
Discovery of Uracil Derivatives as Potent Inhibitors of Fatty Acid Amide Hydrolase

Abstract
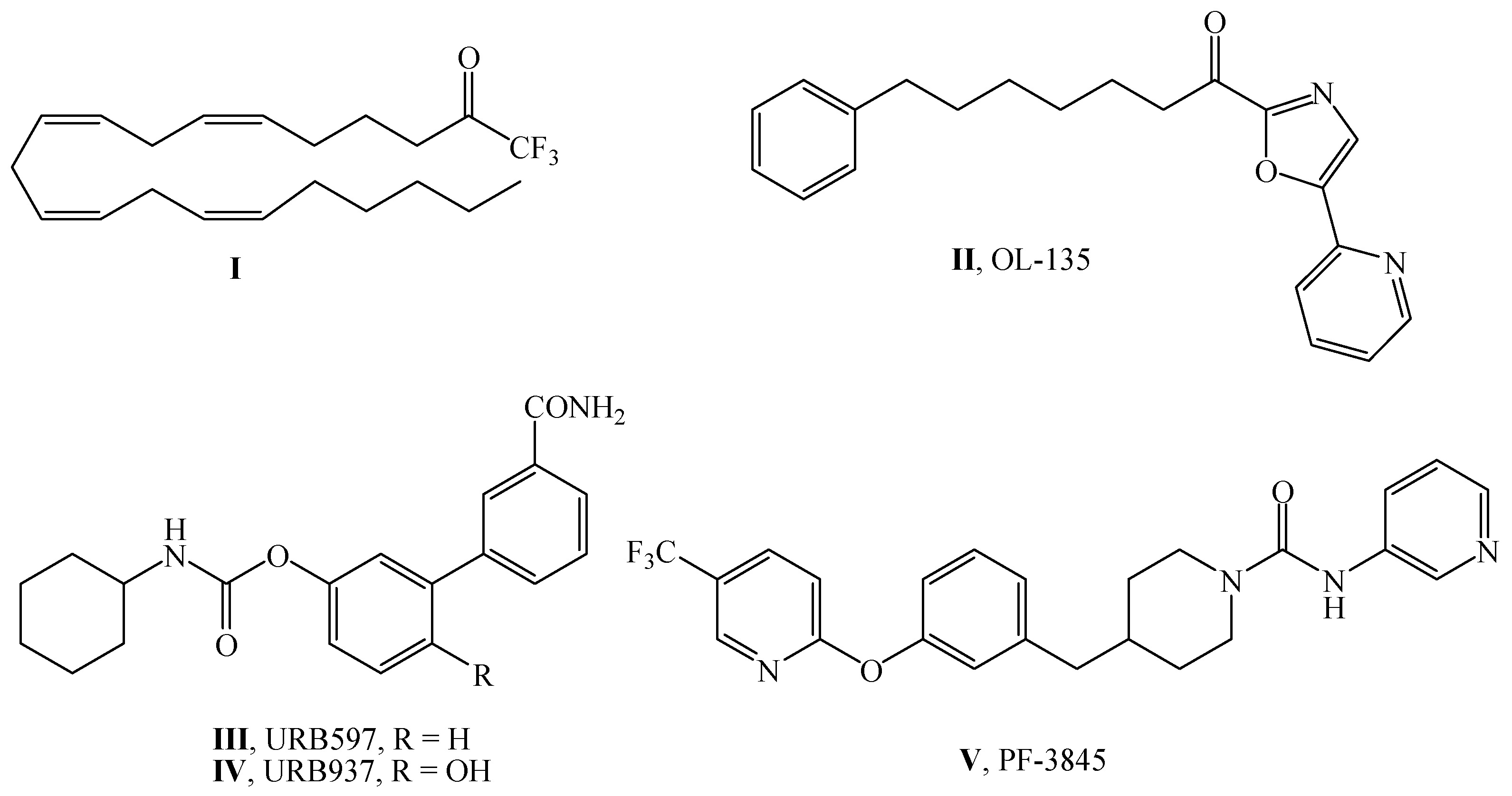
:1. Introduction
2. Results
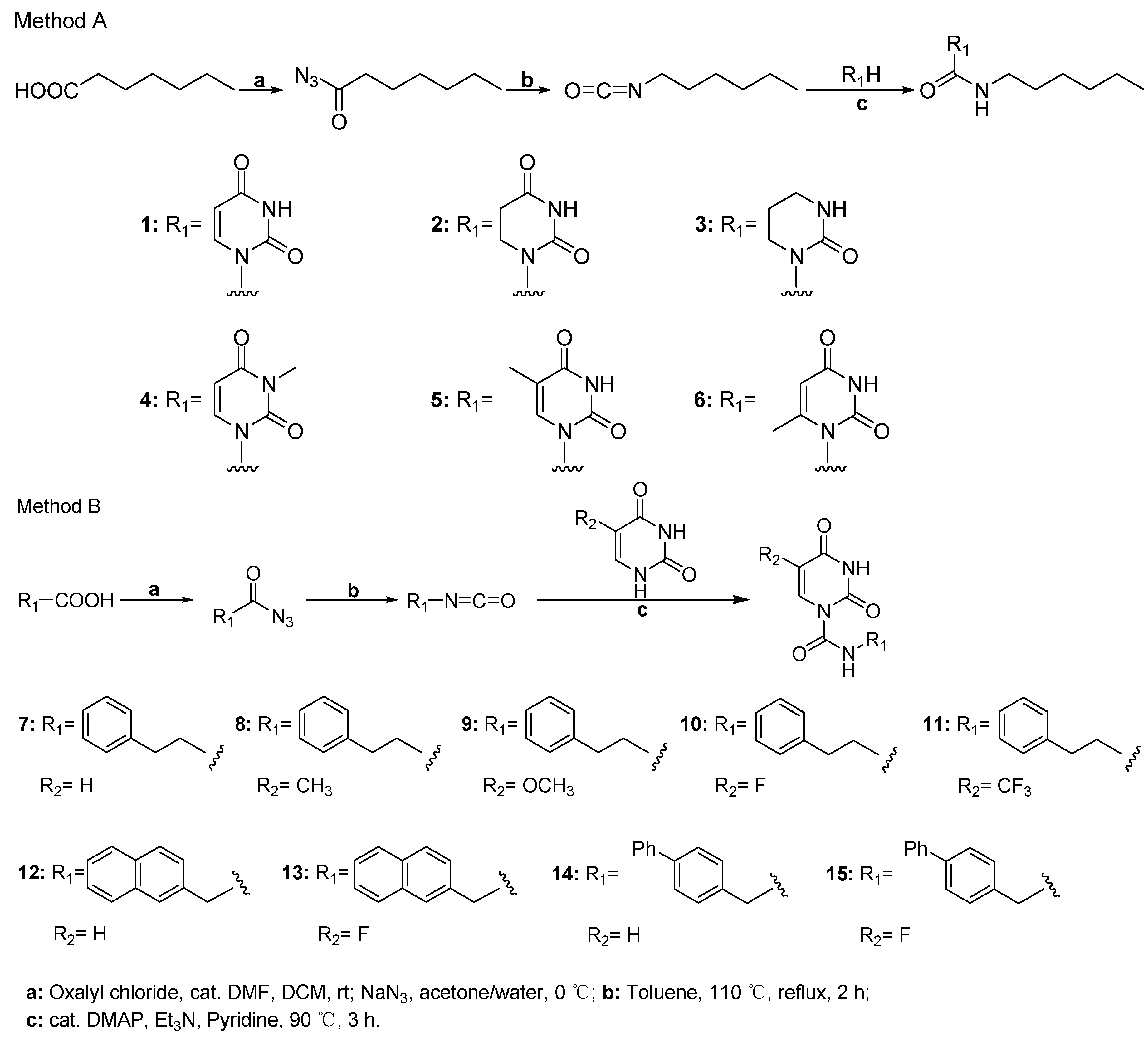
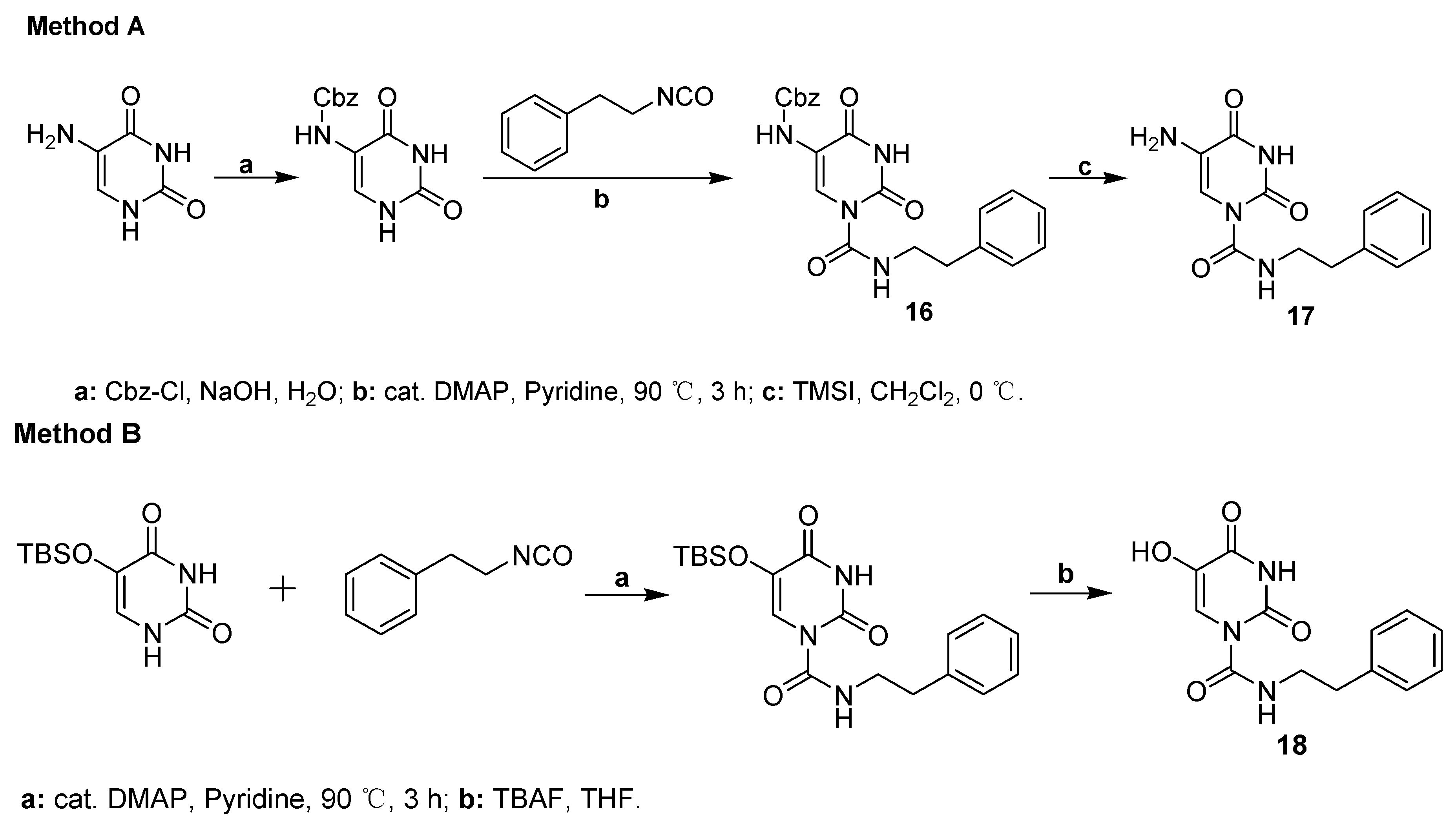
2.1. Chemistry
2.2. FAAH Inhibition
3. Discussion
4. Experimental Section
4.1. General
4.2. General Procedure for the Synthesis of Uracil Derivatives 1–15
4.3. Synthesis of Uracil Derivatives 16–17
4.4. Synthesis of Compound 18
4.5. General Procedure for the Synthesis of Uracil Derivatives 19–21
4.6. Enzyme Activity Assay
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Piomelli, D.; Sasso, O. Peripheral gating of pain signals by endogenous lipid mediators. Nat. Neurosci. 2014, 17, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.M.; Huang, S.M.; Strangman, N.M.; Tsou, K.; Sañudo-Peña, M.C. Pain modulation by release of the endogenous cannabinoid anandamide. Proc. Natl. Acad. Sci. USA 1999, 96, 12198–12203. [Google Scholar] [CrossRef] [PubMed]
- Alvheim, A.R.; Torstensen, B.E.; Lin, Y.H.; Lillefosse, H.H.; Lock, E.-J.; Madsen, L.; Hibbeln, J.R.; Malde, M.K. Dietary linoleic acid elevates endogenous 2-arachidonoylglycerol and anandamide in Atlantic salmon (Salmo salar L.) and mice, and induces weight gain and inflammation in mice. Brit. J. Nutr. 2013, 109, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Hernangómez, M.; Mestre, L.; Correa, F.G.; Loría, F.; Mecha, M.; Iñigo, P.M.; Docagne, F.; Williams, R.O.; Borrell, J.; Guaza, C. CD200-CD200R1 interaction contributes to neuroprotective effects of anandamide on experimentally induced inflammation. Glia 2012, 60, 1437–1450. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Cordaro, M.; Cuzzocrea, S. Roles of fatty acid ethanolamides (FAE) in traumatic and ischemic brain injury. Pharmacol. Res. 2014, 86, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, Y.; Ren, J.; Zhu, C.; Fu, J.; Lin, D.; Qiu, Y. Celastrol attenuates inflammatory and neuropathic pain mediated by cannabinoid receptor type 2. Int. J. Mol. Sci. 2014, 15, 13637–13648. [Google Scholar] [CrossRef] [PubMed]
- Cravatt, B.F.; Demarest, K.; Patricelli, M.P.; Bracey, M.H.; Giang, D.K.; Martin, B.R.; Lichtman, A.H. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. USA 2001, 98, 9371–9376. [Google Scholar] [CrossRef] [PubMed]
- Fezza, F.; Bari, M.; Florio, R.; Talamonti, E.; Feole, M.; Maccarrone, M. Endocannabinoids, related compounds and their metabolic routes. Molecules 2014, 19, 17078–17106. [Google Scholar] [PubMed]
- Kathuria, S.; Gaetani, S.; Fegley, D.; Valiño, F.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; La Rana, G.; Calignano, A. Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 2003, 9, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Fichna, J.; Sałaga, M.; Stuart, J.; Saur, D.; Sobczak, M.; Zatorski, H.; Timmermans, J.P.; Bradshaw, H.; Ahn, K.; Storr, M. Selective inhibition of FAAH produces antidiarrheal and antinociceptive effect mediated by endocannabinoids and cannabinoid-like fatty acid amides. Neurogastroent. Motil. 2014, 26, 470–481. [Google Scholar] [CrossRef] [PubMed]
- McKinney, M.K.; Cravatt, B.F. Structure and function of fatty acid amide hydrolase. Annu. Rev. Biochem. 2005, 74, 411–432. [Google Scholar] [CrossRef] [PubMed]
- McKinney, M.K.; Cravatt, B.F. Evidence for distinct roles in catalysis for residues of the serine-serine-lysine catalytic triad of fatty acid amide hydrolase. J. Biol. Chem. 2003, 278, 37393–37399. [Google Scholar] [CrossRef] [PubMed]
- Seierstad, M.; Breitenbucher, J.G. Discovery and development of fatty acid amide hydrolase (FAAH) inhibitors. J. Med. Chem. 2008, 51, 7327–7343. [Google Scholar] [CrossRef] [PubMed]
- Romero, F.A.; Du, W.; Hwang, I.; Rayl, T.J.; Kimball, F.S.; Leung, D.; Hoover, H.S.; Apodaca, R.L.; Breitenbucher, J.G.; Cravatt, B.F. Potent and selective α-ketoheterocycle-based inhibitors of the anandamide and oleamide catabolizing enzyme, fatty acid amide hydrolase. J. Med. Chem. 2007, 50, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Mileni, M.; Kamtekar, S.; Wood, D.C.; Benson, T.E.; Cravatt, B.F.; Stevens, R.C. Crystal structure of fatty acid amide hydrolase bound to the carbamate inhibitor URB597: Discovery of a deacylating water molecule and insight into enzyme inactivation. J. Mol. Biol. 2010, 400, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Clapper, J.R.; Moreno-Sanz, G.; Russo, R.; Guijarro, A.; Vacondio, F.; Duranti, A.; Tontini, A.; Sanchini, S.; Sciolino, N.R.; Spradley, J.M. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat. Neurosci. 2010, 13, 1265–1270. [Google Scholar] [CrossRef] [PubMed]
- Blankman, J.L.; Cravatt, B.F. Chemical probes of endocannabinoid metabolism. Pharmacol. Rev. 2013, 65, 849–871. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Johnson, D.S.; Mileni, M.; Beidler, D.; Long, J.Z.; McKinney, M.K.; Weerapana, E.; Sadagopan, N.; Liimatta, M.; Smith, S.E.; et al. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem. Biol. 2009, 16, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Realini, N.; Solorzano, C.; Pagliuca, C.; Pizzirani, D.; Armirotti, A.; Luciani, R.; Costi, M.P.; Bandiera, T.; Piomelli, D. Discovery of highly potent acid ceramidase inhibitors with in vitro tumor chemosensitizing activity. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Pizzirani, D.; Pagliuca, C.; Realini, N.; Branduardi, D.; Bottegoni, G.; Mor, M.; Bertozzi, F.; Scarpelli, R.; Piomelli, D.; Bandiera, T. Discovery of a new class of highly potent inhibitors of acid ceramidase: Synthesis and structure–activity relationship (SAR). J. Med. Chem. 2013, 56, 3518–3530. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, L.; Chen, L.; Li, Y.; Chen, H.; Li, Y.; Ji, G.; Lin, D.; Liu, Z.; Qiu, Y. Potential analgesic effects of a novel N-acylethanolamine acid amidase inhibitor F96 through PPAR-α. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, S.; Ike, Y.; Mizuno, H.; Ishikawa, K.; Mori, H. 5-Fluorouracil derivatives. I. The synthesis of 1-carbamoyl-5-fluorouracils. B Chem. Soc. Jpn. 1977, 50, 2406–2412. [Google Scholar] [CrossRef]
- Pischel, H.; Holý, A.; Veselý, J.; Wagner, G.; Cech, D. Synthesis and biological activity of N-substituted 5-fluorouracil-1-acetamides. Collect. Czechoslov. Chem. Commun. 1982, 47, 2806–2813. [Google Scholar] [CrossRef]
- Li, Y.; Yang, L.; Chen, L.; Zhu, C.; Huang, R.; Zheng, X.; Qiu, Y.; Fu, J. Design and synthesis of potent N-acylethanolamine-hydrolyzing acid amidase (NAAA) inhibitor as anti-inflammatory compounds. PLoS ONE 2012, 7, e43023. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 1–21 are available from the corresponding author.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 of FAAH Inhibition (μM) | Compound | IC50 of FAAH Inhibition (μM) |
|---|---|---|---|
| 1 | 0.11 ± 0.03 | 12 | 0.81 ± 0.10 |
| 2 | >10 | 13 | 1.21 ± 0.22 |
| 3 | >10 | 14 | 0.053 ± 0.006 |
| 4 | 0.86 ± 0.08 | 15 | 0.10 ± 0.01 |
| 5 | 0.25 ± 0.03 | 16 | 0.12 ± 0.03 |
| 6 | 1.27 ± 0.21 | 17 | 1.33 ± 0.13 |
| 7 | 0.30 ± 0.07 | 18 | 0.54 ± 0.07 |
| 8 | 1.07 ± 0.14 | 19 | >100 |
| 9 | 0.531 ± 0.08 | 20 | >100 |
| 10 | 0.098 ± 0.01 | 21 | >100 |
| 11 | 0.021 ± 0.004 | URB597 | 0.035 ± 0.008 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, Y.; Zhang, Y.; Li, Y.; Ren, J. Discovery of Uracil Derivatives as Potent Inhibitors of Fatty Acid Amide Hydrolase. Molecules 2016, 21, 229. https://doi.org/10.3390/molecules21020229
Qiu Y, Zhang Y, Li Y, Ren J. Discovery of Uracil Derivatives as Potent Inhibitors of Fatty Acid Amide Hydrolase. Molecules. 2016; 21(2):229. https://doi.org/10.3390/molecules21020229
Chicago/Turabian StyleQiu, Yan, Yang Zhang, Yuhang Li, and Jie Ren. 2016. "Discovery of Uracil Derivatives as Potent Inhibitors of Fatty Acid Amide Hydrolase" Molecules 21, no. 2: 229. https://doi.org/10.3390/molecules21020229
APA StyleQiu, Y., Zhang, Y., Li, Y., & Ren, J. (2016). Discovery of Uracil Derivatives as Potent Inhibitors of Fatty Acid Amide Hydrolase. Molecules, 21(2), 229. https://doi.org/10.3390/molecules21020229