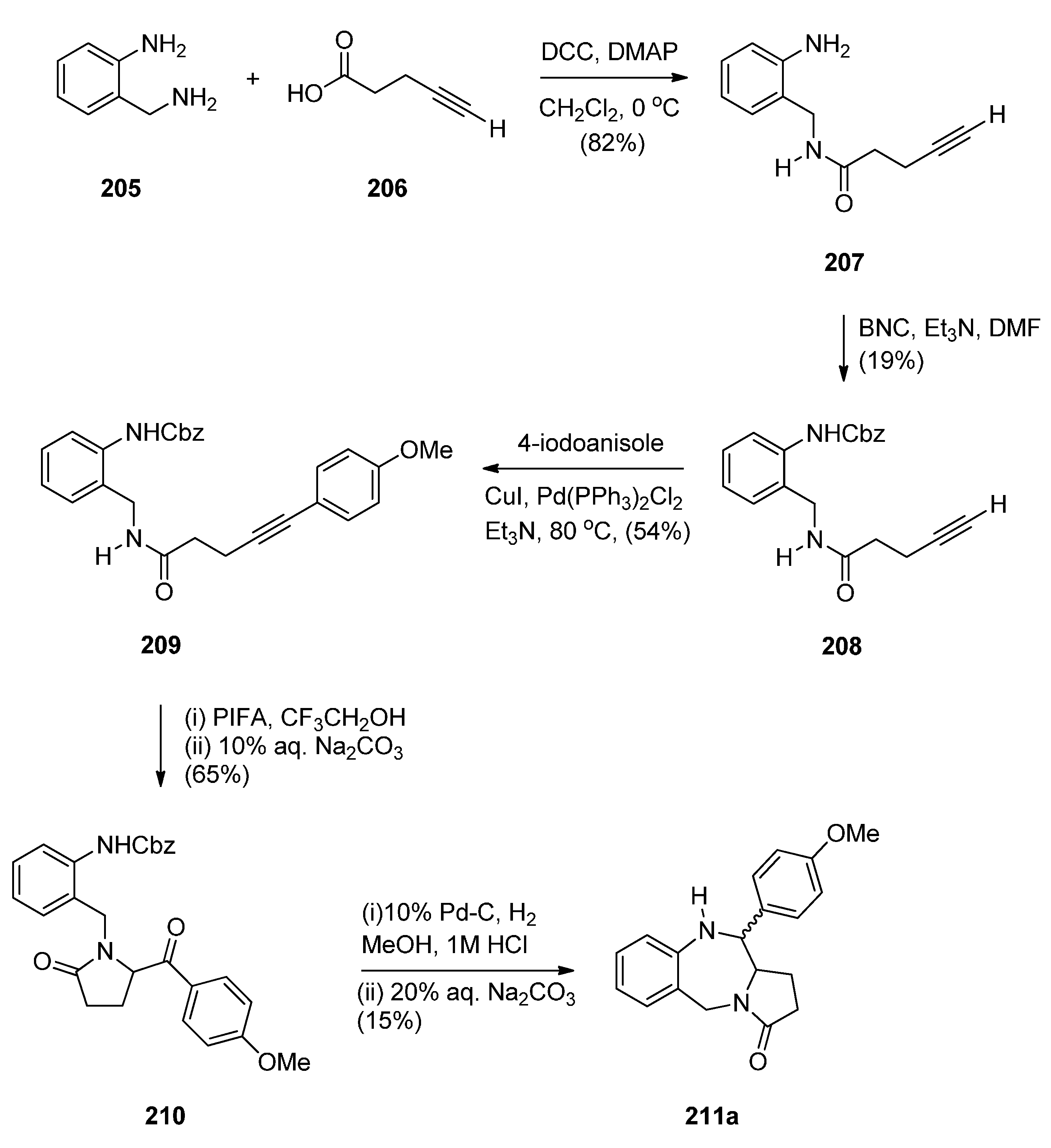
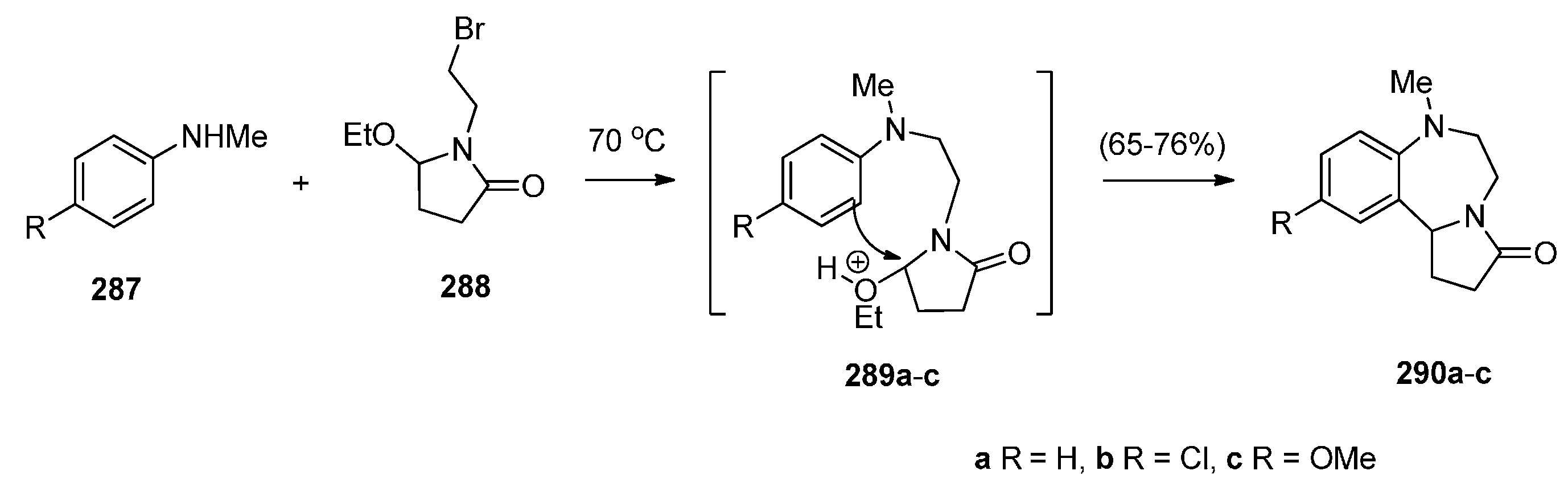
Cyclisation of a 2-Fluoro-N-(pyrrolidin-2-ylmethyl)benzamide Precursor

Rearrangement of Intermediate 3,4′-Dioxospiro[pyrrolidine-1,1′-quinazoline]ylides



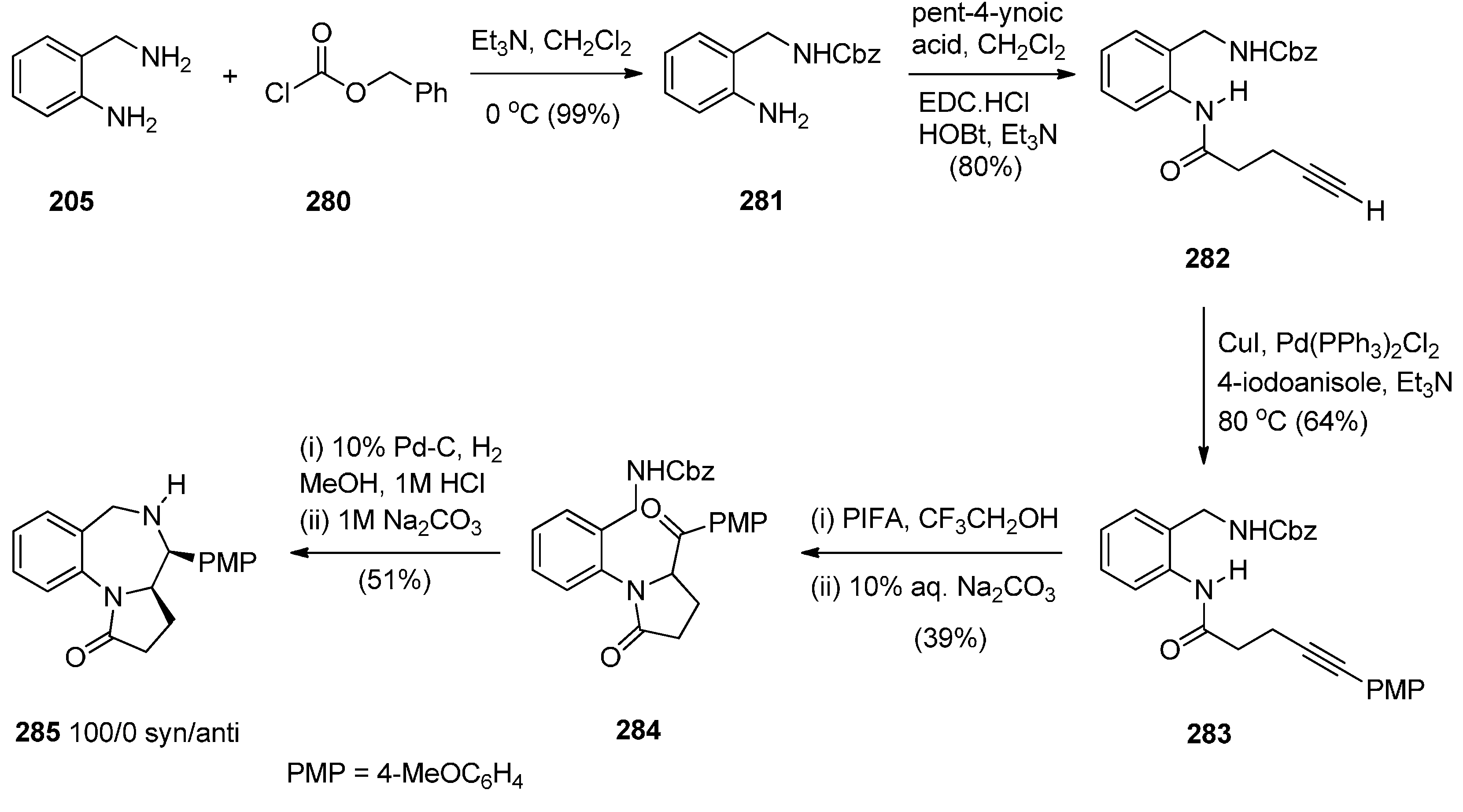
Reductive Cyclisation of 1-[2-(Aminomethyl)phenyl]-5-(4-methoxybenzoyl)pyrrolidin-2-one

2.3. Pyrrolo[1,2-d][1,4]benzodiazepines
2.3.1. Pyrrolo[1,2-d][1,4]benzodiazepines with a Non-Aromatic Pyrrole Ring
Rearrangement of Pyrrolo[2,1-a]isoquinolinediones

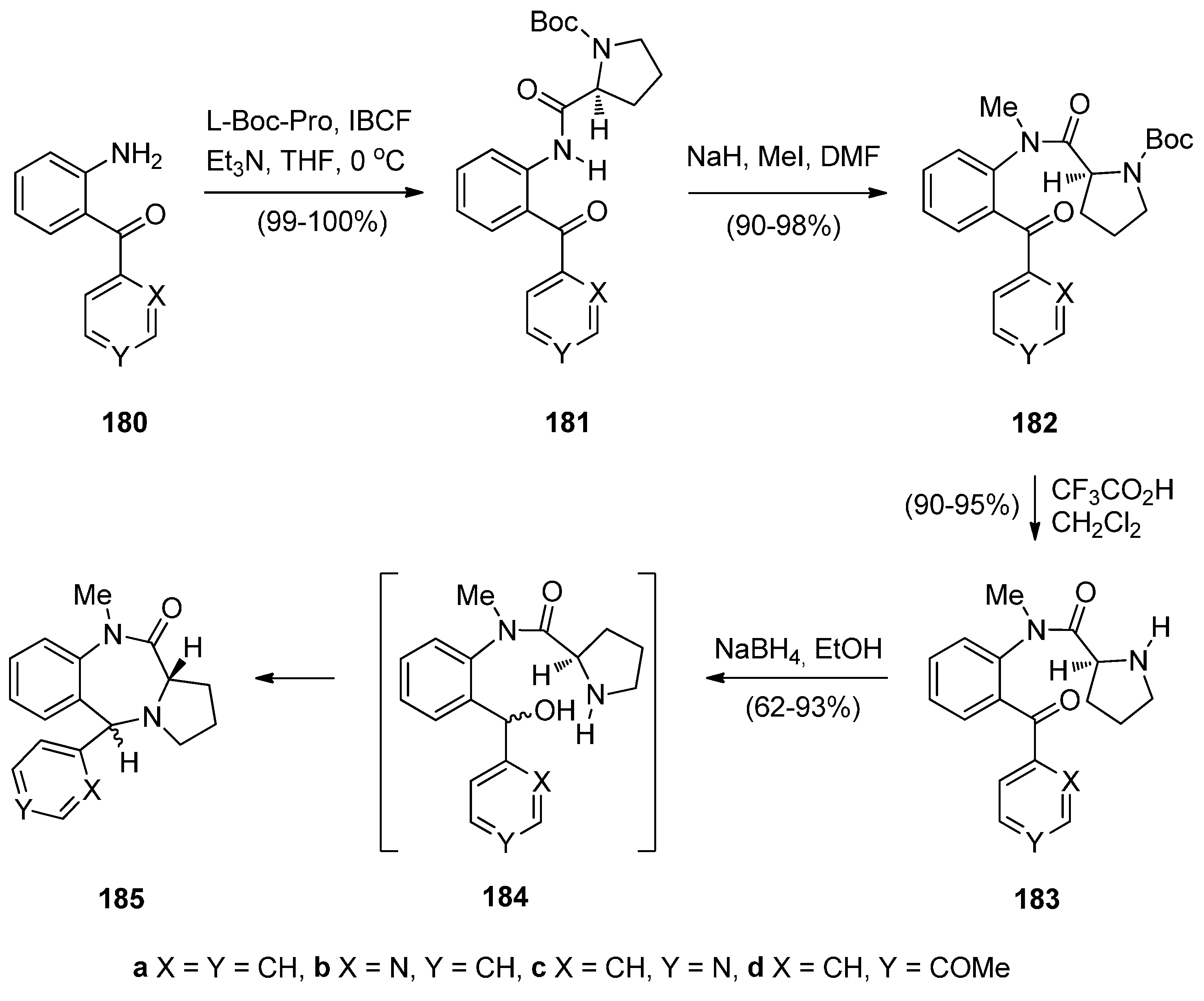
Cyclisation of 1-(2-Anilinoethyl)-5-ethoxypyrrolidin-2-ones

2.3.2. Pyrrolo[1,2-d][1,4]benzodiazepines with an Aromatic Pyrrole Ring
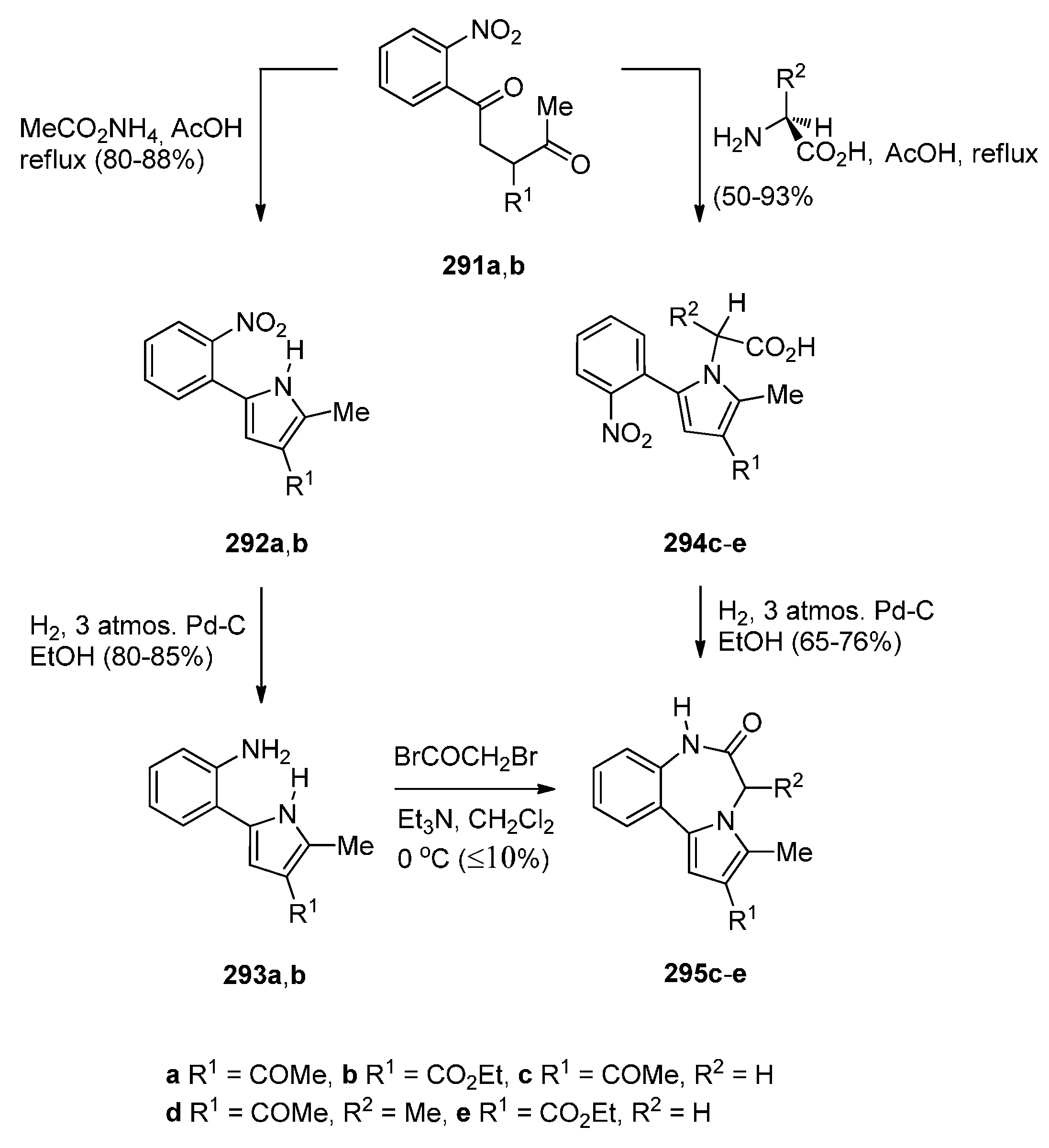
Reductive Cyclisation of [2-Methyl-5-(2-nitrophenyl)pyrrol-1-yl]acetic Acids

Cyclisation of N-(2-Carbonylphenyl)glycinamide Precursors Then Formation of Pyrrole Ring



3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Leimgruber, W.; Batcho, A.D.; Czajkowski, R.C. Total synthesis of anthramycin. J. Am. Chem. Soc. 1968, 90, 5641–5643. [Google Scholar] [CrossRef] [PubMed]
- Cheeseman, G.W.H.; Rafiq, M. Further cyclisation reactions of 1-arylpyrroles. J. Chem. Soc. C 1971. [Google Scholar] [CrossRef]
- Yamawaki, Y.; Watanabe, M.; Yamamura, S.; Saito, S. Studies on synthetic drugs. II. Syntheses of pyrrolo[1,2-d][1,4]benzodiazepine derivatives. Yakugaku Zasshi 1977, 97, 135–142. [Google Scholar]
- Leimgruber, W.; Stefanovic, V.; Shenker, F.; Karr, A.; Berger, J. Isolation and characterization of anthramycin, a new antitumour antibiotic. J. Am. Chem. Soc. 1965, 87, 5791–5793. [Google Scholar] [CrossRef] [PubMed]
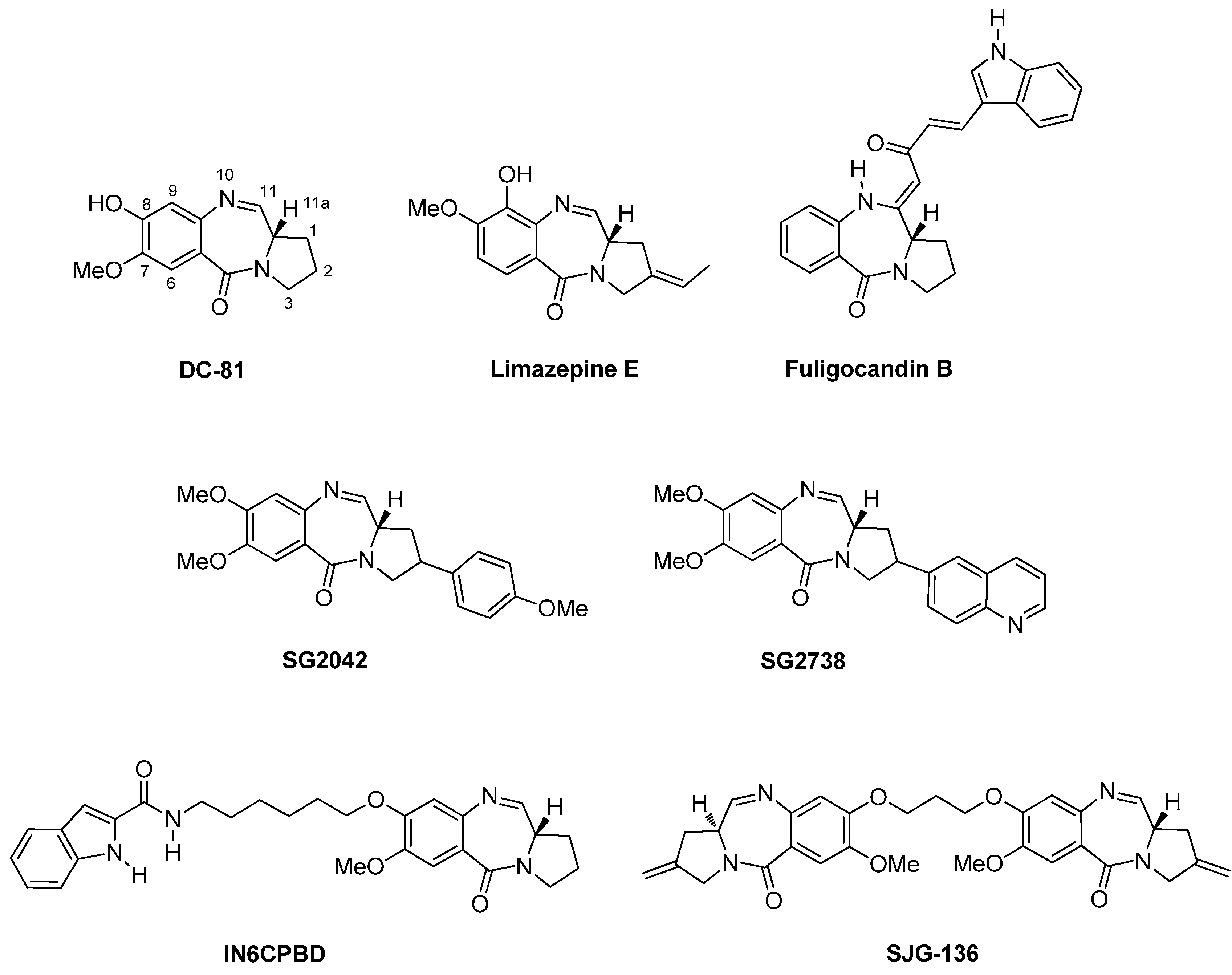
- Thurston, D.E. Advances in the study of pyrrolo[2,1-c][1,4]benzodiazepine (PBD) antitumour antibiotics. In Molecular Aspects of Anticancer Drug-DNA Interactions; Neidle, S., Waring, M.J., Eds.; The Macmillan Press Ltd.: London, UK, 1993; pp. 54–88. [Google Scholar]
- Arora, S.K. Structural investigations of mode of action of drugs. II. Molecular structure of anthramycin methyl ether monohydrate. Acta Crystallogr. B 1979, 35, 2945–2948. [Google Scholar] [CrossRef]
- Hurley, L.H.; Reck, T.; Thurston, D.E.; Langley, D.R.; Holden, K.G.; Hertzberg, R.P.; Hoover, J.R.E.; Gallagher, G., Jr.; Faucette, L.F. Pyrrolo[1,4]benzodiazepine antitumour antibiotics: Relationship of DNA alkylation and sequence specificity to the biological activity of natural and synthetic compounds. Chem. Res. Toxicol. 1988, 1, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Fotso, S.; Zabriskie, T.M.; Proteau, P.J.; Flatt, P.M.; Santosa, D.A.; Mahmud, T. Limazepines A–F, pyrrolo[1,4]benzodiazepine antibiotics from an Indonesian Micrococcus sp. J. Nat. Prod. 2009, 72, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, S.; Yamamoto, Y.; Hayashi, M.; Komiyama, K.; Ishibashi, M. Cycloanthranilylproline-Derived Constituents from a Myxomycete Fuligo candida. Chem. Pharm. Bull. 2004, 52, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.-P.; Yu, H.-S.; Sung, P.-J.; Tsai, F.-Y.; Shen, Y.-K.; Long-Sen Chang, L.-S.; Wang, J.-J. DC-81-Indole Conjugate Agent Induces Mitochondria Mediated Apoptosis in Human Melanoma A375 Cells. Chem. Res. Toxicol. 2007, 20, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Thurston, D.E.; Bose, D.S. Synthesis of DNA interactive pyrrolo[2,1-c][1,4]benzodiazepines. Chem. Rev. 1994, 94, 433–465. [Google Scholar] [CrossRef]
- Antonow, D.; Thurston, D.E. Synthesis of DNA-interactive pyrrolo[2,1-c][1,4]benzodiazepines (PBDs). Chem. Rev. 2011, 111, 2815–2864. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.A. The development of pyrrolobenzodiazepines as antitumour agents. Expert Opin. Investig. Drugs 2011, 20, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Gerratana, B. Biosynthesis, synthesis, and biological activities of pyrrolobenzodiazepines. Med. Res. Rev. 2012, 32, 254–293. [Google Scholar] [CrossRef] [PubMed]
- Kaliszczak, M.; Antonow, D.; Patel, K.I.; Howard, P.; Jodrell, D.I.; Thurston, D.E.; Guichard, S.M. Optimization of the antitumor activity of sequence-specific pyrrolobenzodiazepine derivatives based on their affinity for ABC transporters. Am. Assoc. Pharm. Sci. J. 2010, 12, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.M.; Vassoler, H.; James, C.H.; Thurston, D.E. DNA sequence preference and adduct orientation of pyrrolo[2,1-c][1,4]benzodiazepine antitumor agents. ACS Med. Chem. Lett. 2010, 1, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Janjigian, Y.Y.; Lee, W.; Kris, M.G.; Miller, V.A.; Krug, L.M.; Azzoli, C.G.; Senturk, E.; Calcutt, M.W.; Rizvi, N.A. A phase I trial of SJG-136 (NSC#694501) in advanced solid tumours. Cancer Chemother. Pharmacol. 2010, 65, 833–838. [Google Scholar] [PubMed]
- Hartley, J.A.; Hamaguchi1, A.; Coffils, M.; Martin, C.R.H.; Suggitt, M.; Chen, Z.; Gregson, S.J.; Masterson, L.A.; Tiberghien, A.C.; Hartley, J.M.; et al. SG2285, a novel C2-aryl-substituted pyrrolobenzodiazepine dimer pro-drug that cross-links DNA and exerts highly potent antitumour activity. Cancer Res. 2010, 70, 6849–6858. [Google Scholar] [CrossRef] [PubMed]
- Reid, J.M.; Buhrow, S.A.; Kuffel, M.J.; Jia, L.; Spanswick, V.J.; Hartley, J.A.; Thurston, D.E.; Tomaszewski, J.E.; Ames, M.M. Pharmacokinetics, pharmacodynamics and metabolism of the dimeric pyrrolobenzodiazepine SJG-136 in rats. Cancer Chemother. Pharmacol. 2011, 68, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Rosado, H.; Rahman, K.M.; Feuerbaum, E.-A.; Hinds, J.; Thurston, D.E.; Taylor, P.W. The minor groove-binding agent ELB-21 forms multiple interstrand and intrastrand covalent cross-links with duplex DNA and displays potent bactericidal activity against methicillin-resistant Staphylococcus aureus. J. Antimicrob. Chemother. 2011, 66, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.M.; James, C.H.; Bui, T.T.T.; Drake, A.F.; Thurston, D.E. Observation of a single-stranded DNA/pyrrolobenzodiazepine adduct. J. Am. Chem. Soc. 2011, 133, 19376–19385. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.M.; James, C.H.; Thurston, D.E. Effect of base sequence on the DNA cross-linking properties of pyrrolobenzodiazepine (PBD) dimers. Nucleic Acids Res. 2011, 39, 5800–5812. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.M.; James, C.H.; Thurston, D.E. Observation of the reversibility of a covalent pyrrolobenzodiazepine (PBD) DNA adduct by HPLC/MS and CD spectroscopy. Org. Biomol. Chem. 2011, 9, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Yonemoto, I.T.; Li, W.; Khullar, A.; Reixach, N.; Gerratana, B. Mutasynthesis of a potent anticancer sibiromycin analogue. ACS Chem. Biol. 2012, 7, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.A.; Hochhauser, D. Small molecule drugs—Optimizing DNA damaging agent-based therapeutics. Curr. Opin. Pharm. 2012, 12, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.A.; Hamaguchi, A.; Suggitt, M.; Gregson, S.J.; Thurston, D.E.; Howard, P.W. DNA interstrand cross-linking and in vivo antitumor activity of the extended pyrrolo[2,1-c][1,4]benzodiazepine dimer SG2057. Investig. New Drugs 2012, 30, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.M.; Rosado, H.; Moreira1, J.B.; Feuerbaum, E.-A.; Fox, K.R.; Stecher, E.; Howard, P.W.; Gregson, S.J.; James, C.H.; de la Fuente, M.; et al. Antistaphylococcal activity of DNA-interactive pyrrolobenzodiazepine (PBD) dimers and PBD-biaryl conjugates. J. Antimicrob. Chemother. 2012, 67, 1683–1696. [Google Scholar] [CrossRef] [PubMed]
- Thurston, D.E.; Vassoler, H.; Jackson, P.J.M.; James, C.H.; Rahman, K.M. Effect of hairpin loop structure on reactivity, sequence preference and adduct orientation of a DNA-interactive pyrrolo[2,1-c][1,4]benzodiazepine (PBD) antitumour agent. Org. Biomol. Chem. 2015, 13, 4031–4040. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, S.C.; Burke, P.J.; Lyon, R.P.; Meyer, D.W.; Sussman, D.; Anderson, M.; Hunter, J.H.; Leiske, C.I.; Miyamoto, J.B.; Nicholas, N.D.; et al. A potent anti-CD70 antibody-drug conjugate combining a dimeric pyrrolobenzodiazepine drug with site-specific conjugation technology. Bioconjug. Chem. 2013, 24, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, M.S.K.; Walter, R.B.; Jeffrey, S.C.; Burke, P.J.; Yu, C.; Kostner, H.; Stone, S.; Ryan, M.C.; Sussman, D.; Lyon, R.P.; et al. SGN-CD33A: A novel CD33-targeting antibody–drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood 2013, 122, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Flygare, J.A.; Thomas, H.; Pillow, T.H.; Aristoff, P. Antibody-drug Conjugates for the treatment of cancer. Chem. Biol. Drug Des. 2013, 81, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.A. Antibody-Drug Conjugates Delivering DNA Cytotoxics. In Cancer Drug Design and Discovery, 2nd ed.; Neidle, S., Ed.; Academic Press: London, UK, 2014; pp. 479–490. [Google Scholar]
- Rahman, K.M.; Corcoran, D.B.; Bui, T.T.T.; Jackson, P.J.M.; David, E.; Thurston, D.E. Pyrrolobenzodiazepines (PBDs) do not bind to DNA G-quadruplexes. PLoS ONE 2014, 9, e105021. [Google Scholar] [CrossRef] [PubMed]
- Schneditz, G.; Rentner, J.; Roier, S.; Pletz, J.; Herzog, K.A.T.; Troeger, R.B.H.; Schild, S.; Weber, H.; Breinbauer, R.; Gorkiewicz, G.; et al. Enterotoxicity of a nonribosomal peptide causes antibiotic-associated colitis. Proc. Natl. Acad. Sci. USA 2014, 111, 13181–13186. [Google Scholar] [CrossRef] [PubMed]
- Rettig, M.; Langel, W.; Kamal, A.; Weisz, K. NMR structural studies on the covalent DNA binding of a pyrrolobenzodiazepine–naphthalimide conjugate. Org. Biomol. Chem. 2010, 8, 3179–3187. [Google Scholar] [CrossRef] [PubMed]
- Hopton, S.R.; Thompson, A.S. Nuclear Magnetic Resonance Solution Structures of Inter- and Intrastrand Adducts of DNA Cross-Linker SJG-136. Biochemistry 2011, 50, 4720–4732. [Google Scholar] [CrossRef] [PubMed]
- Raju, G.; Srinivas, R.; Reddy, V.S.; Idris, M.M.; Kamal, A.; Nagesh, N. Interaction of pyrrolobenzodiazepine (PBD) ligands with parallel intermolecular G-quadruplex complex using spectroscopy and ESI-MS. PLoS ONE 2012, 7, e35920. [Google Scholar] [CrossRef]
- Jackson, P.J.M.; James, C.H.; Jenkins, T.C.; Rahman, K.M.; David, E.; Thurston, D.E. Computational studies support the role of the C7-sibirosamine sugar of the pyrrolobenzodiazepine (PBD) sibiromycin in transcription factor inhibition. ACS Chem. Biol. 2014, 9, 2432–2440. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.; Jang, J.-H.; Choo, S.-J.; Kim, S.-O.; Kim, J.W.; Ko, S.-K.; Soung, N.-K.; Lee, J.-S.; Kim, C.J.; Oh, H.; et al. Boseongazepines A–C, pyrrolobenzodiazepine derivatives from a Streptomyces sp. 11A057. Bioorg. Med. Chem. Lett. 2014, 24, 1802–1804. [Google Scholar] [CrossRef] [PubMed]
- Corelli, F.; Massa, S.; Stefancich, G.; Ortenzi, G.; Artico, M.; Pantaleoni, G.C.; Palumbo, G.; Fanini, D.; Giorgi, R. Benzodiazepines with Both Sedative and Analgesic Activities. Eur. J. Med. Chem 1986, 21, 445–449. [Google Scholar]
- Mai, A.; di Santo, R.; Massa, S.; Artico, M.; Pantaleoni, G.C.; Giorgi, R.; Coppolino, M.F.; Barracchini, A. Pyrrolobenzodiazepines with antinociceptive activity: Synthesis and pharmacological activities. Eur. J. Med. Chem. 1995, 30, 593–601. [Google Scholar] [CrossRef]
- Massa, S.; Artico, M.; Mai, A.; Corelli, F.; Pantaleoni, G.C.; Giorgi, R.; Ottaviani, D.; Cagnotto, A. Pyrrolobenzodiazepine and related systems. I. Synthesis and pharmacological evaluation of new 5,6-dihydro-4H-pyrrolo[1,2-a][1,4]benzodiazepine derivatives. Farmaco 1990, 45, 1265–1281. [Google Scholar] [PubMed]
- Meerpoel, L.; van Gestel, J.; van Gerven, F.; Woestenborghs, F.; Marichal, P.; Sipido, V.; Terence, G.; Nash, R.; Corens, D.; Richards, R.D. Pyrrolo[1,2-a][1,4]benzodiazepine: A novel class of non-azole anti-dermatophyte anti-fungal agents. Bioorg. Med. Chem. Lett. 2005, 15, 3453–3458. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Kayama, Y.; Mori, T.; Itoh, K.; Fujimori, H.; Sunami, T.; Hashimoto, Y.; Ishimoto, S. Diazepines. 5. Synthesis and Biological Action of 6-Phenyl-4H-pyrrolo[1,2-a][1,4]benzodiazepines. J. Med. Chem. 1978, 21, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Massa, S.; Corelli, F.; Artico, M.; Mai, A.; Silvestri, R.; Pantaleoni, G.C.; Palumbo, G.; Fanini, D.; Giorgi, R. 5-Aroyl-5,6-dihydro-4H-pyrrolo[1,2-a][1,4]benzodiazepin-4-carboxylic acids: Synthesis and analgesic and neurobehavioral activity. Farmaco 1989, 44, 109–123. [Google Scholar] [PubMed]
- Massa, S.; Artico, M.; Mai, A.; Corelli, F.; Botta, M.; Tafi, A.; Pantaleoni, G.C.; Giorgi, R.; Coppolino, M.F.; Cagnotto, A.; et al. Pyrrolobenzodiazepines and related systems. 2. Synthesis and biological properties of isonoraptazepine derivatives. J. Med. Chem. 1992, 35, 4533–4541. [Google Scholar] [CrossRef] [PubMed]
- De Lucca, G.V.; Otto, M.J. Synthesis and anti-HIV activity of pyrrolo[1,2-d][1,4]benzodiazepine-6-ones. Bioorg. Med. Chem. Lett. 1992, 2, 1639–1644. [Google Scholar] [CrossRef]
- Kamal, A.; Rajender; Reddy, R.; Reddy, M.K.; Balakishan, G.; Shaik, T.B.; Chourasia, M.; Sastry, G.N. Remarkable enhancement in the DNA-binding ability of C2-fluoro substituted pyrrolo[2,1-c]-[1,4]benzodiazepines and their anticancer potential. Bioorg. Med. Chem. 2009, 17, 1557–1572. [Google Scholar] [CrossRef] [PubMed]
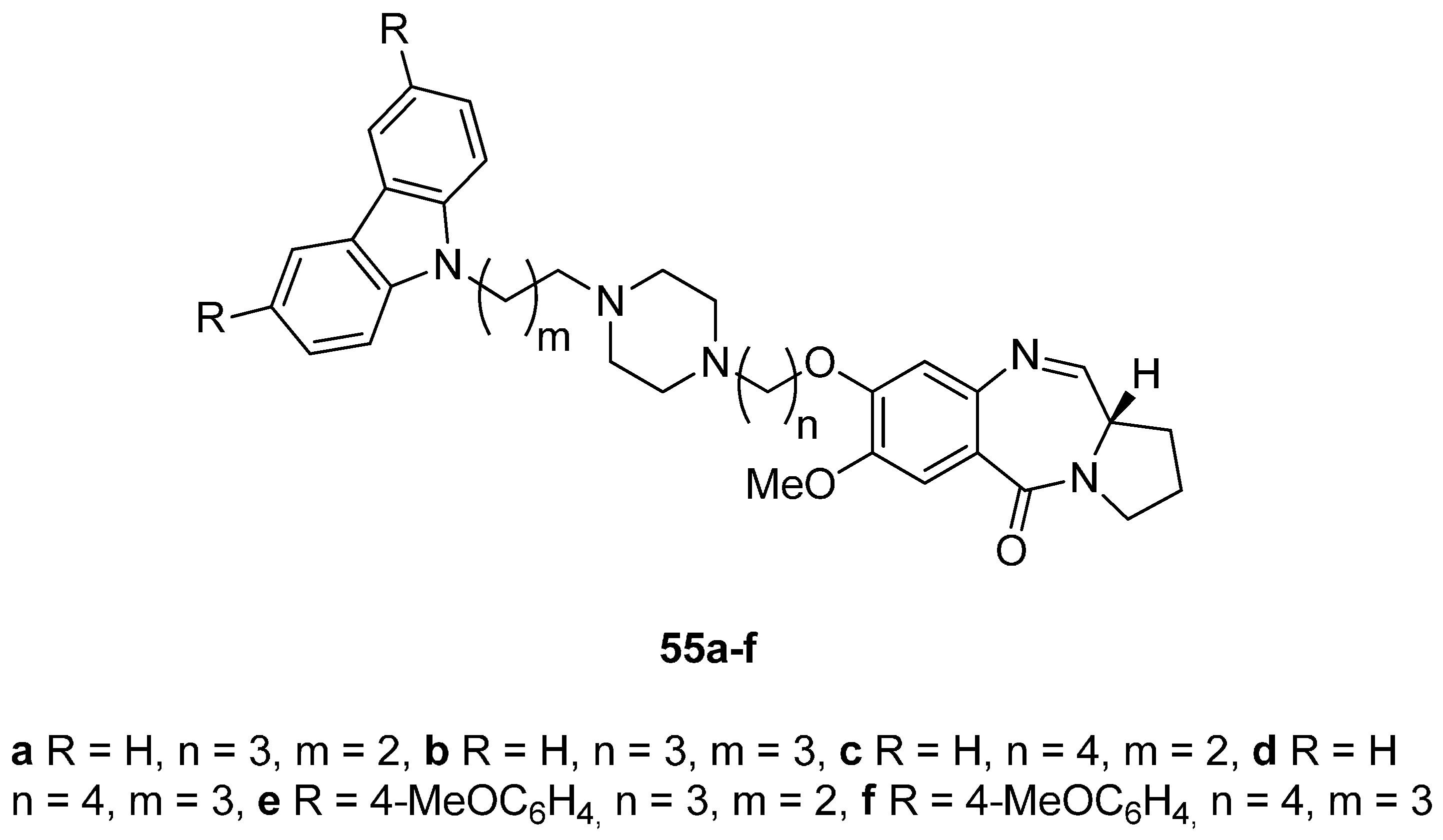
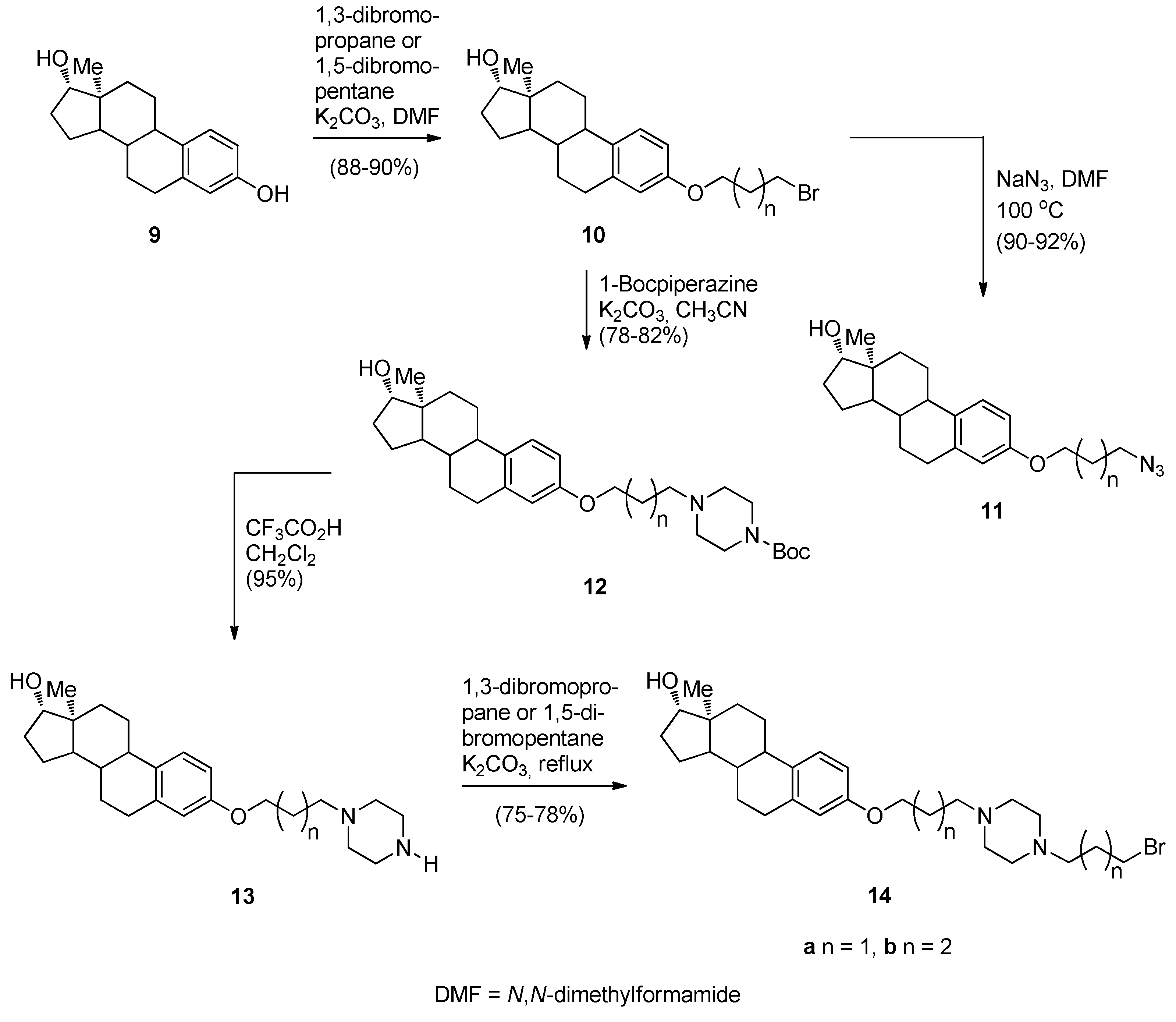
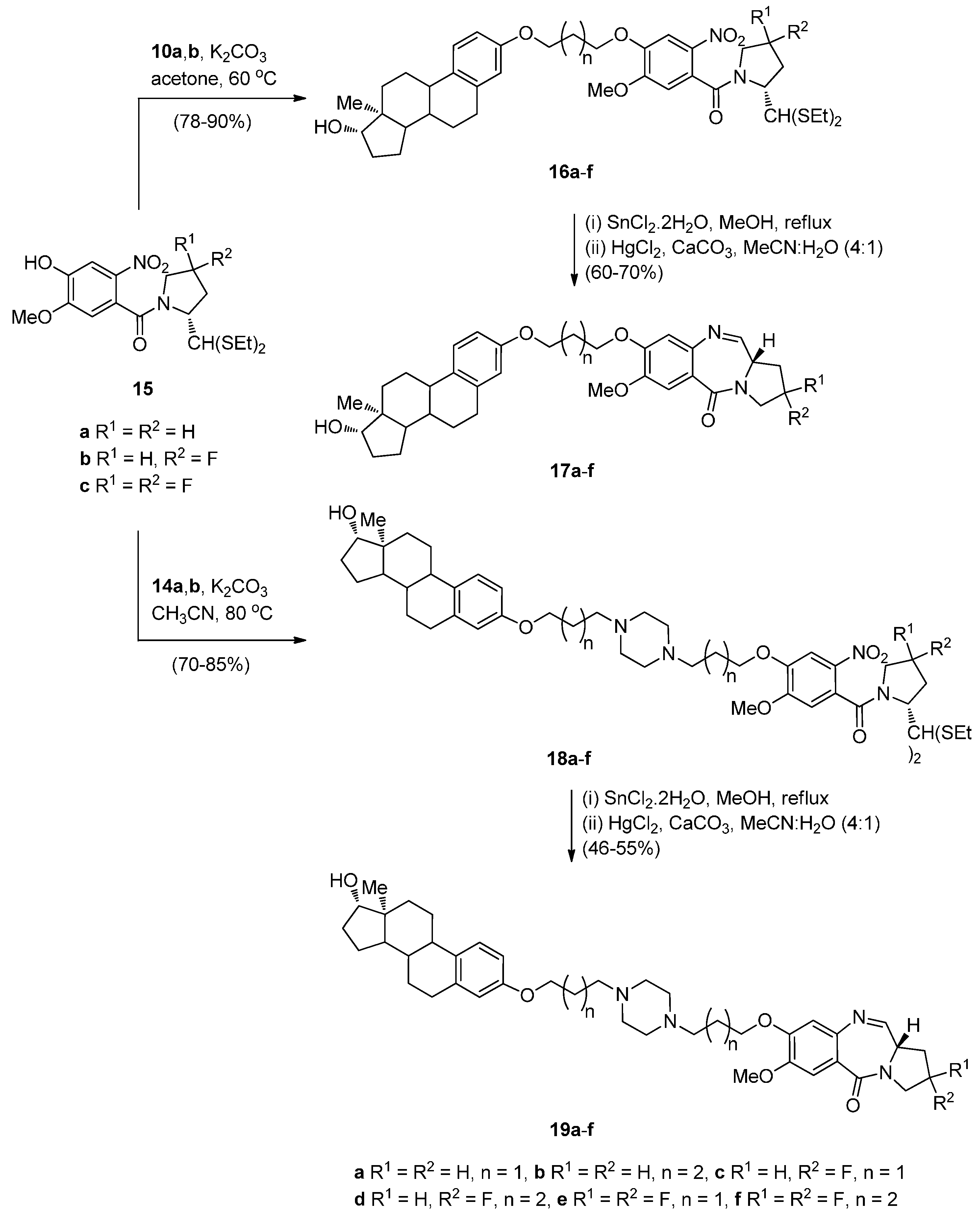
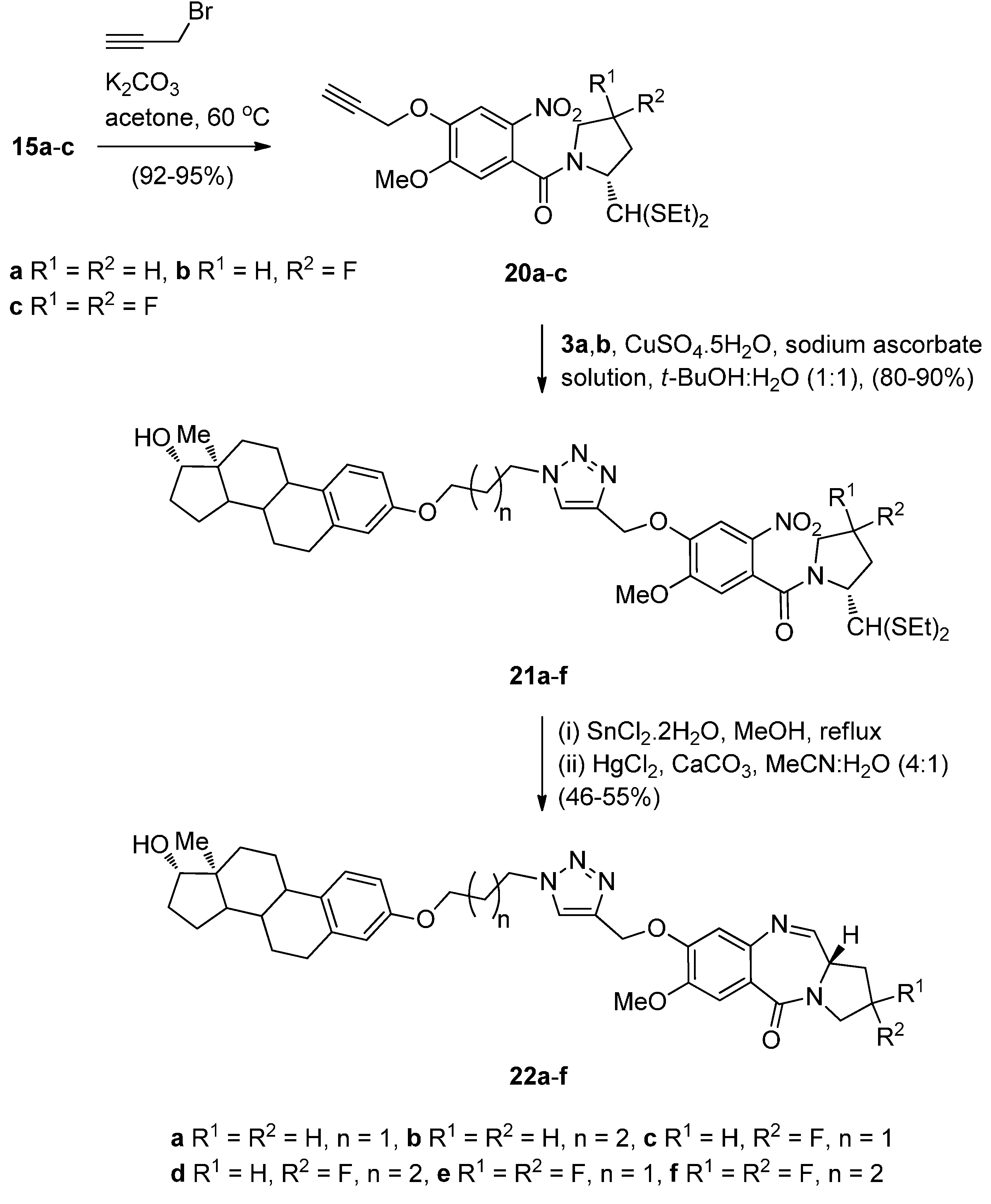
- Kamal, A.; Reddy, M.K.; Ramaiah, M.J.; Reddy, R.J.S.; Srikanth, Y.V.V.; Dastagiri, D.; Bharathi, E.V.; Pushpavalli, S.N.; Sarma, P.; Pal-Bhadra, M. Synthesis and biological evaluation of estradiol linked pyrrolo[2,1-c][1,4]benzodiazepine (PBD) conjugates as potential anticancer agents. Bioorg. Med. Chem. 2011, 19, 2565–2581. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Reddy, M.K.; Ramaiah, M.J.; Srikanth, Y.V.V.; Reddy, R.V.S.; Kumar, G.B.; Pushpavalli, S.N.; Bag, I.; Juvekar, A.; Sen, S.; et al. Synthesis of aryl-substituted naphthalene-linked pyrrolobenzodiazepine conjugates as potential anticancer agents with apoptosis-inducing ability. ChemMedChem 2011, 6, 1665–1679. [Google Scholar] [CrossRef] [PubMed]
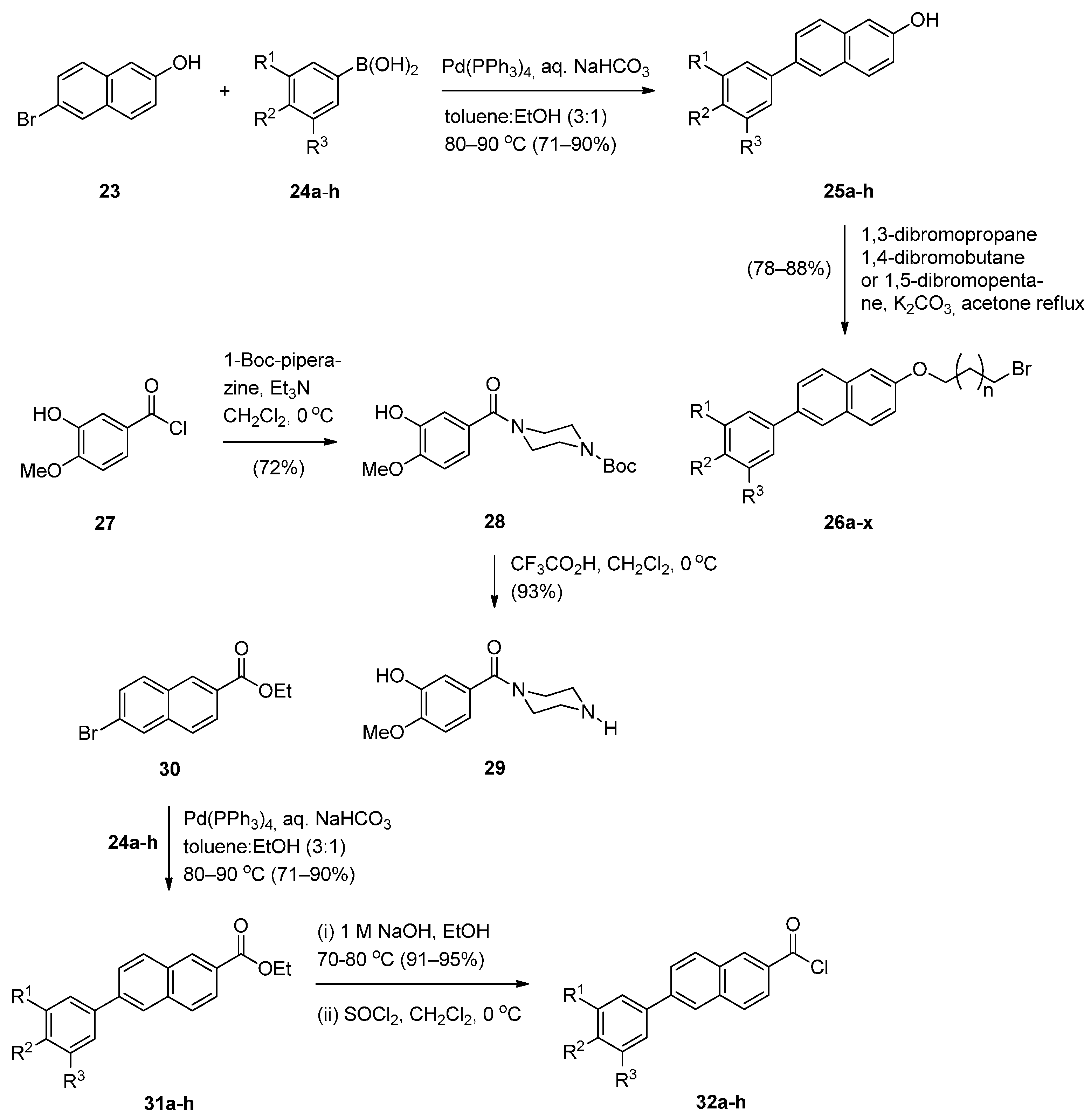
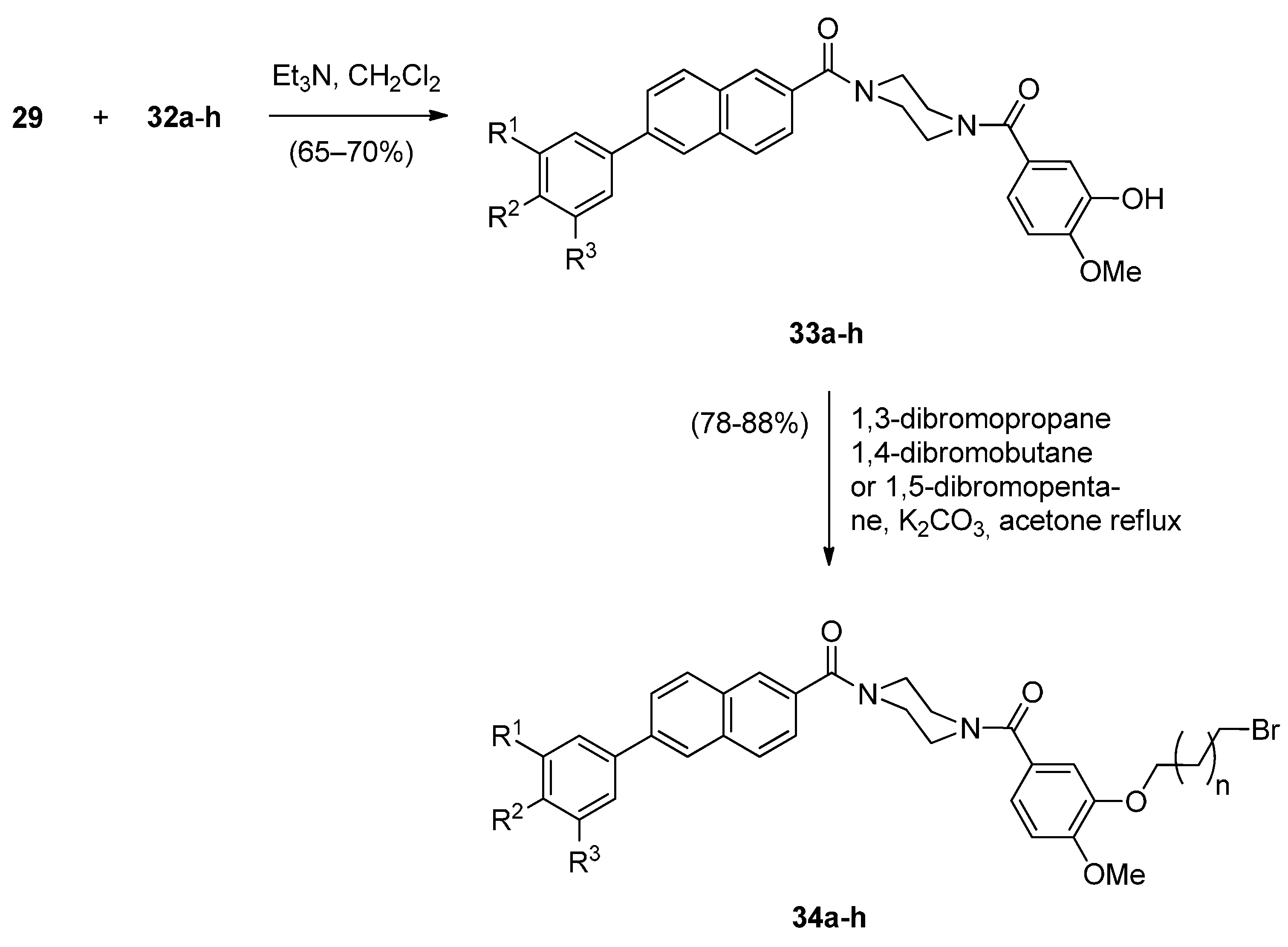
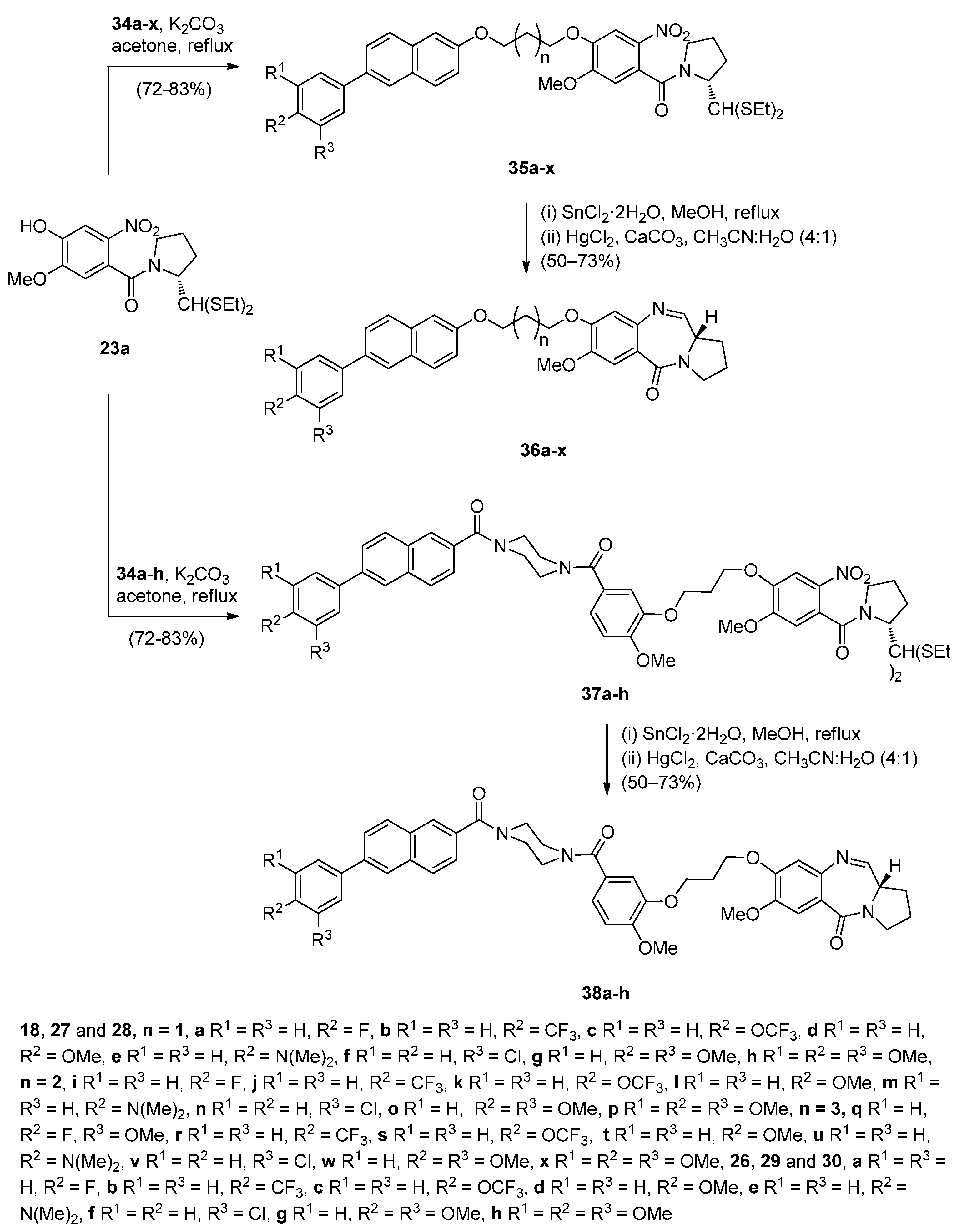
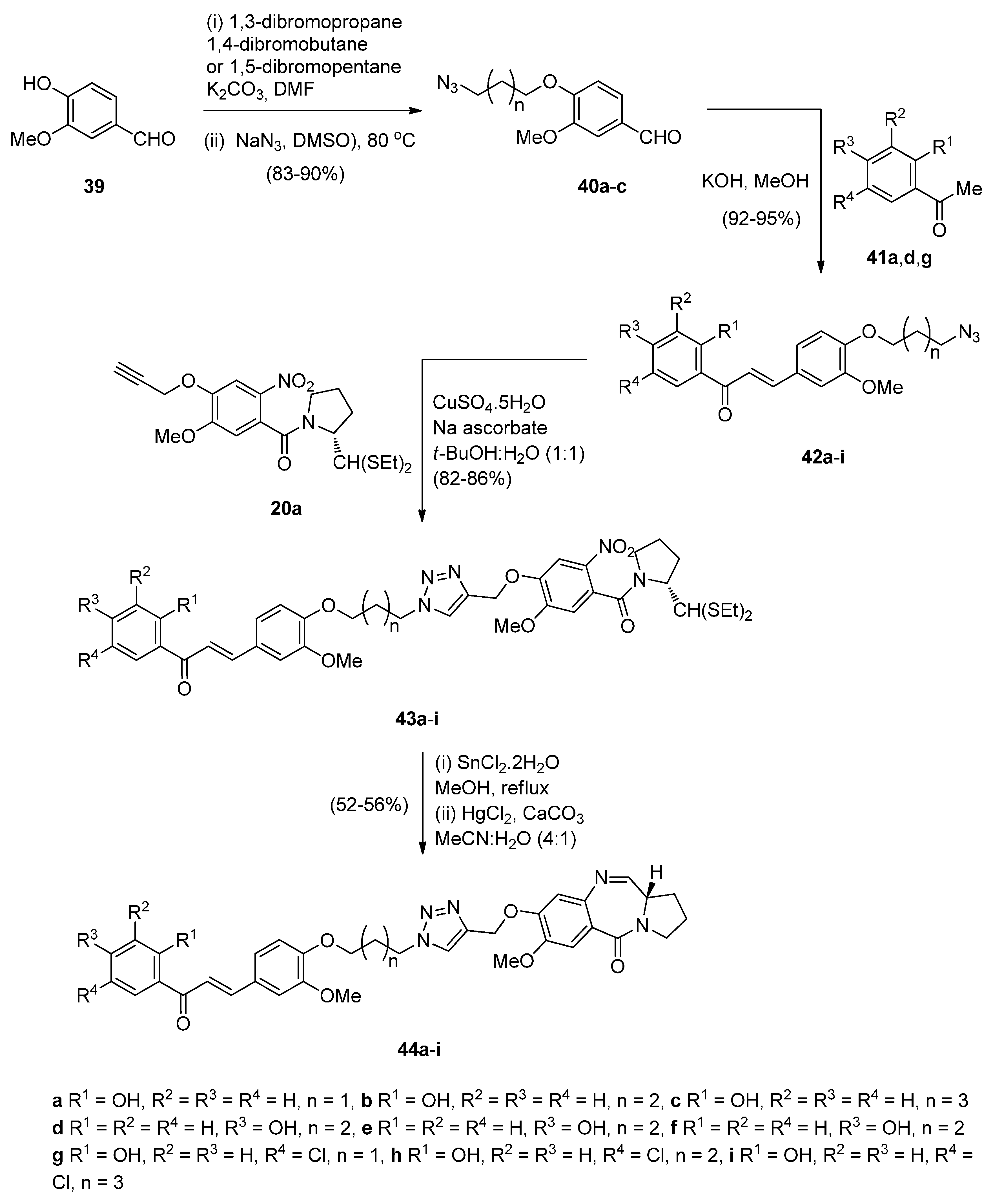
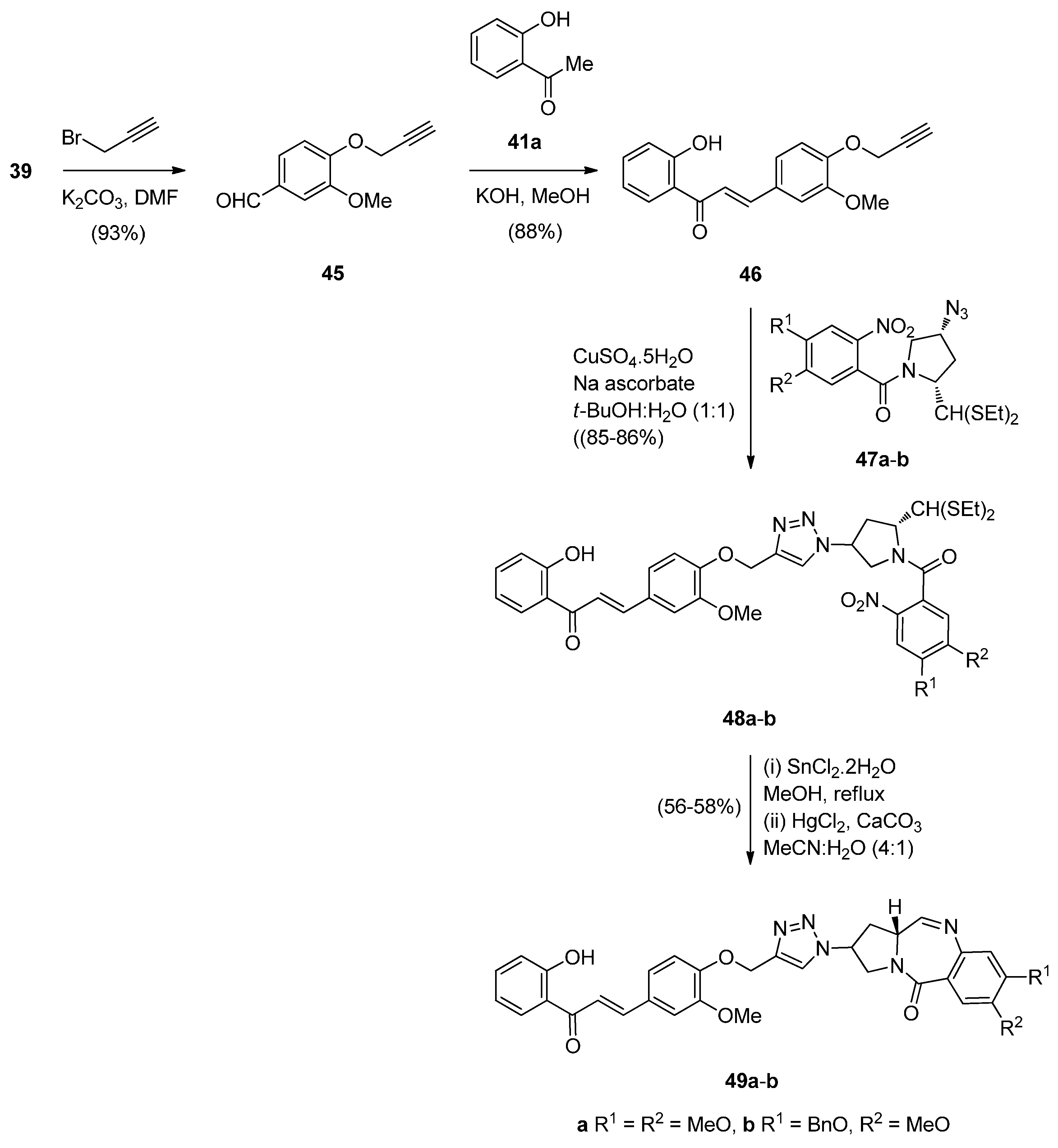
- Kamal, A.; Prabhakar, S.; Ramaiah, M.J.; Reddy, P.V.; Reddy, C.R.; Mallareddy, A.; Shankaraiah, N.; Reddy, T.L.M.; Pushpavalli, S.N.; Pal-Bhadra, M. Synthesis and anticancer activity of chalcone-pyrrolobenzodiazepine conjugates linked via 1,2,3-triazole ring side-armed with alkane spacers. Eur. J. Med. Chem. 2011, 46, 3820–3831. [Google Scholar] [CrossRef] [PubMed]
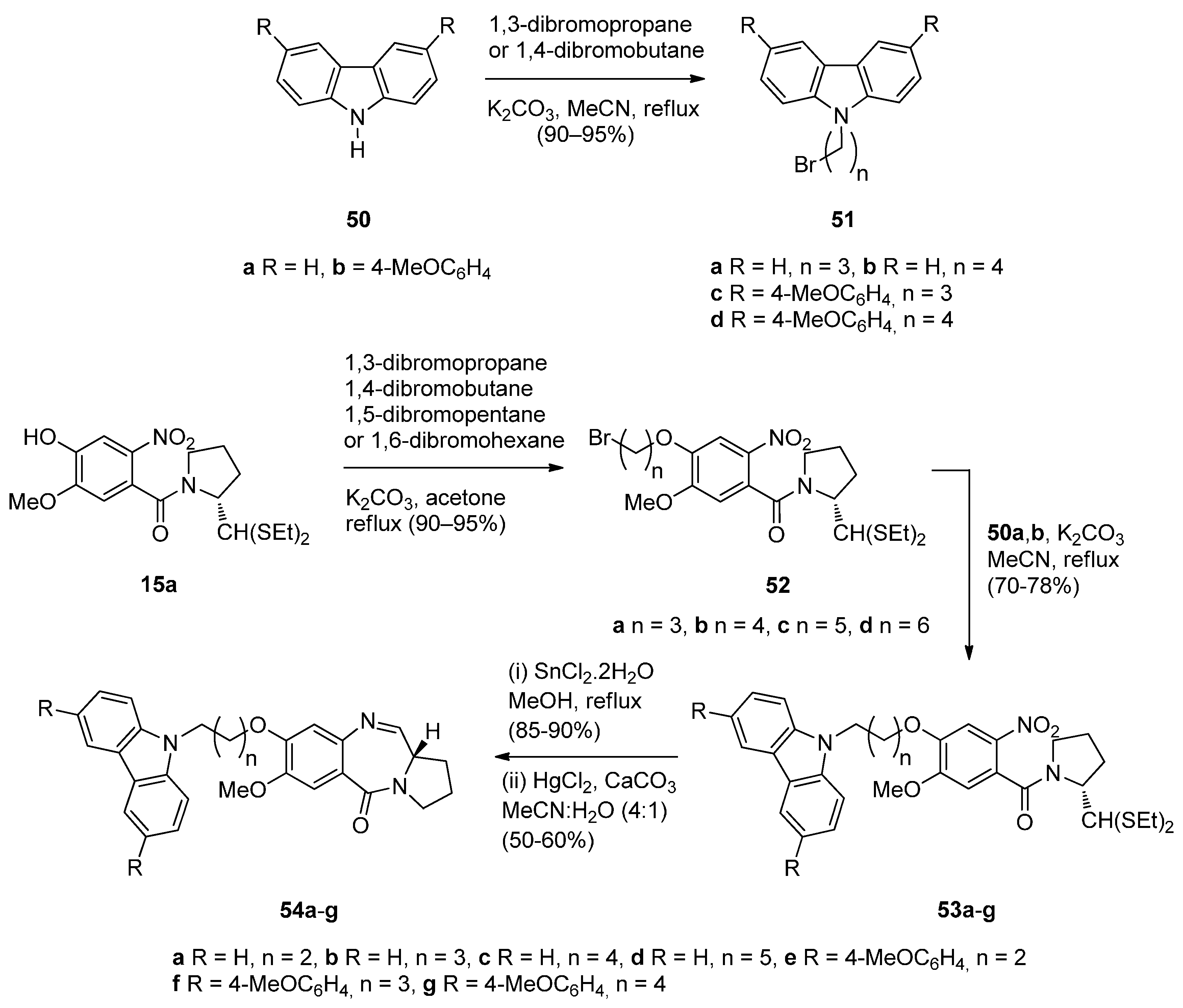
- Kamal, A.; Shetti, R.V.; Ramaiah, M.J.; Swapna, P.; Reddy, K.S.; Mallareddy, A.; Rao, M.P.N.; Chourasia, M.; Sastry, G.N.; Juvekar, A.; et al. Carbazole-pyrrolo[2,1-c][1,4]benzodiazepine conjugates: Design, synthesis, and biological evaluation. Med. Chem. Commun. 2011, 2, 780–788. [Google Scholar] [CrossRef]
- Thurston, D.E.; Murty, V.S.; Langley, D.R.; Jones, G.B. O-Debenzylation of a pyrrolo[2,1-c][1,4]benzodiazepine in the presence of a carbinolamine functionality: Synthesis of DC-81. Synthesis 1990, 1, 81–84. [Google Scholar] [CrossRef]
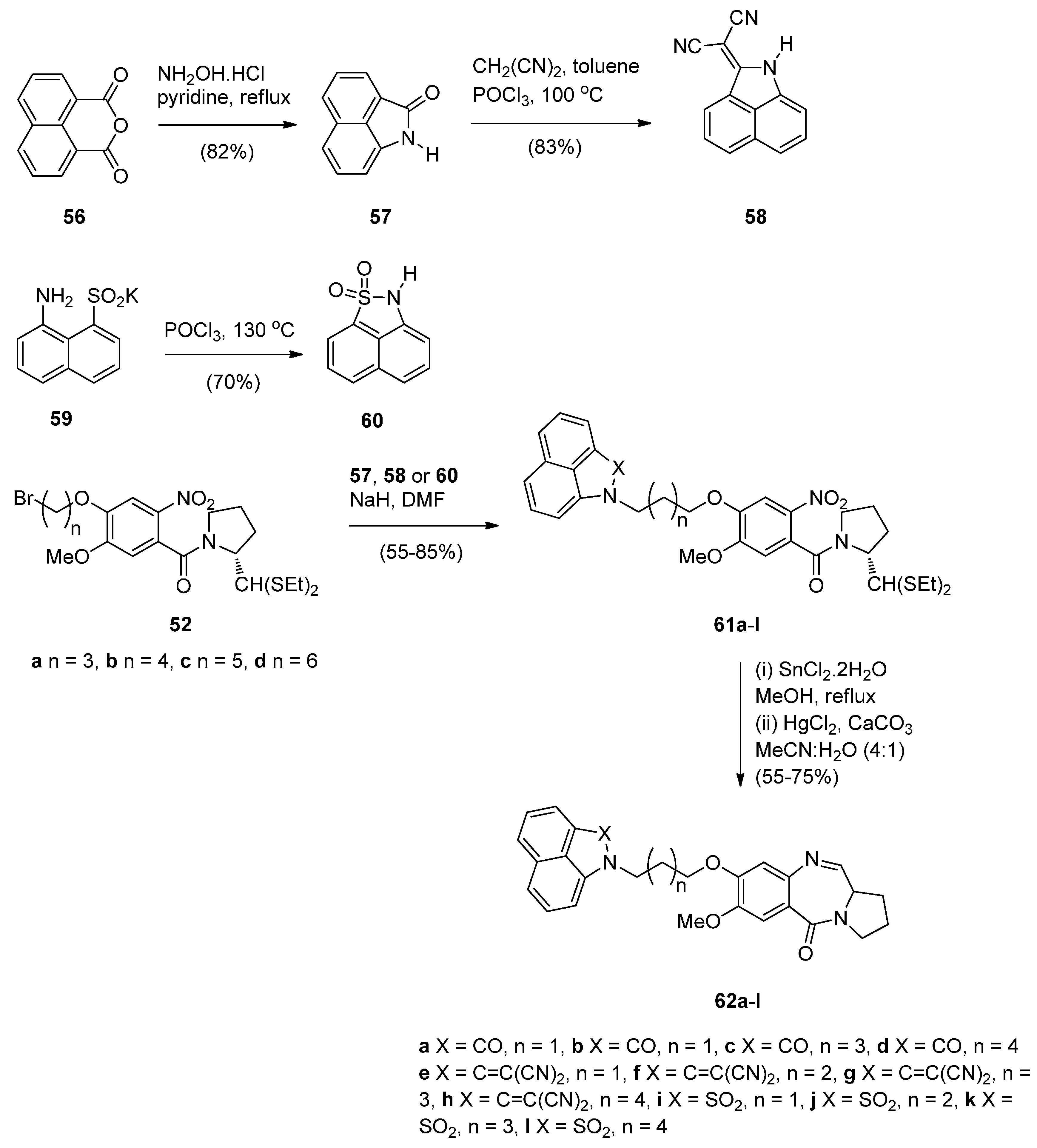
- Kamal, A.; Ramakrishna, G.; Nayak, V.L.; Raju, P.; Rao, A.V.S.; Viswanath, A.; Vishnuvardhan, M.V.P.S.; Ramakrishna, S.; Srinivas, G. Design and synthesis of benzo[c,d]indolone-pyrrolobenzodiazepine conjugates as potential anticancer agents. Bioorg. Med. Chem. 2012, 20, 789–800. [Google Scholar] [CrossRef] [PubMed]
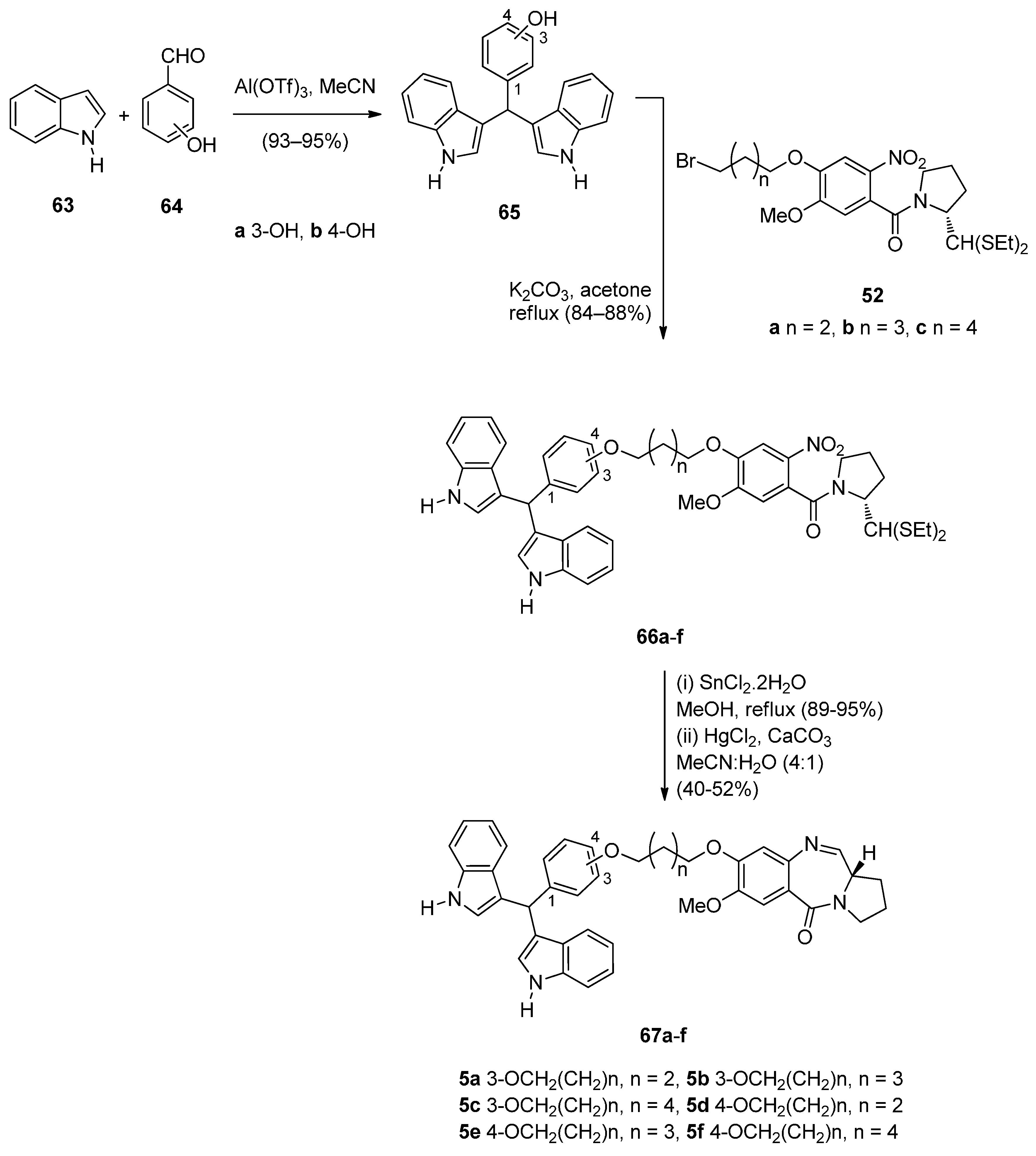
- Kamal, A.; Srikanth, Y.V.V.; Ramaiah, M.J.; Khan, M.N.A.; Reddy, M.K.; Ashraf, Md.; Lavanya, A.; Pushpavalli, S.N.; Pal-Bhadra, M. Synthesis, anticancer activity and apoptosis inducing ability of bisindole linked pyrrolo[2,1-c][1,4]benzodiazepine conjugates. Bioorg. Med. Chem. Lett. 2012, 22, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Ramakrishna, G.; Ramaiah, M.J.; Viswanath, A.; Ayinampudi, V.S.R.; Bagul, C.; Mukhopadyay, D.; Pushpavalli, S.N.; Pal-Bhadra, M. Design, synthesis and biological evaluation of imidazo[1,5-a]pyridine–PBD conjugates as potential DNA-directed alkylating agents. Med. Chem. Commun. 2013, 4, 697–703. [Google Scholar] [CrossRef]
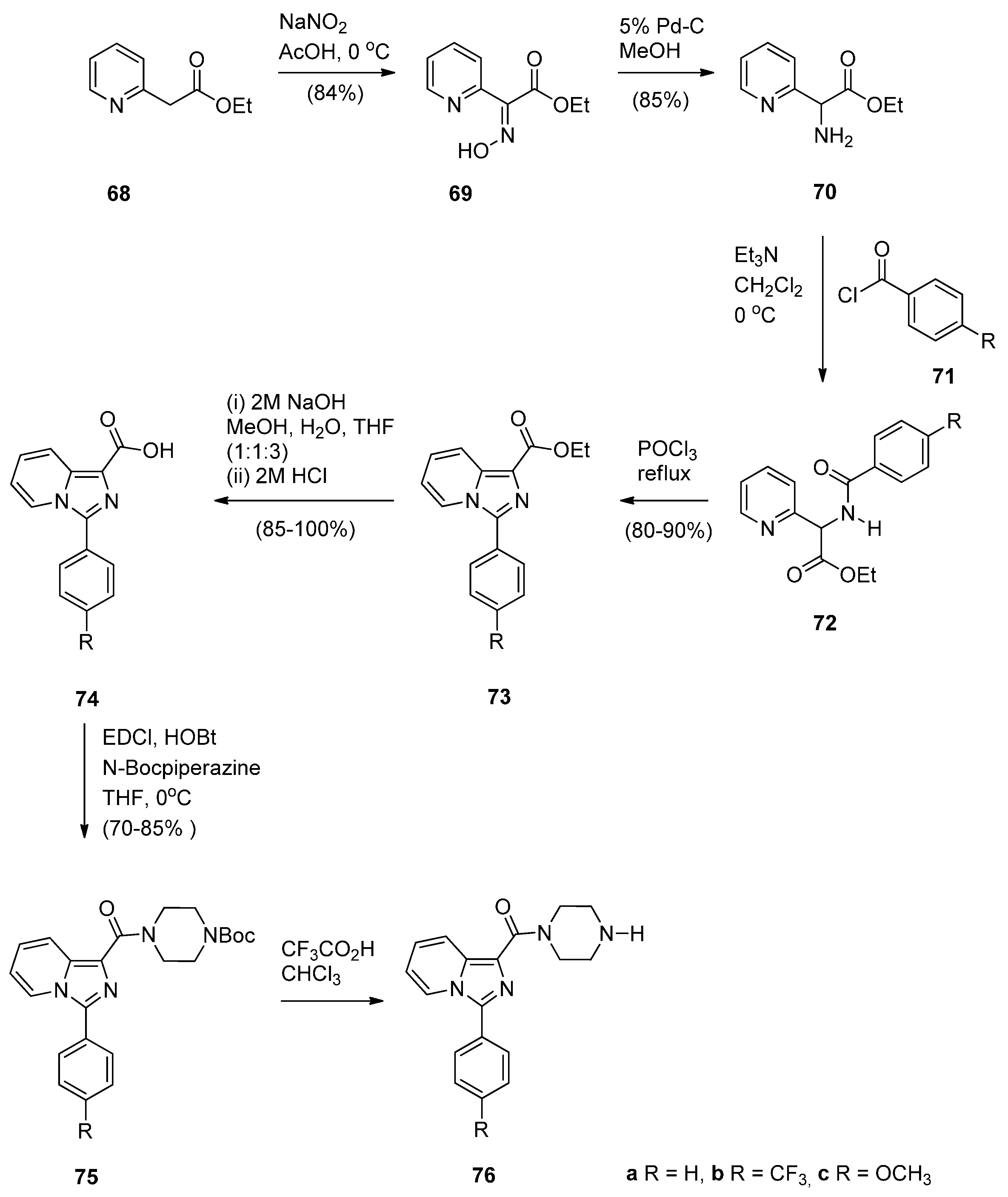
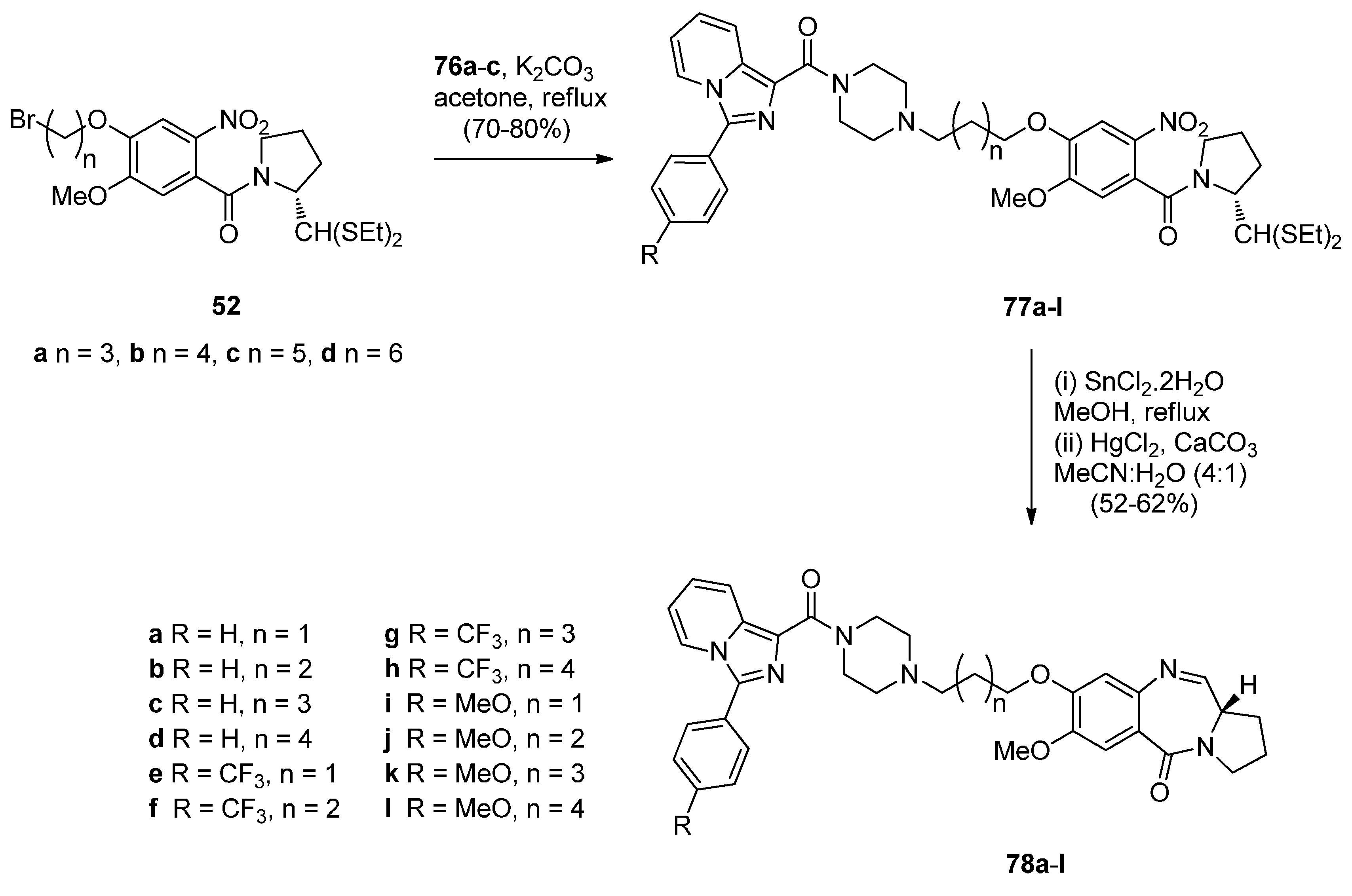
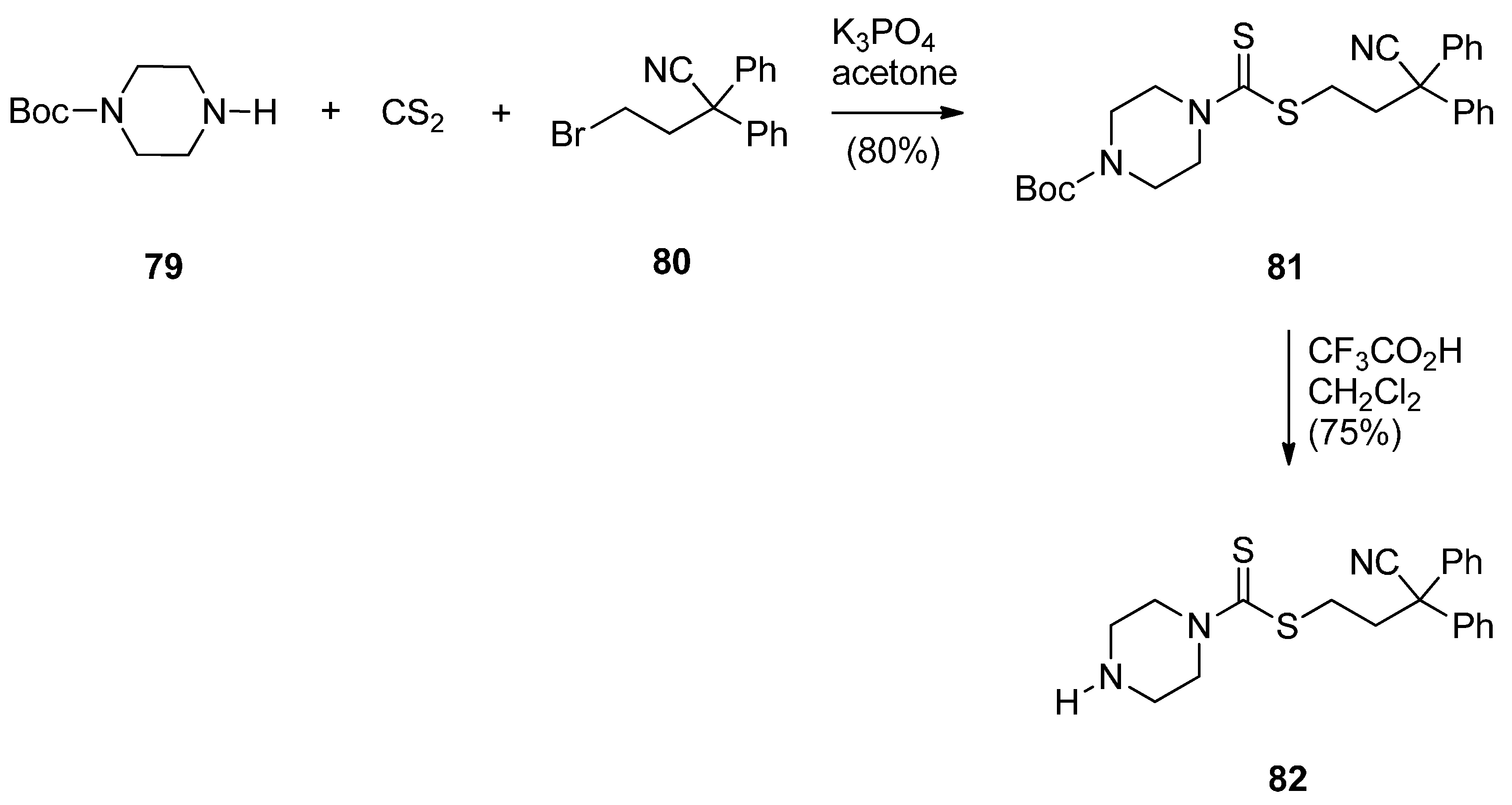
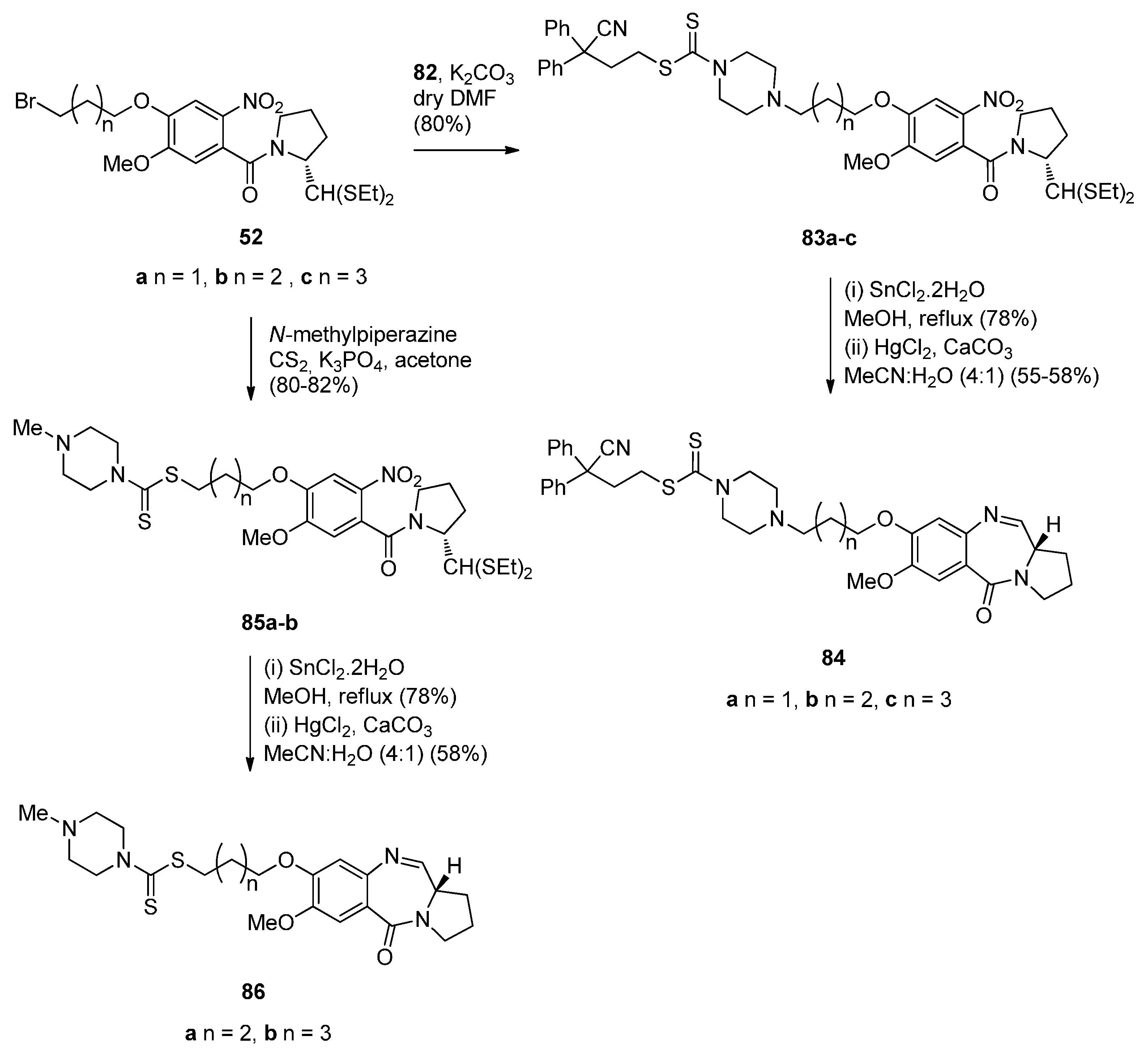
- Kamal, A.; Sreekanth, K.; Shankaraiah, N.; Sathish, M.; Nekkanti, S.; Srinivasulu, V. Dithiocarbamate/piperazine bridged pyrrolobenzodiazepines as DNA-minor groove binders: Synthesis, DNA-binding affinity and cytotoxic activity. Bioorg. Chem. 2015, 59, 23–30. [Google Scholar] [CrossRef] [PubMed]
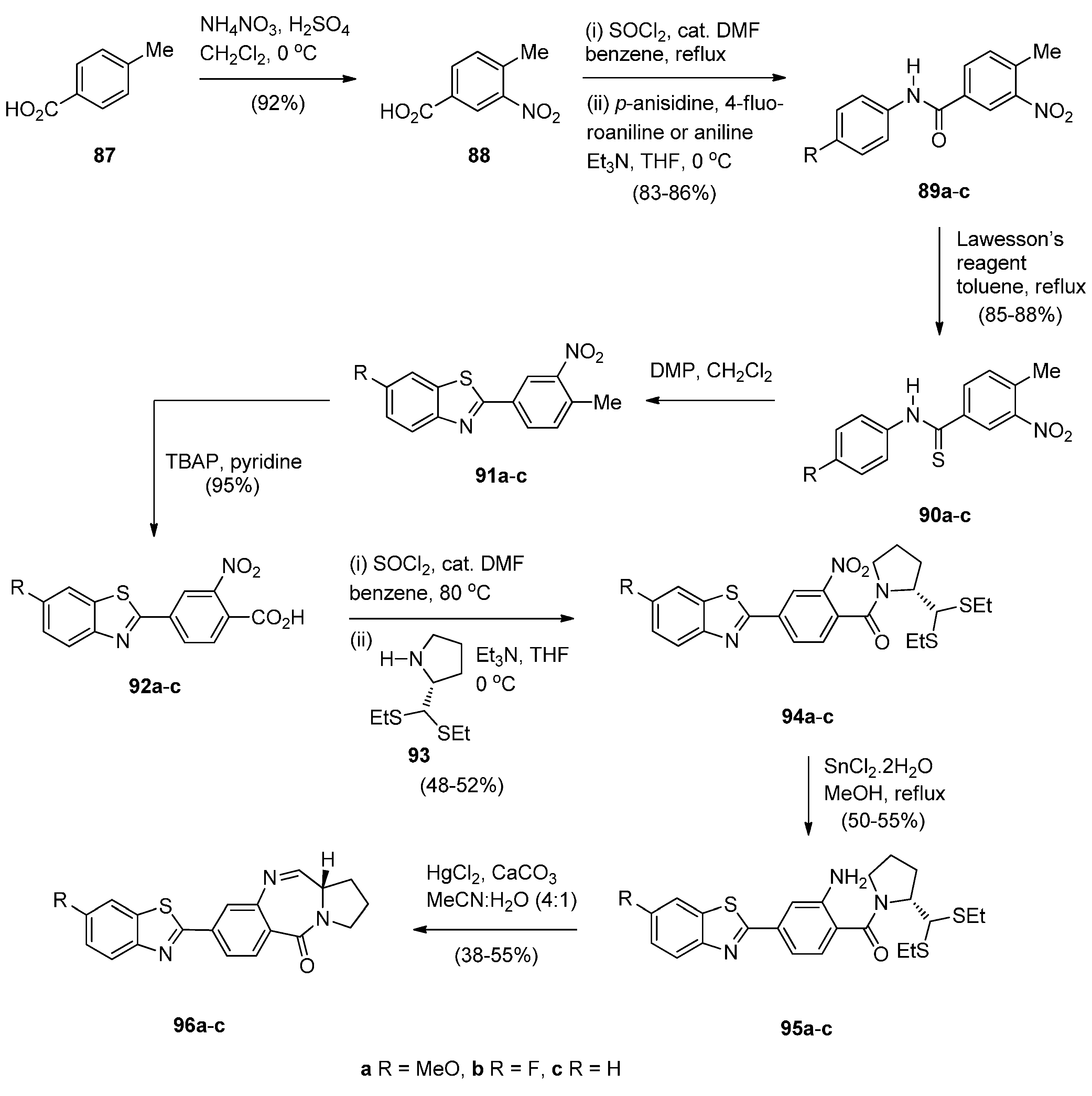
- Bose, D.S.; Idrees, M.; Todewale, I.K.; Jakka, N.M.; Rao, J.V. Hybrids of privileged structures benzothiazoles and pyrrolo[2,1-c][1,4]benzodiazepin-5-one, and diversity-oriented synthesis of benzothiazoles. Eur. J. Med. Chem. 2012, 50, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Langley, D.R.; Thurston, D.E. A versatile and efficient synthesis of carbinolamine-containing pyrrolo[1,4]benzodiazepines via the cyclisation of N-(2-aminobenzoyl)-pyrrolidine-2-carboxaldehyde diethyl thioacetals: total synthesis of prothracarcin. J. Org. Chem. 1987, 52, 91–97. [Google Scholar] [CrossRef]
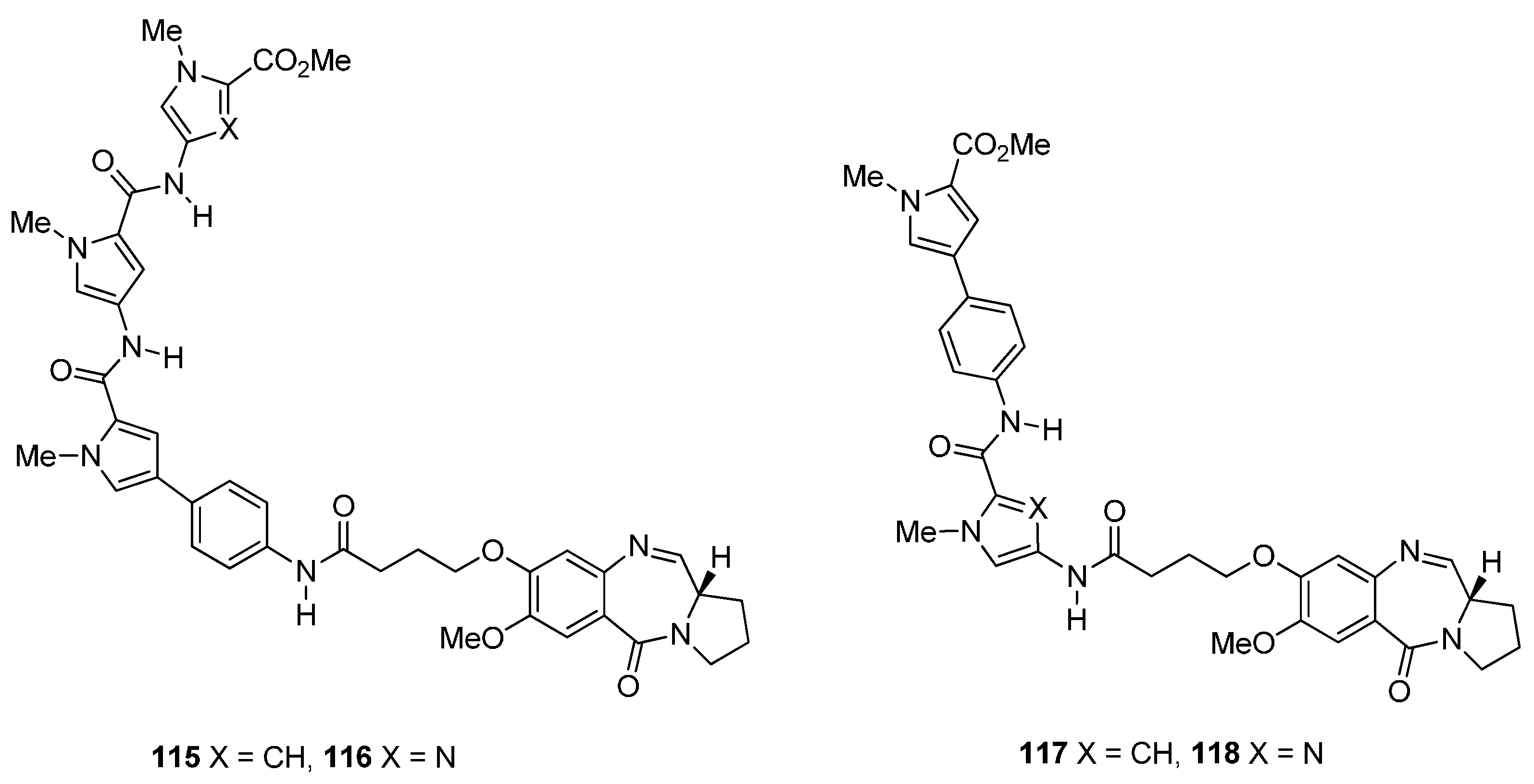
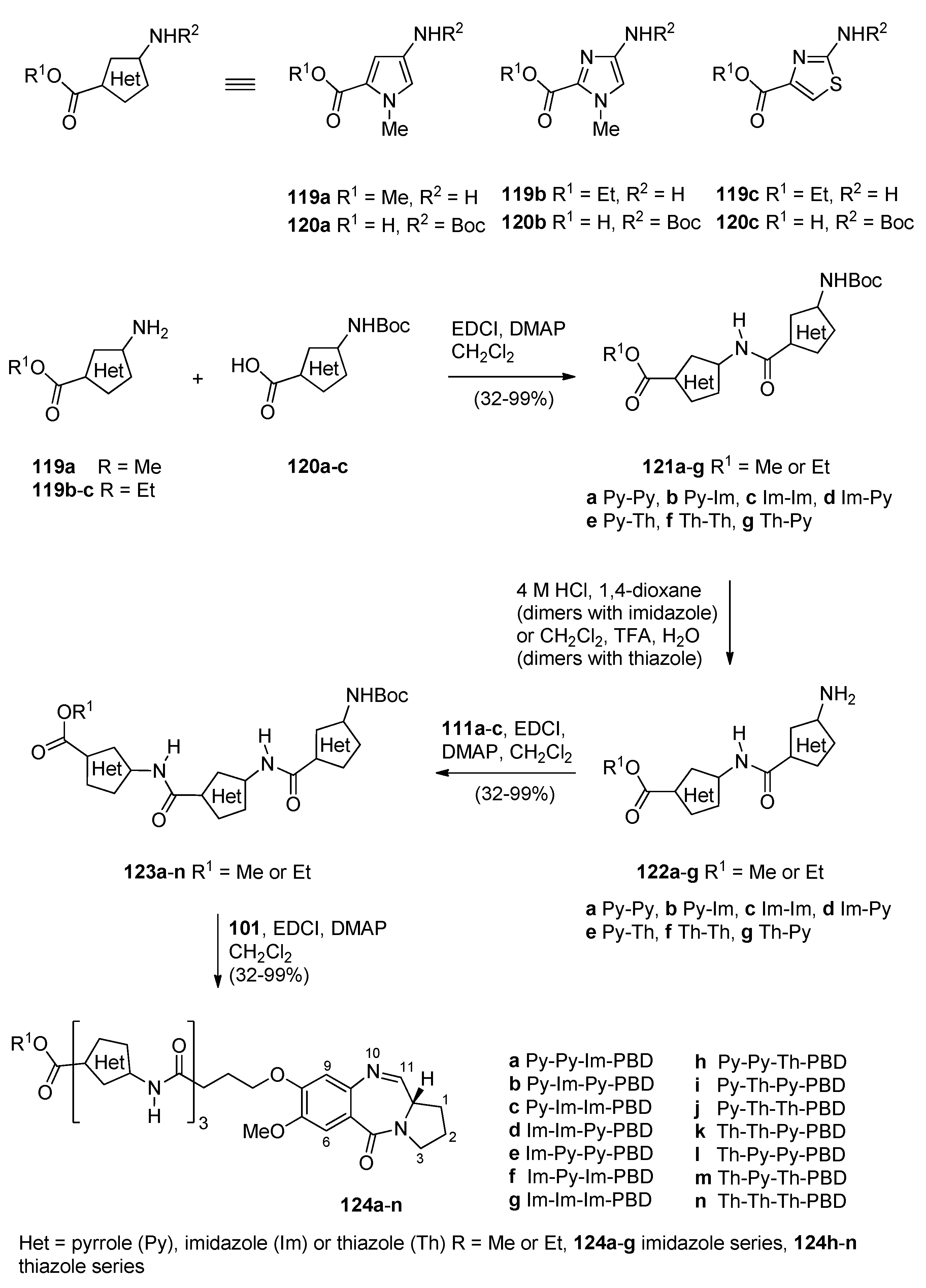
- Wells, G.; Martin, C.R.H.; Howard, P.W.; Sands, Z.A.; Laughton, C.A.; Tiberghien, A.; Woo, C.K.; Masterson, L.A.; Stephenson, M.J.; Hartley, J.A.; et al. Design, synthesis, and biophysical and biological evaluation of a series of pyrrolobenzodiazepine-poly(N-methylpyrrole) conjugates. J. Med. Chem. 2006, 49, 5442–5461. [Google Scholar] [CrossRef] [PubMed]
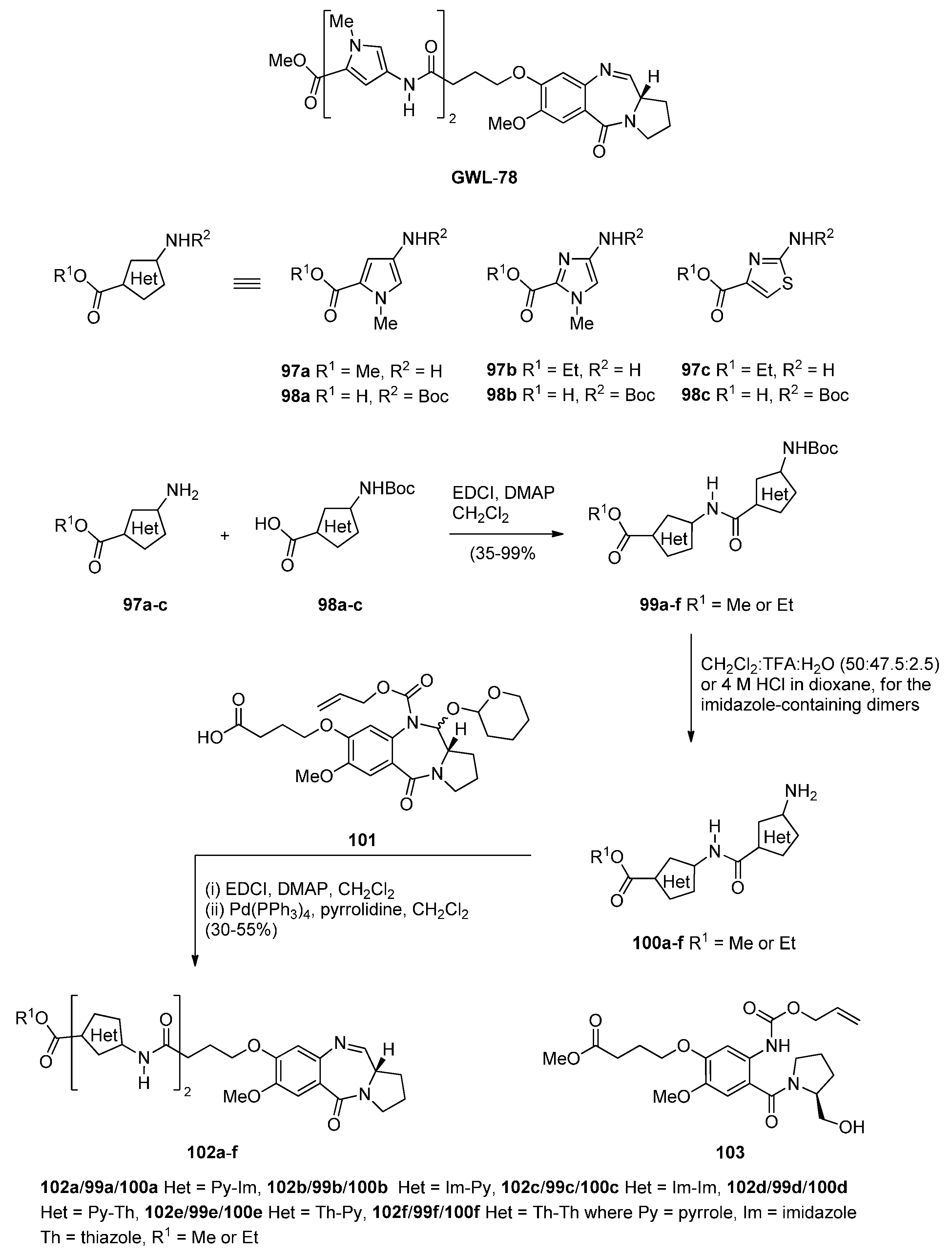
- Kotecha, M.; Kluza, J.; Wells, G.; O’Hare, C.C.; Forni, C.; Mantovani, R.; Howard, P.W.; Morris, P.; Thurston, D.E.; Hartley, J.A.; et al. Inhibition of DNA binding of the NF-Y transcription factor by the pyrrolobenzodiazepine-polyamide conjugate GWL-78. Mol. Cancer Ther. 2008, 7, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
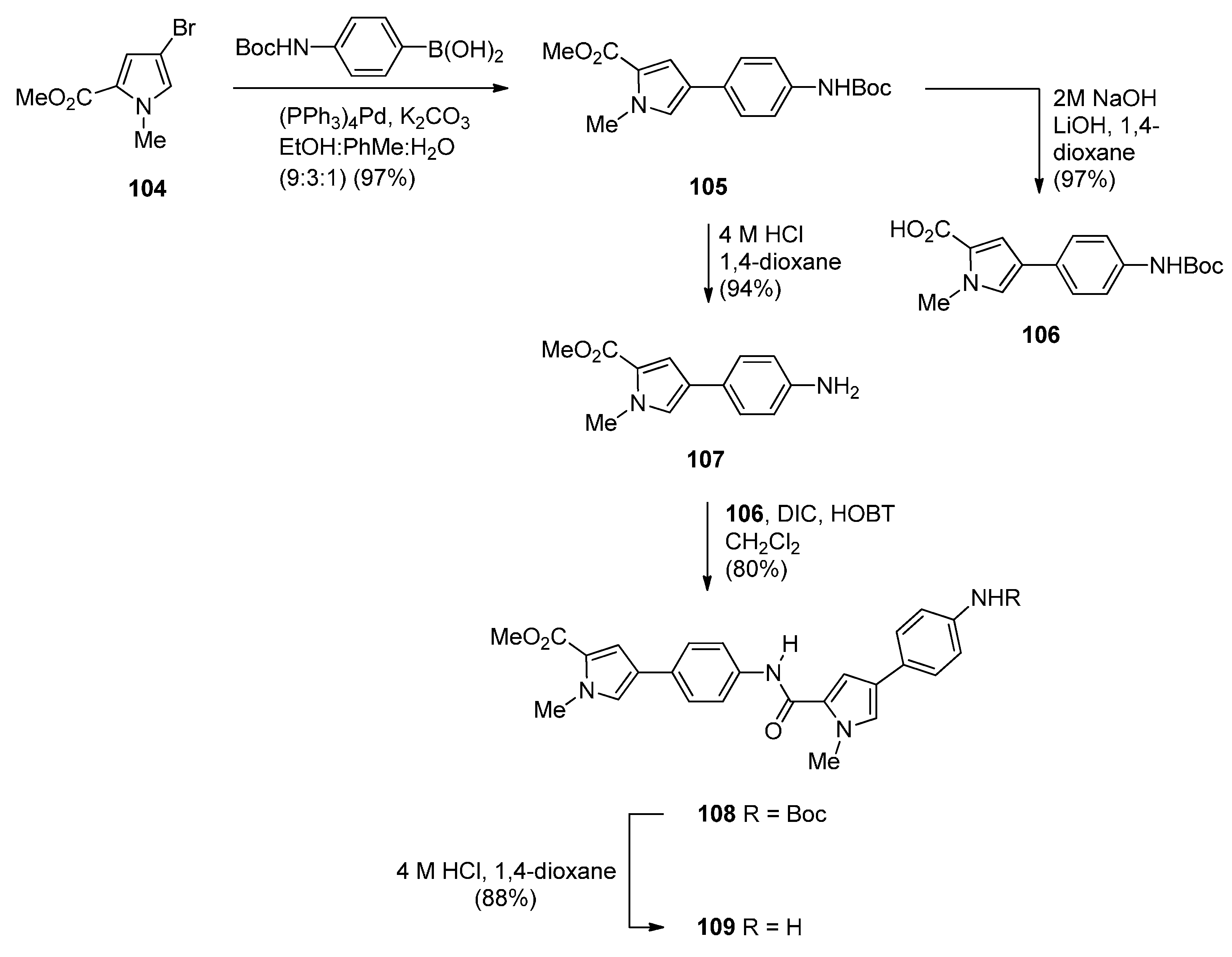
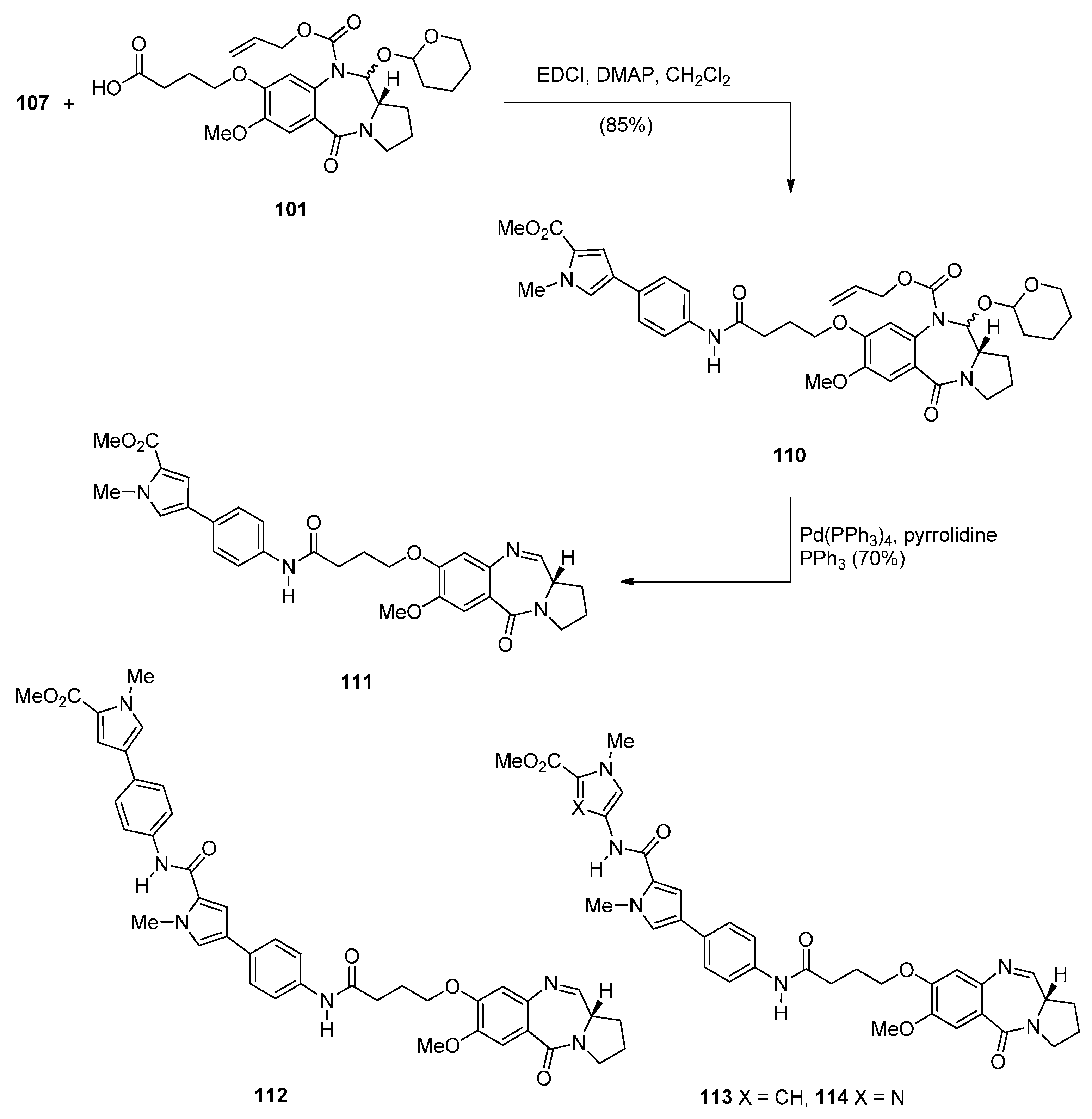
- Brucoli, F.; Hawkins, R.M.; James, C.H.; Wells, G.; Jenkins, T.C.; Ellis, T.; Hartley, J.A.; Howard, P.W.; Thurston, D.E. Novel C8-linked pyrrolobenzodiazepine (PBD)–heterocycle conjugates that recognize DNA sequences containing an inverted CCAAT box. Bioorg. Med. Chem. Lett. 2011, 21, 3780–3783. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.M.; Jackson, P.J.M.; James, C.H.; Basu, B.P.; Hartley, J.A.; de la Fuente, M.; Schatzlein, A.; Robson, M.; Pedley, R.B.; Pepper, C.; et al. GC-targeted C8-linked pyrrolobenzodiazepine-biaryl conjugates with femtomolar in vitro cytotoxicity and in vivo antitumor activity in mouse models. J. Med. Chem. 2013, 56, 2911–2935. [Google Scholar] [CrossRef] [PubMed]
- Brucoli, F.; Hawkins, R.M.; James, C.H.; Jackson, P.J.M.; Wells, G.; Jenkins, T.C.; Ellis, T.; Kotecha, M.; Hochhauser, D.; Hartley, J.A.; et al. An extended pyrrolobenzodiazepine–polyamide conjugate with selectivity for a DNA sequence containing the ICB2 transcription factor binding site. J. Med. Chem. 2013, 56, 6339–6351. [Google Scholar] [CrossRef] [PubMed]
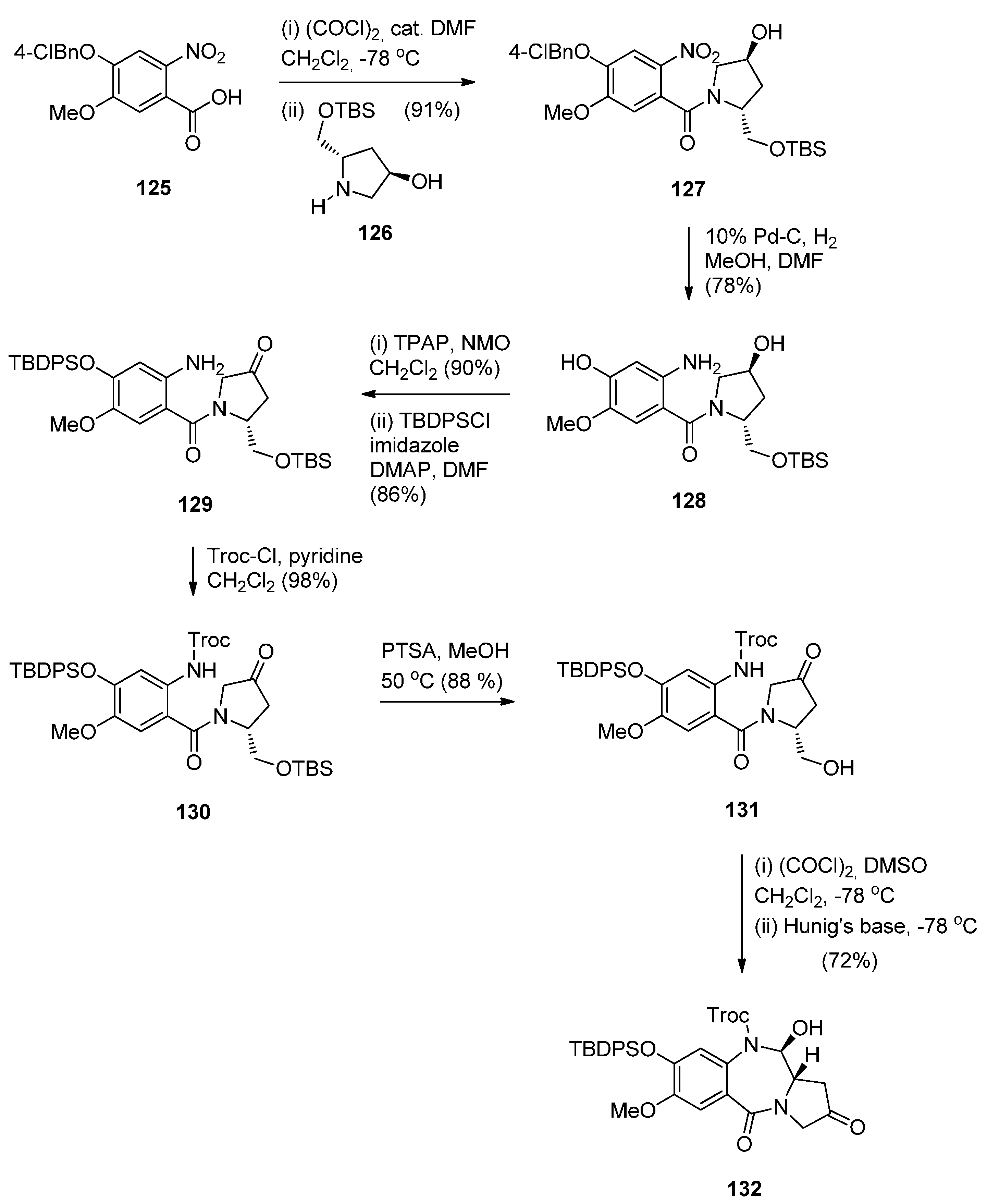
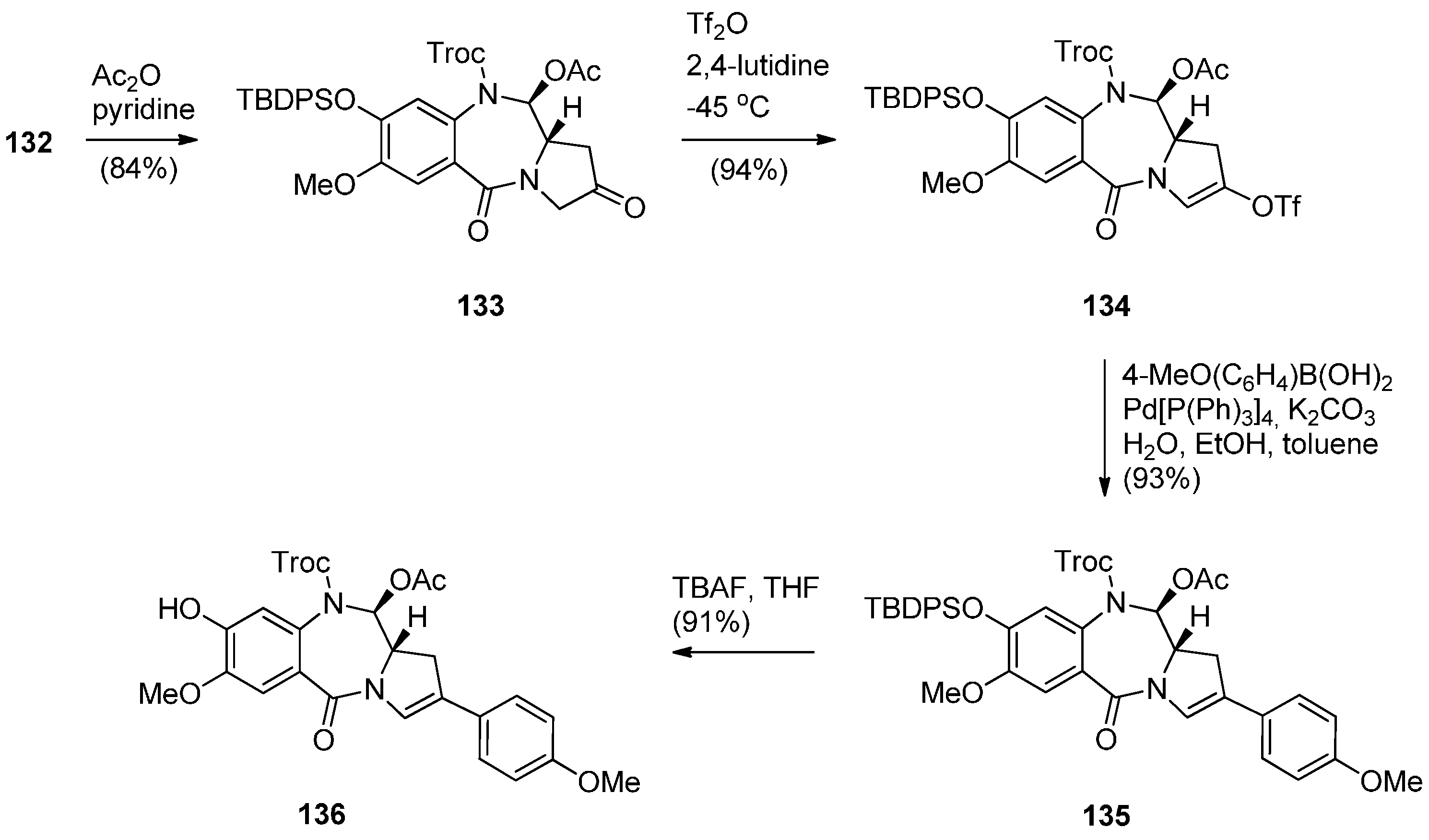
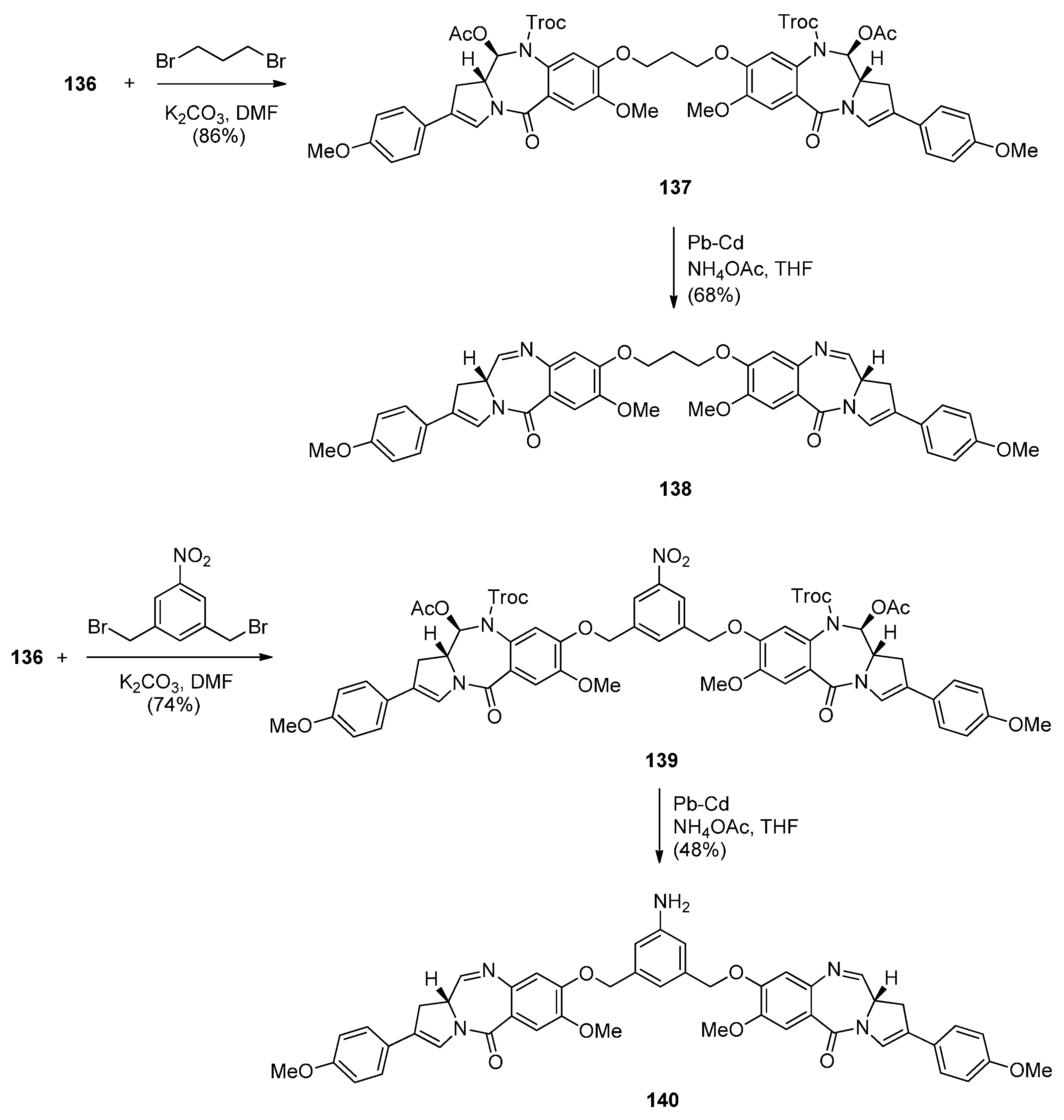
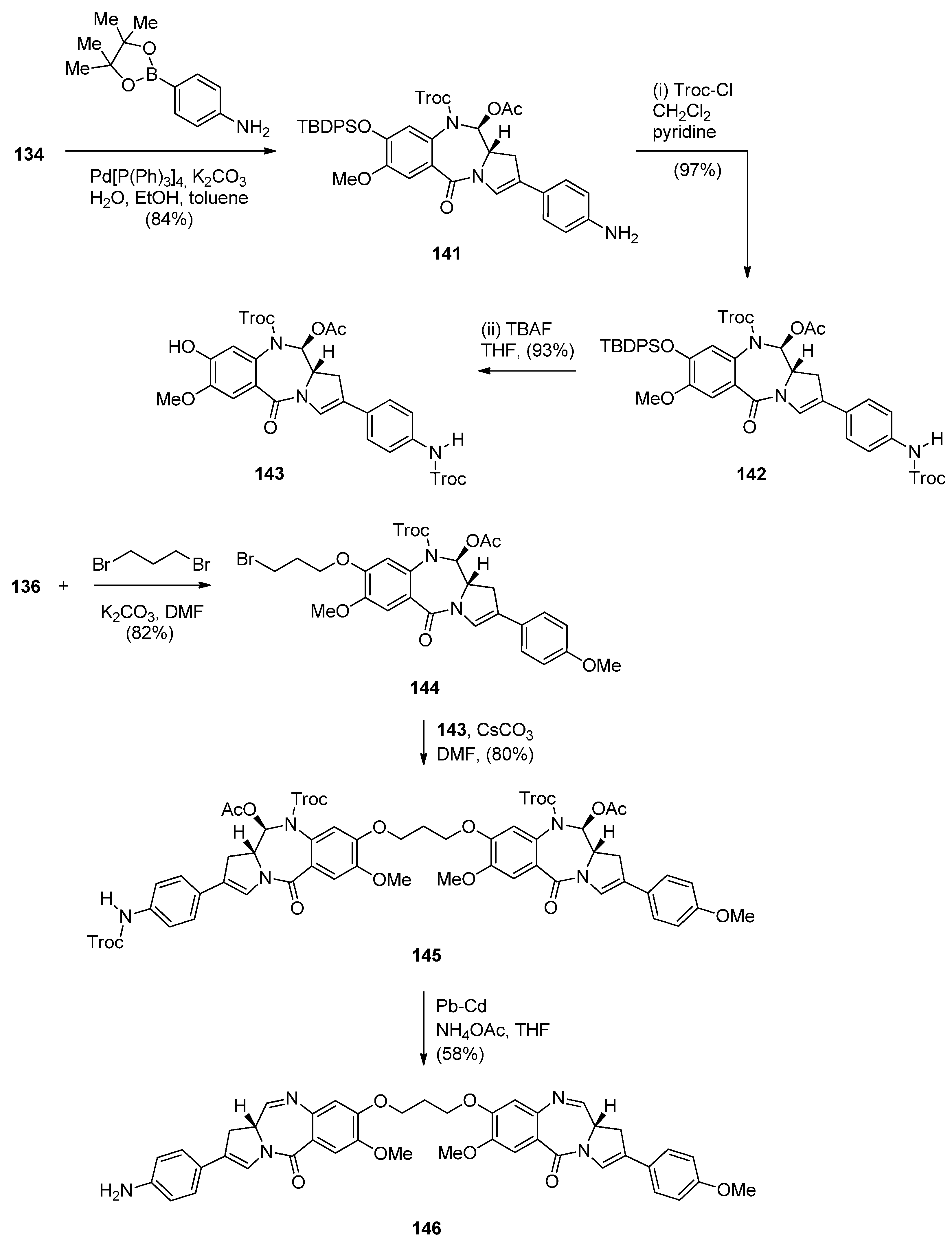
- Kolakowski, R.V.; Young, T.D.; Howard, P.W.; Jeffrey, S.C.; Senter, P.D. Synthesis of a C2-arylpyrrolo[2,1-c][1,4]benzodiazepine monomer enabling the convergent construction of symmetrical and non-symmetrical dimeric analogs. Tetrahedron Lett. 2015, 56, 4512–4515. [Google Scholar] [CrossRef]
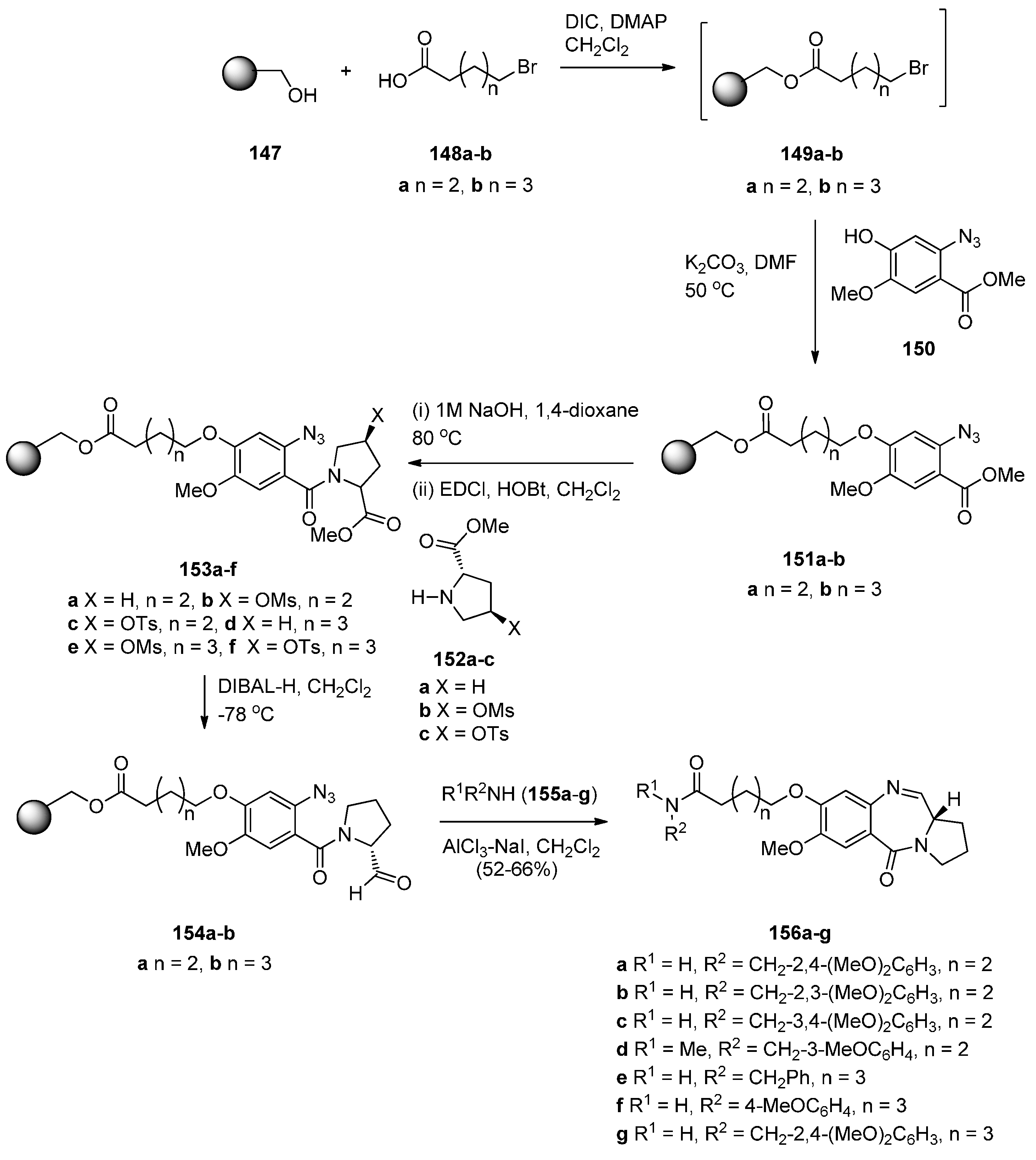
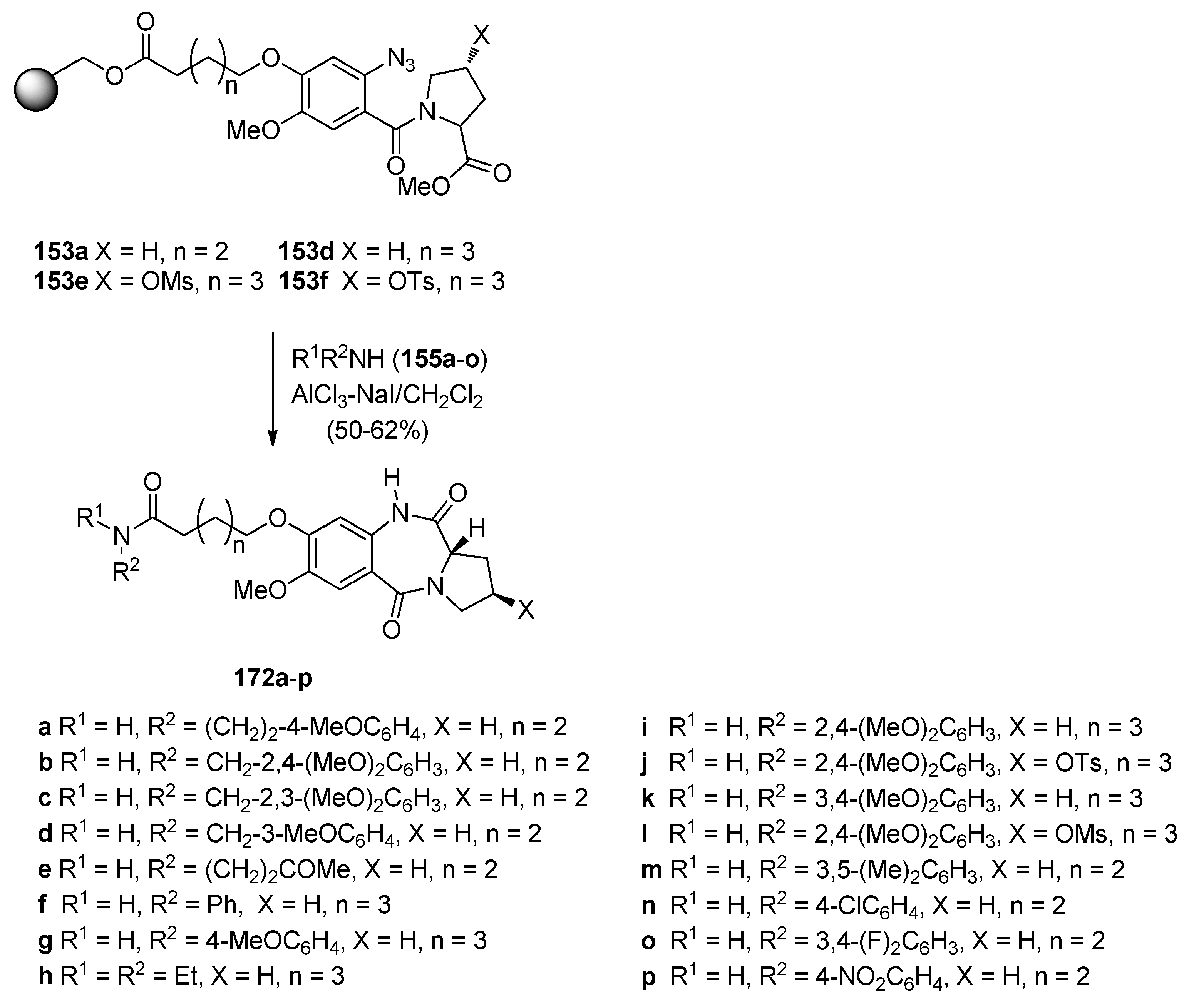
- Kamal, A.; Prabhakar, S.; Shankaraiah, N.; Markandey, N.; Reddy, P.V.; Srinivasulu, V.; Sathish, M. AlCl3–NaI assisted cleavage of polymer-bound esters with concomitant amine coupling and azido-reductive cyclisation: Synthesis of pyrrolobenzodiazepine derivatives. Tetrahedron Lett. 2013, 54, 4435–4441. [Google Scholar] [CrossRef]
- Kamal, A.; Shankaraiah, N.; Devaiah, V.; Reddy, K.L. Solid-phase synthesis of fused [2,1-b]-quinazolinone alkaloids. Tetrahedron Lett. 2006, 47, 9025–9028. [Google Scholar]
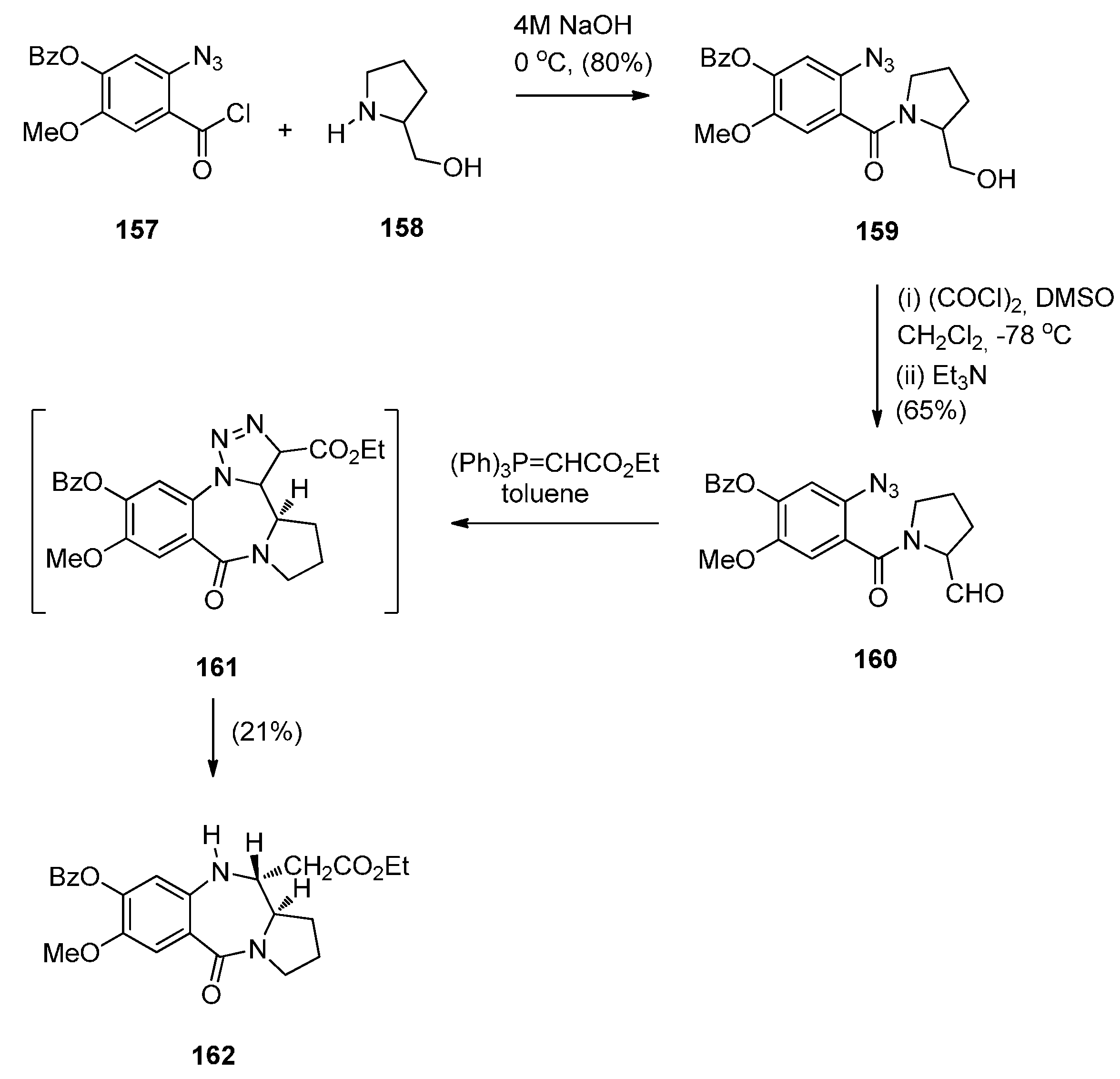
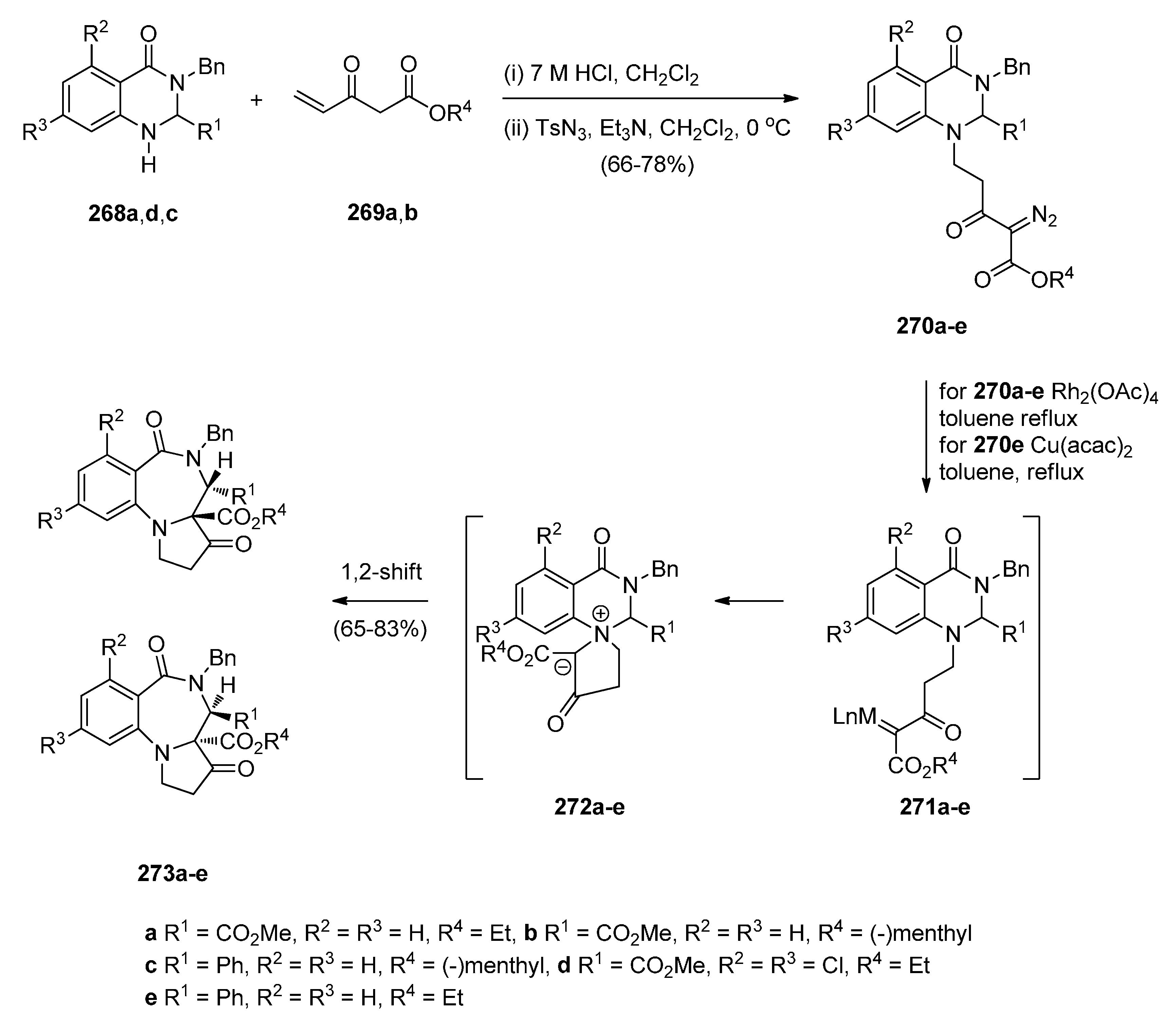
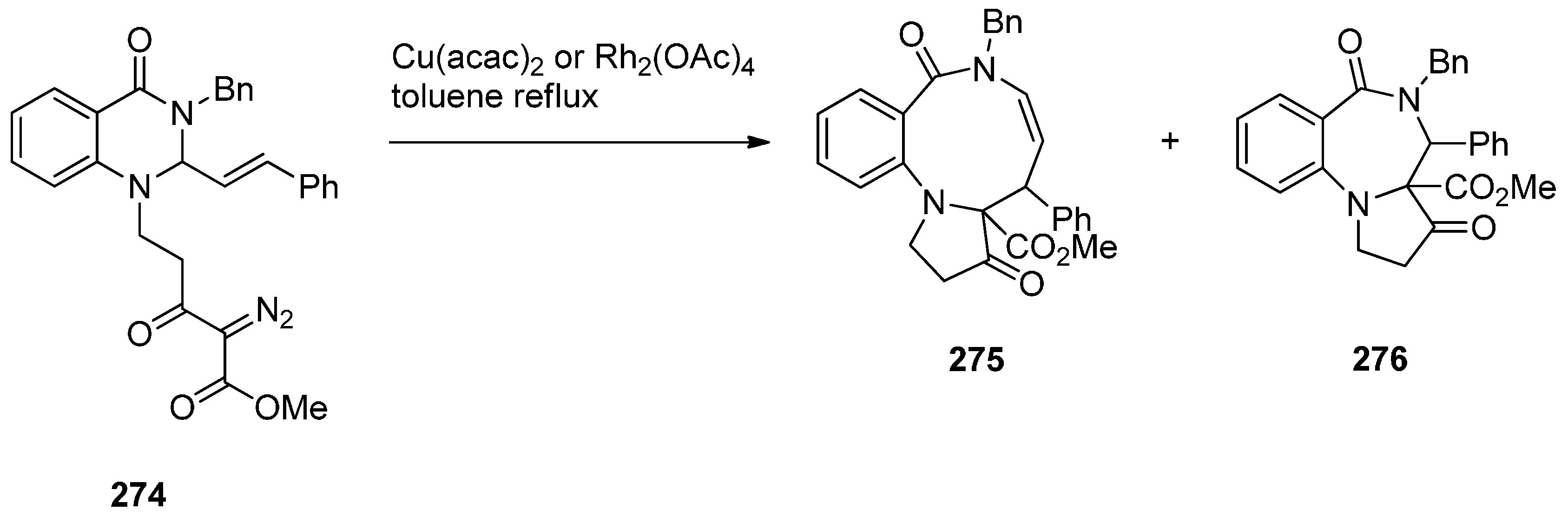
- Hemming, K.; Chambers, C.S.; Jamshaid, F.; O’Gorman, P.A. Intramolecular azide to alkene cycloadditions for the construction of pyrrolobenzodiazepines and azetidinobenzodiazepines. Molecules 2014, 19, 16737–16756. [Google Scholar] [CrossRef] [PubMed]
- Hemming, K.; Chambers, C.S.; Hamasharif, M.S.; João, H.; Khan, M.N.; Nilesh Patel, N.; Airley, R.; Day, S. Azide based routes to tetrazolo and oxadiazolo derivatives of pyrrolobenzodiazepines and pyrrolobenzothiadiazepines. Tetrahedron 2014, 70, 7306–7317. [Google Scholar] [CrossRef]
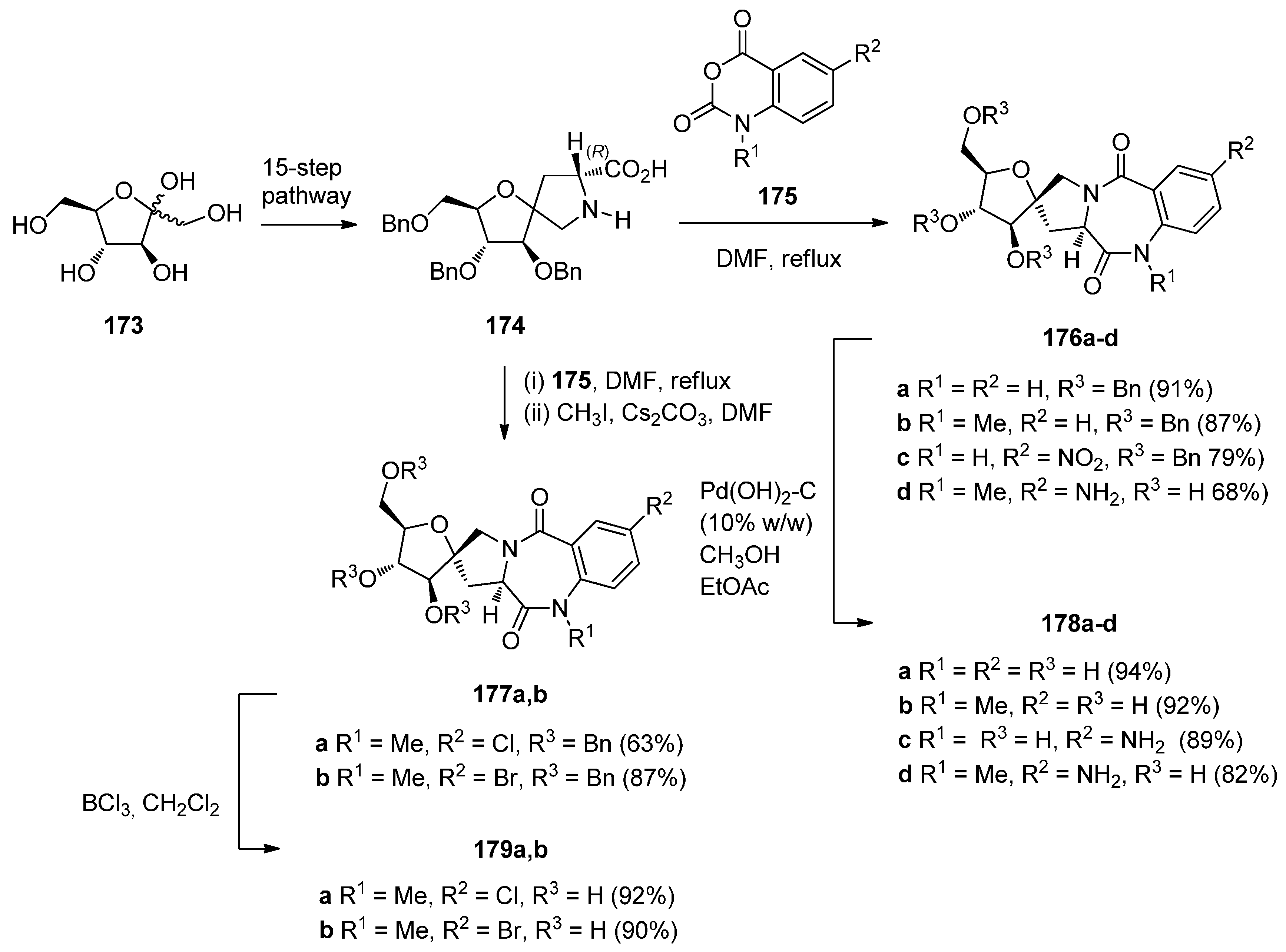
- Araújo, A.C.; Rauter, A.P.; Nicotra, F.; Airoldi, C.; Costa, B.; Cipolla, L. Sugar-based enantiomeric and conformationally constrained pyrrolo[2,1-c][1,4]benzodiazepines as potential GABAA ligands. J. Med. Chem. 2011, 54, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, L.; Redaelli, C.; Nicotra, F. Synthesis of a spiro d-proline analogue bearing d-fructose. Lett. Drug Des. Discov. 2005, 2, 291–293. [Google Scholar] [CrossRef]
- Legerén, L.; Domínguez, D. Synthesis of 5-arylpyrrolo[2,1-c][1,4]benzodiazepines under mild cyclodehydration conditions. Tetrahedron 2010, 66, 2718–2722. [Google Scholar] [CrossRef]
- Sharma, S.K.; Mandadapu, A.K.; Kumaresan, K.; Arora, A.; Gauniyal, H.M.; Kundu, B. Efficient synthesis of naturally occurring skeleton 5–7–6 tricyclic pyrrolo[2,1-c][1,4]benzodiazepin-5-one and its derivatives via cationic π-cyclisation. Synthesis 2010, 23, 4087–4095. [Google Scholar]
- Leber, J.D.; Hoover, J.R.E.; Holden, K.G.; Johnson, R.K.; Hecht, S.M. A revised structure of sibiromycin. J. Am. Chem. Soc. 1988, 110, 2992–2993. [Google Scholar] [CrossRef]
- Gao, K.; Wu, B.; Yu, C.-B.; Chen, Q.-A.; Ye, Z.-S.; Zhou, Y.G. Iridium catalyzed asymmetric hydrogenation of cyclic imines of benzodiazepinones and benzodiazepines. Org. Lett. 2012, 14, 3890–3893. [Google Scholar] [CrossRef] [PubMed]
- Molinari, A.J.; Trybulski, E.J.; Bagli, J.; Croce, S.; Considine, J.; Qi, J.; Ali, K.; Demaio, W.; Lihotz, L.; Cochran, D. Identification and synthesis of major metabolites of Vasopressin V2-receptor agonist WAY-151932, and antagonist, Lixivaptan®. Bioorg. Med. Chem. Lett. 2007, 17, 5796–5800. [Google Scholar] [CrossRef] [PubMed]
- Roszkowski, P.; Maurin, J.K.; Czarnocki, Z. First enantioselective synthesis of aptazepine. Synthesis 2012, 2, 241–246. [Google Scholar]
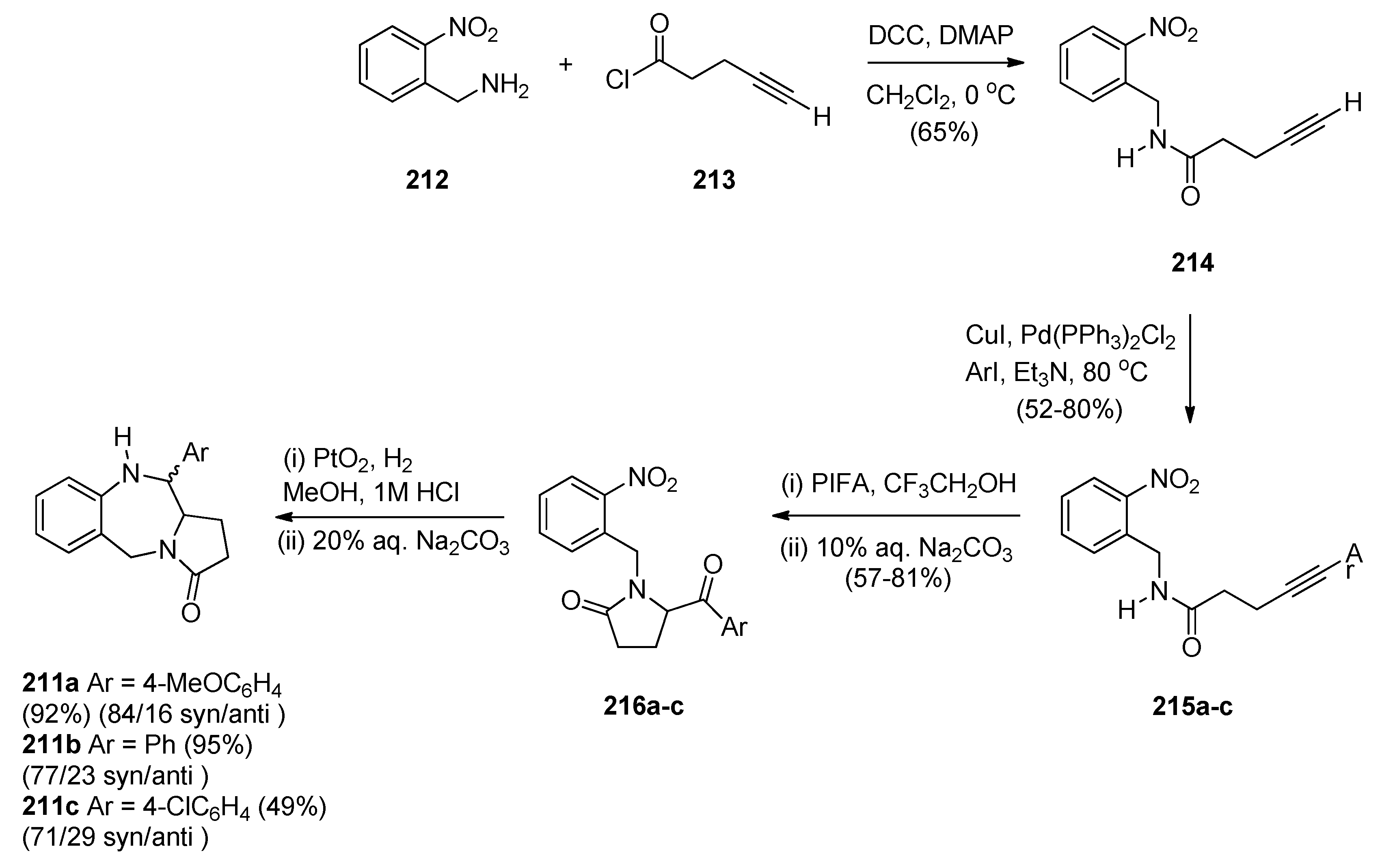
- Pardo, L.M.; Tellitu, I.; Esther Domínguez, E. A versatile PIFA-mediated approach to structurally diverse pyrrolo(benzo)diazepines from linear alkynylamides. Tetrahedron 2010, 66, 5811–5818. [Google Scholar] [CrossRef]
- Raines, S.; Chai, S.Y.; Palopoli, F.P. Mannich reactions. Synthesis of 4,5-dihydropyrrolo[l,2-a]quinoxalines, 2,3,4,5-tetrahydro-lH-pyrrolo[l,2-a][l,4]diazepines and 5,6-dihydro-4H-pyrrolo[1,2-a][1,4]benzodiazepines. J. Heterocycl. Chem. 1976, 13, 711–716. [Google Scholar] [CrossRef]
- Cheeseman, G.W.H.; Greenberg, S.G. Synthesis of 5,6-dihydro-4H-pyrrolo[1,2-a][1,4]benzodiazepines. J. Heterocycl. Chem. 1979, 16, 241–244. [Google Scholar] [CrossRef]
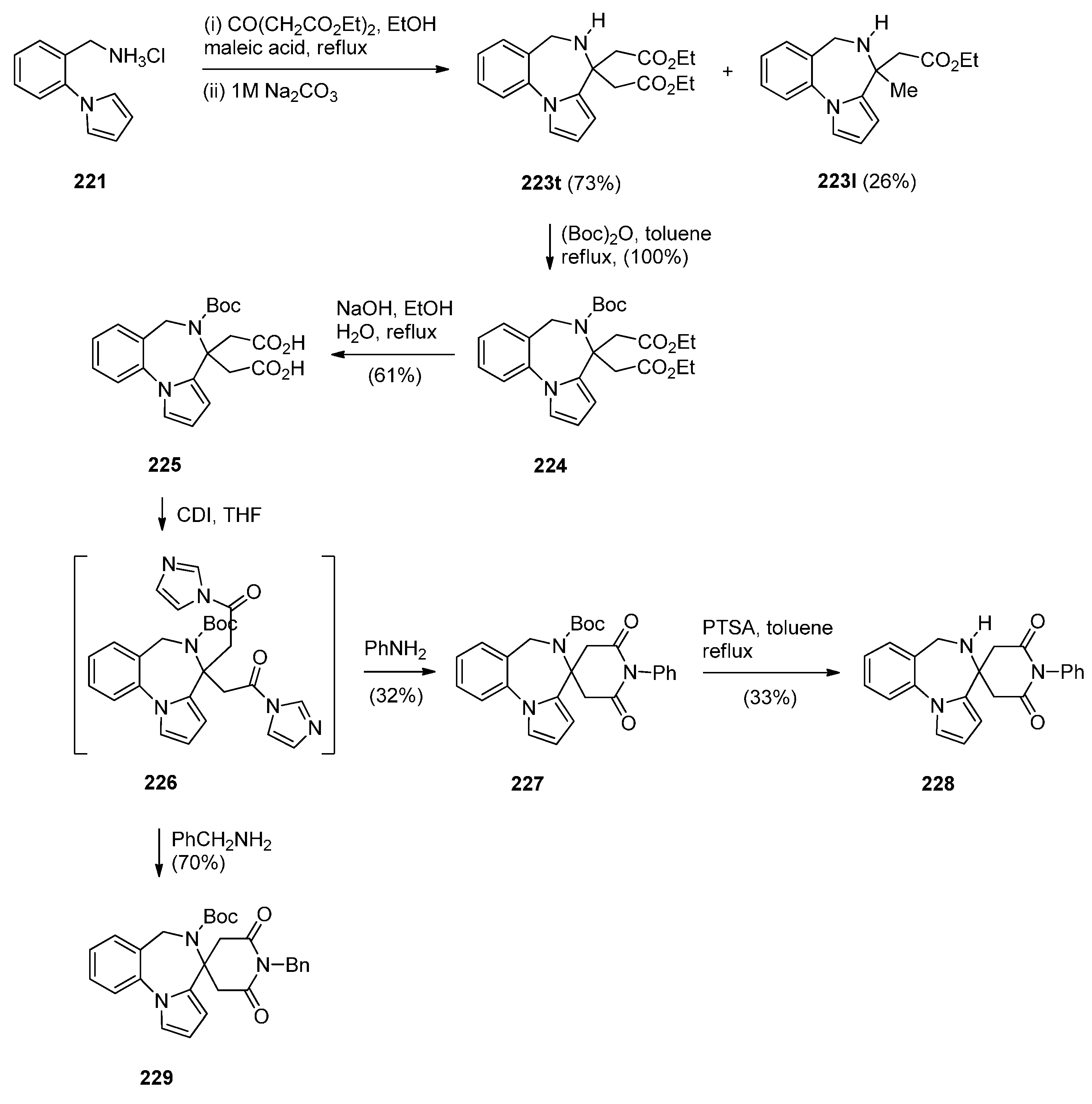
- Massa, S.; Mai, A.; di Santo, R.; Artico, M. 5,6-Dihydro-4H-pyrrolo[1,2-a][1,4]benzodiazepine-4,4-diacetic acid diethyl ester, an useful synthon for the synthesis of new polycyclic nitrogen systems of pharmacological interest. J. Heterocycl. Chem. 1993, 30, 897–903. [Google Scholar] [CrossRef]
- Massa, S.; Mai, A.; Artico, M.; Corelli, F.; Botta, M. Synthesis of 3b,4,6,7-tetrahydro-5H,9H-pyrazino[2,l-c]-pyrrolo[1,2-a][l,4]benzodiazepine, a valuable precursor of potential central nervous system agents. Tetrahedron 1989, 45, 2763–2772. [Google Scholar] [CrossRef]
- Massa, S.; di Santo, R.; Costi, R.; Artico, M. Research on nitrogen containing heterocyclic compounds. XX. Synthesis of 8H-Imidazo[5,1-c]pyrrolo[1,2-a][1,4]benzodiazepine and its 6-derivatives. J. Heterocycl. Chem. 1993, 30, 749–753. [Google Scholar] [CrossRef]
- Voskressensky, L.G.; Borisova, T.N.; Babakhanova, M.I.; Titov, A.A.; Chervyakova, T.M.; Novikov, R.A.; Butin, A.S.; Khrustalev, V.N.; Varlamov, A.V. Transformation of 4-substituted tetrahydropyrrolo-benzodiazepines in a three-component reaction with methyl propiolate and indole. Chem. Heterocycl. Compd. 2014, 49, 1785–1794. [Google Scholar] [CrossRef]
- Voskressensky, L.G.; Borisova, T.N.; Babakhanova, M.I.; Chervyakova, T.M.; Titov, A.A.; Butin, A.S.; Nevolina, T.A.; Khrustalev, V.N.; Varlamov, A.V. Synthesis of pyrrolo[1,2-a][1,6]benzodiazonines from pyrrolo[1,2-a][1,4]benzodiazepines and alkynes containing electron-acceptor substituents. Chem. Heterocycl. Compd. 2013, 49, 1024–1032. [Google Scholar] [CrossRef]
- Duceppe, J.S.; Gauthier, J. Synthesis of 5H-imidazo[2,1-c]pyrrolo[1,2-a][1,4]benzodiazepine. J. Heterocycl. Chem. 1984, 21, 1685–1687. [Google Scholar] [CrossRef]
- Kayama, Y.; Hara, T.; Itoh, K.; Sunami, T. Diazepines II. A new synthesis of 4H-pyrrolo[1,2-a][1,4]-benzodiazepine. J. Heterocycl. Chem. 1977, 14, 171–172. [Google Scholar] [CrossRef]
- Korakas, D.; Varvounis, G. A convenient synthesis of 2-aminomethyl-1-arylpyrroles. Synthesis 1994, 1994, 164–166. [Google Scholar] [CrossRef]
- Korakas, D.; Varvounis, G. Synthesis of 5,6-dihydro-4H-pyrrolo[1,2-a][1,4]benzodiazepine and 10,11-dihydro-5H,12H-pyrrolo[2,1-c][1,4]benzodiazocine derivatives via cyclisation of 2-aminomethylpyrroles. J. Heterocycl. Chem. 1994, 31, 1317–1320. [Google Scholar] [CrossRef]
- Butin, A.V.; Nevolina, T.A.; Shcherbinin, V.A.; Trushkov, I.V.; Cheshkov, D.A.; Krapivin, G.D. Furan ring opening-pyrrole ring closure: A new synthetic route to aryl(heteroaryl)-annulated pyrrolo[1,2-a][1,4]diazepines. Org. Biomol. Chem. 2010, 8, 3316–3327. [Google Scholar] [CrossRef] [PubMed]
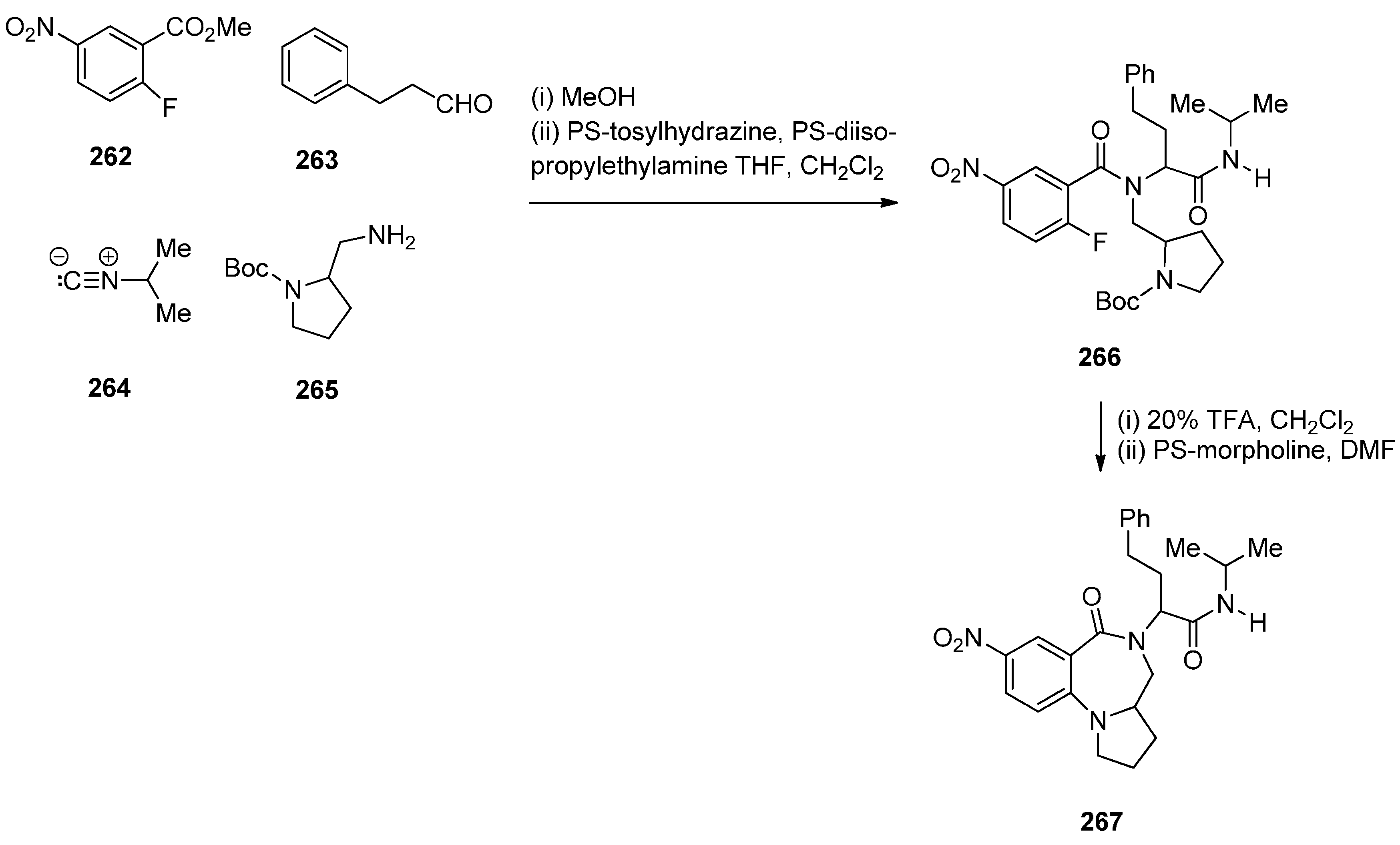
- Tempest, P.; Ma, V.; Kelly, M.G.; Jones, W.; Hulme, C. MCC/SNAr methodology. Part 1: Novel access to a range of heterocyclic cores. Tetrahedron Lett. 2001, 42, 4963–4968. [Google Scholar] [CrossRef]
- Mucedda, M.; Muroni, D.; Saba, A.; Manassero, C. Concise diastereospecific pyrrolo[1,2-a][1,4]-benzodiazepinone synthesis. Tetrahedron 2007, 63, 12232–12238. [Google Scholar] [CrossRef]
- Muroni, D.; Mucedda, M.; Saba, A. Efficient synthesis of 1-azatricyclic ring systems from anthranylamide. Heterocycles 2009, 78, 635–644. [Google Scholar]
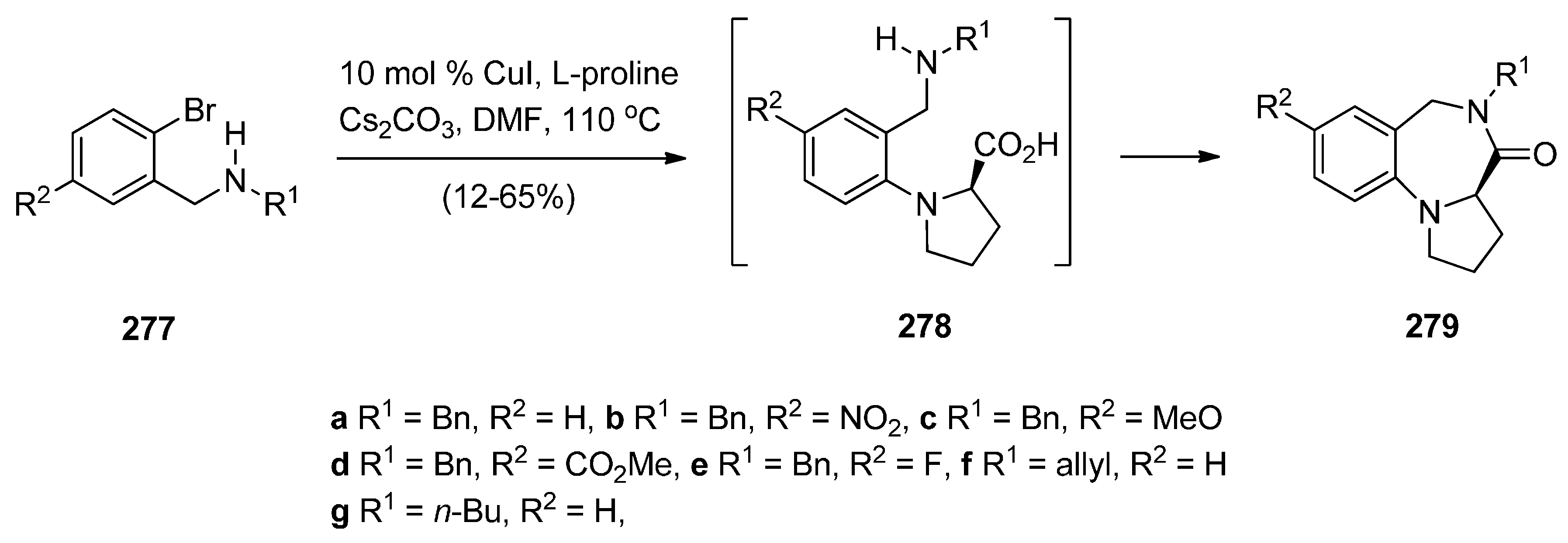
- Wang, H.; Jiang, Y.; Gao, K.; Ma, D. Facile synthesis of 1,4-benzodiazepin-3-ones from o-bromobenzylamines and amino acids via a cascade coupling/condensation process. Tetrahedron 2009, 65, 8956–8960. [Google Scholar] [CrossRef]
- Kraus, G.A.; Yue, S. Amidoalkylation Reactions of Anilines. A direct synthesis of benzodiazepines. J. Org. Chem. 1983, 48, 2936–2937. [Google Scholar] [CrossRef]
- Aiello, E.; Dattolo, G.; Cirrincione, G.; Plescia, S.E.; Daidone, G. Polycondensed nitrogen heterocycles. VII. 5,6-Dihydro-7H-pyrrolo[1,2-d][1,4]benzodiazepin-6-ones. A novel series of annelated 1,4-benzodiazepines. J. Heterocycl. Chem. 1979, 16, 209–211. [Google Scholar] [CrossRef]
- Dattolo, G.; Cirrincione, G.; Aiello, E. Polycondensed nitrogen heterocycles. IX. 5,6-Dihydro-7H-pyrrolo[1,2-d][1,4]benzodiazepin-6-one. J. Heterocycl. Chem. 1980, 17, 701–703. [Google Scholar] [CrossRef]
- Nevolina, T.A.; Shcherbinin, V.A.; Serdyuk, O.V.; Butin, A.V. Furan ring opening-pyrrole ring closure: A new route to pyrrolo[1,2-d][1,4]benzodiazepin-6-ones. Synthesis 2011, 21, 3547–3551. [Google Scholar] [CrossRef]
- Dörr, A.A.; Lubell, W.D. γ-Turn mimicry with benzodiazepinones and pyrrolobenzodiazepinones synthesized from a common amino ketone intermediate. Org. Lett. 2015, 17, 3592–3595. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varvounis, G. An Update on the Synthesis of Pyrrolo[1,4]benzodiazepines. Molecules 2016, 21, 154. https://doi.org/10.3390/molecules21020154
Varvounis G. An Update on the Synthesis of Pyrrolo[1,4]benzodiazepines. Molecules. 2016; 21(2):154. https://doi.org/10.3390/molecules21020154
Chicago/Turabian StyleVarvounis, George. 2016. "An Update on the Synthesis of Pyrrolo[1,4]benzodiazepines" Molecules 21, no. 2: 154. https://doi.org/10.3390/molecules21020154
APA StyleVarvounis, G. (2016). An Update on the Synthesis of Pyrrolo[1,4]benzodiazepines. Molecules, 21(2), 154. https://doi.org/10.3390/molecules21020154