
Compound Library Screening Identified Cardiac Glycoside Digitoxin as an Effective Growth Inhibitor of Gefitinib-Resistant Non-Small Cell Lung Cancer via Downregulation of α-Tubulin and Inhibition of Microtubule Formation

Abstract
:1. Introduction
2. Results
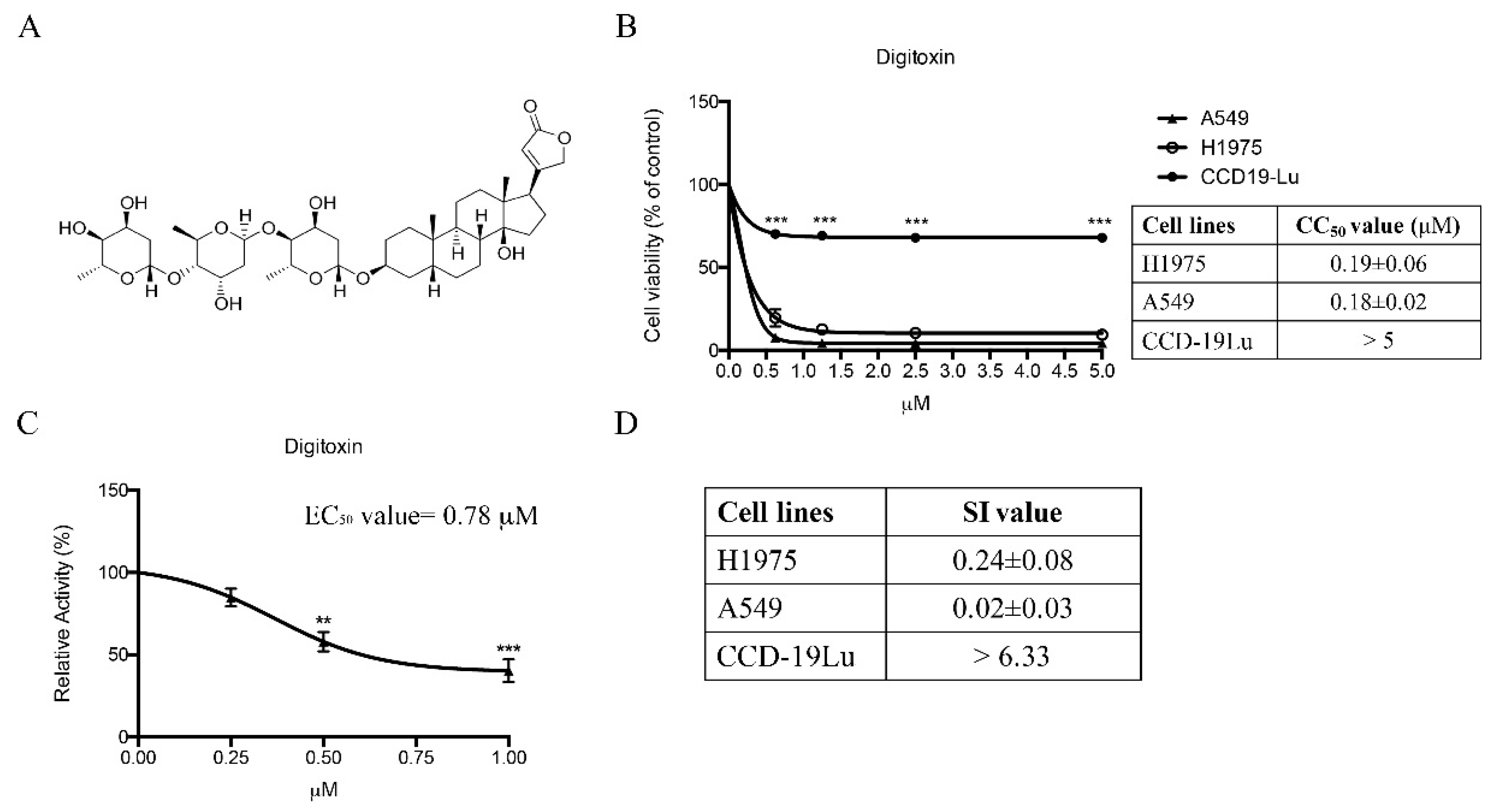
2.1. Twenty-Four Compounds Were Shortlisted from a Natural Product Library Consisting of Compounds by Comparing Their Cytotoxicity in Human NSCLC H1975 and A549 Cells
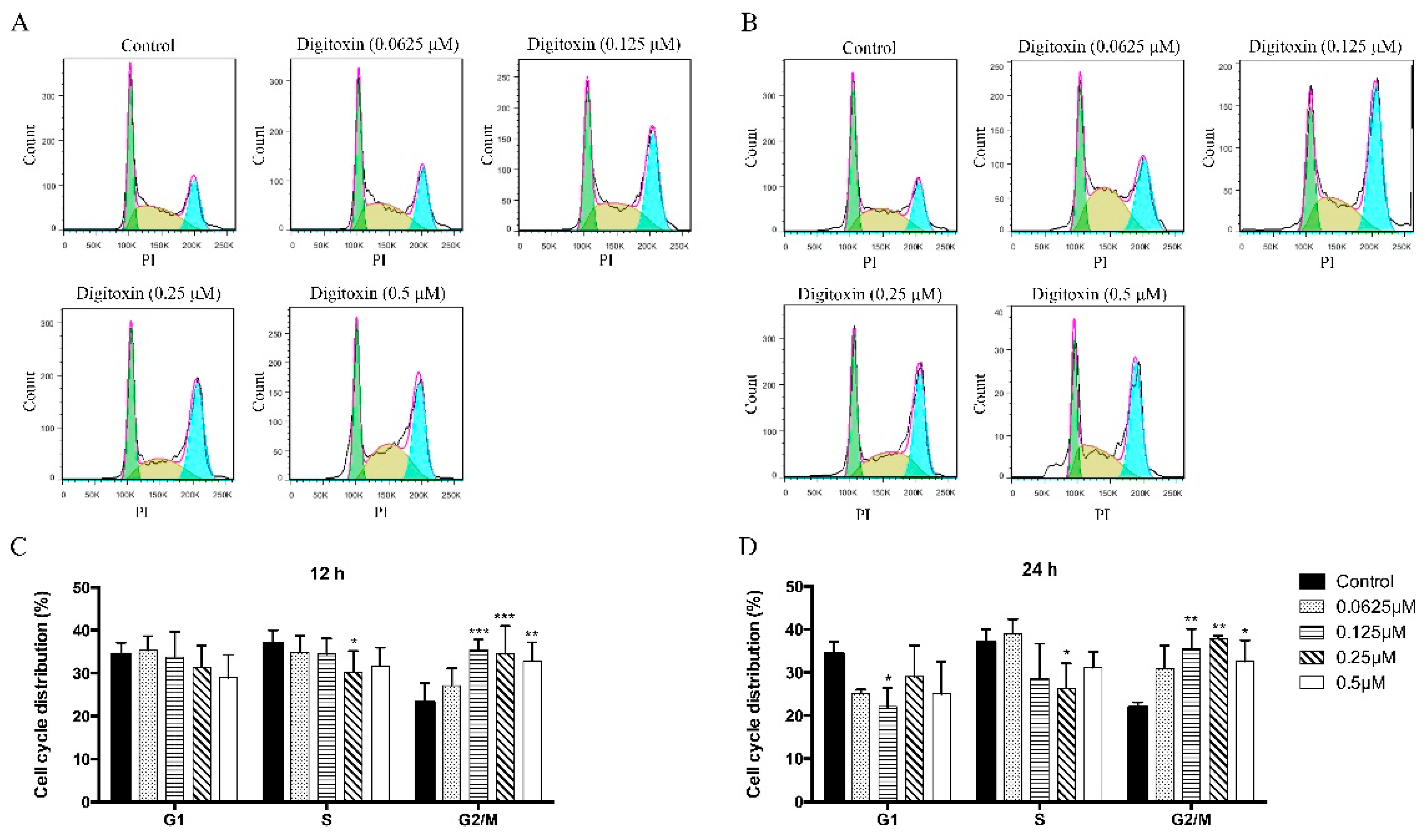
2.2. Digitoxin Induced Cell Cycle Arrest in H1975
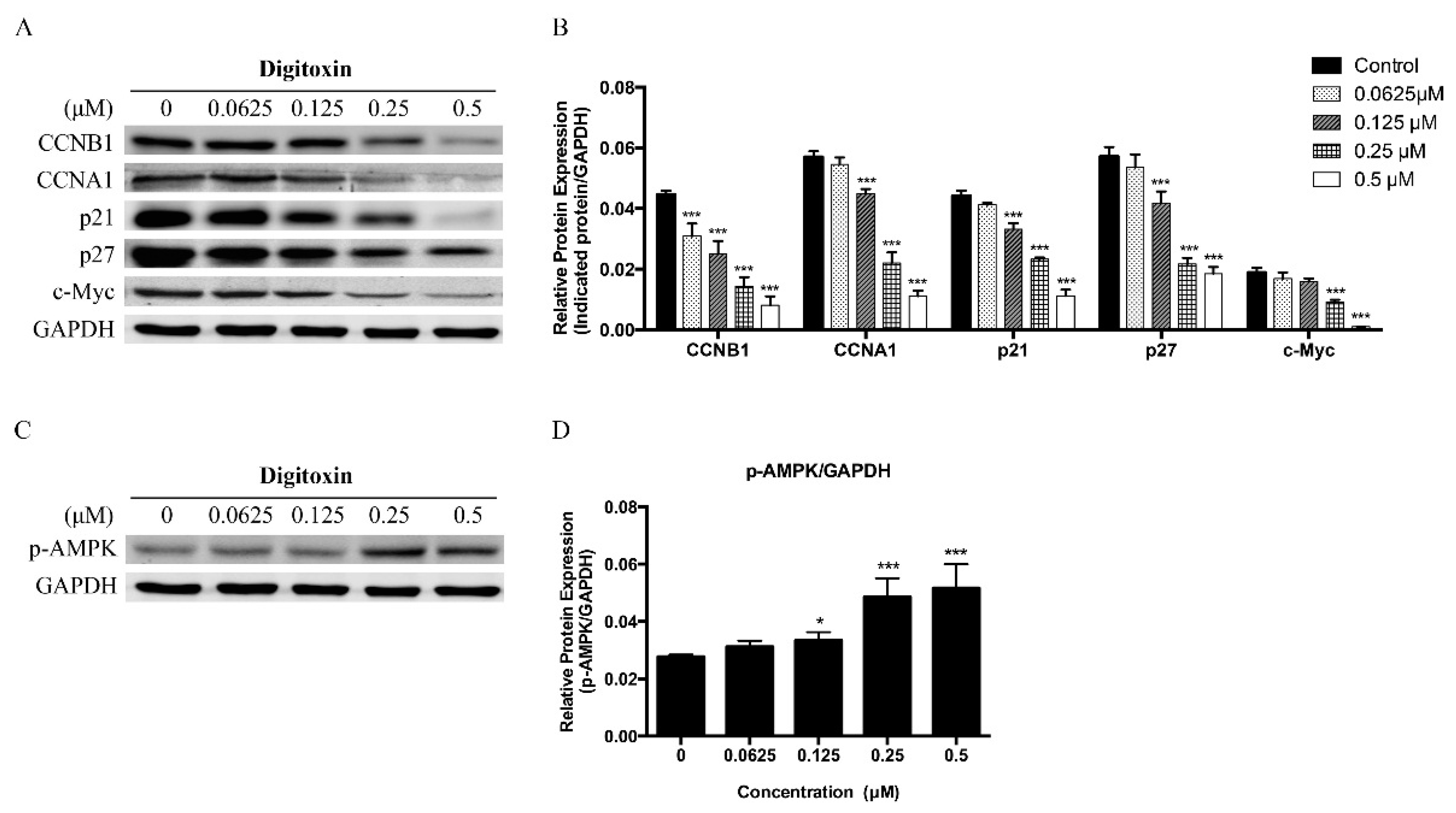
2.3. Effects of Digitoxin on Cell Cycle Regulatory Proteins in H1975
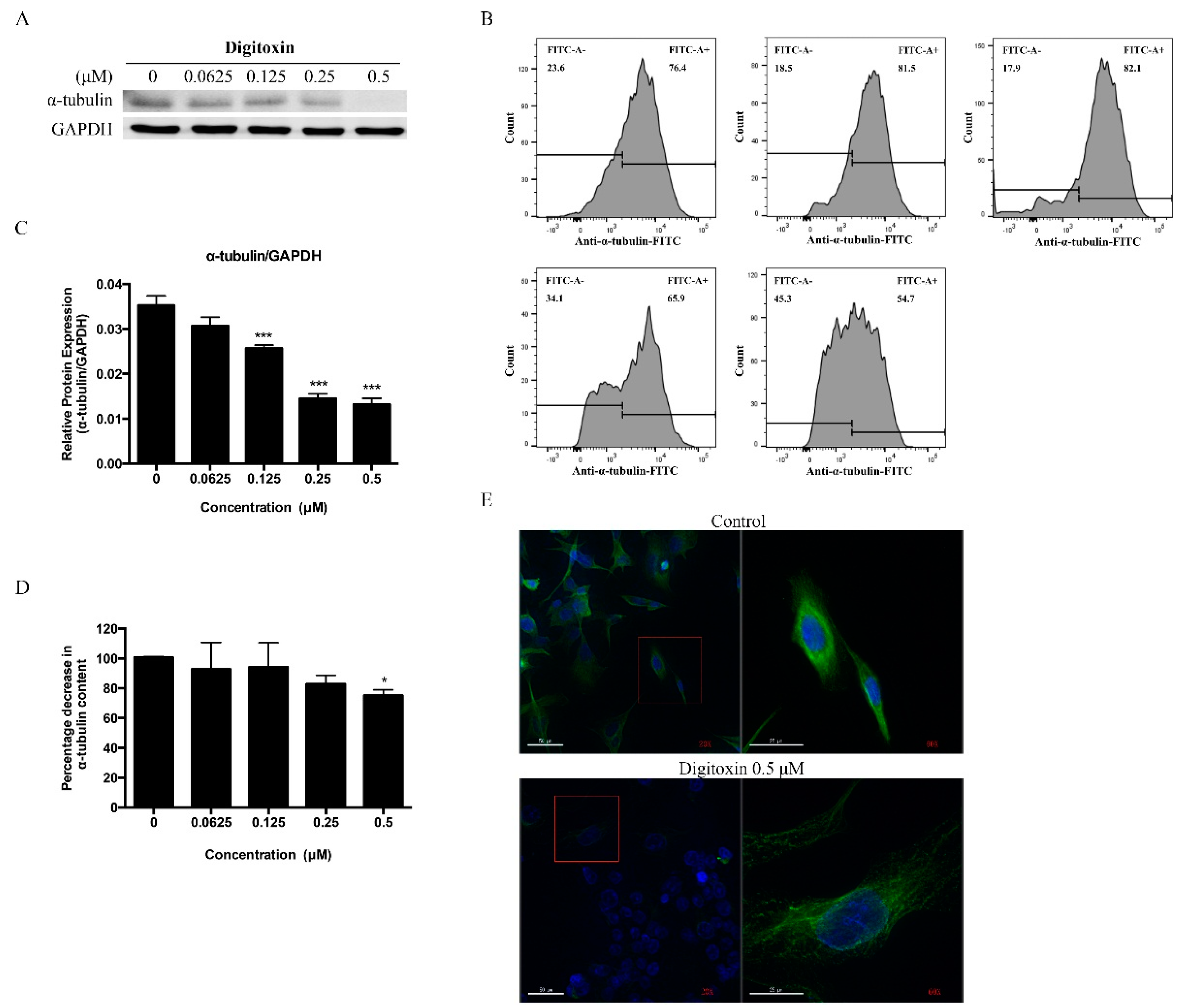
2.4. Digitoxin Inhibited Microtubule Formation
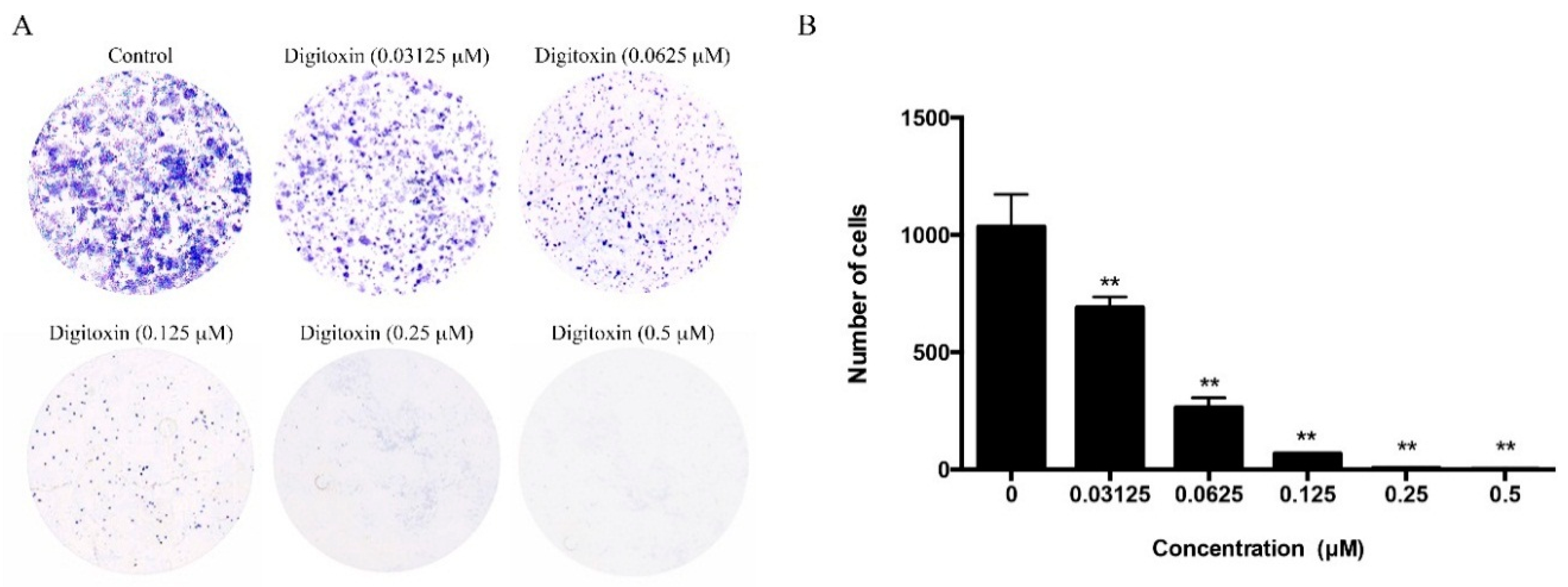
2.5. Effect of Digitoxin on Cell Proliferation
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. MTT Assay
4.4. Na+/K+ ATPase Enzymatic Activity Assay
4.5. Cell Cycle Analysis
4.6. Western Blot Analysis
4.7. Immunofluorescence Flow Cytometry
4.8. Indirect Immunofluorescence Microscopy
4.9. Colony Formation Assay
4.10. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- American Cancer Society. Cancer Facts and Figures; American Cancer Society: Atlanta, GA, USA, 2015. [Google Scholar]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Lam, W.K.; White, N.W.; Chan-Yeung, M.M. Lung cancer epidemiology and risk factors in Asia and Africa. Int. J. Tuberc. Lung Dis. 2004, 8, 1045–1057. [Google Scholar] [PubMed]
- Collins, L.G.; Haines, C.; Perkel, R.; Enck, R.E. Lung cancer: Diagnosis and management. Am. Fam. Phys. 2007, 75, 56–63. [Google Scholar]
- Onishi, H.; Shirato, H.; Nagata, Y.; Hiraoka, M.; Fujino, M.; Gomi, K.; Karasawa, K.; Hayakawa, K.; Niibe, Y.; Takai, Y.; et al. Stereotactic body radiotherapy (SBRT) for operable stage I non-small-cell lung cancer: Can SBRT be comparable to surgery? Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Uzel, E.K.; Abacioglu, U. Treatment of early stage non-small cell lung cancer: Surgery or stereotactic ablative radiotherapy? Balk. Med. J. 2015, 32, 8–16. [Google Scholar]
- Scott, W.J.; Howington, J.; Feigenberg, S.; Movsas, B.; Pisters, K. Treatment of non-small cell lung cancer stage I and stage II: ACCP evidence-based clinical practice guidelines (2nd edition). CHEST 2007, 132, 234S–242S. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, F.R.; Varella-Garcia, M.; Bunn, P.A., Jr.; Di Maria, M.V.; Veve, R.; Bremmes, R.M.; Baron, A.E.; Zeng, C.; Franklin, W.A. Epidermal growth factor receptor in non-small-cell lung carcinomas: Correlation between gene copy number and protein expression and impact on prognosis. J. Clin. Oncol. 2003, 21, 3798–3807. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Fang, R.; Sun, Y.; Han, X.; Li, F.; Gao, B.; Iafrate, A.J.; Liu, X.Y.; Pao, W.; Chen, H.; et al. Spectrum of oncogenic driver mutations in lung adenocarcinomas from East Asian never smokers. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Matsuoka, M.; Sutani, A.; Gemma, A.; Maemondo, M.; Inoue, A.; Okinaga, S.; Nagashima, M.; Oizumi, S.; Uematsu, K.; et al. Frequency of and variables associated with the EGFR mutation and its subtypes. Int. J. Cancer 2010, 126, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Fan, T.; Li, W.; Xing, W.; Huang, H. The anti-inflammatory effects of sanguinarine and its modulation of inflammatory mediators from peritoneal macrophages. Eur. J. Pharmacol. 2012, 689, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Suda, K.; Murakami, I.; Katayama, T.; Tomizawa, K.; Osada, H.; Sekido, Y.; Maehara, Y.; Yatabe, Y.; Mitsudomi, T. Reciprocal and complementary role of MET amplification and EGFR T790M mutation in acquired resistance to kinase inhibitors in lung cancer. Clin. Cancer Res. 2010, 16, 5489–5498. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhu, Q.; Zhu, L.; Pei, D.; Liu, Y.; Yin, Y.; Schuler, M.; Shu, Y. Clinical perspective of afatinib in non-small cell lung cancer. Lung Cancer 2013, 81, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zheng, X.; Huang, H.; Wu, K.; Wang, B.; Chen, X.; Ma, S. Afatinib increases sensitivity to radiation in non-small cell lung cancer cells with acquired EGFR T790M mutation. Oncotarget 2015, 6, 5832–5845. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Berna, G.; Mangas-Sanjuan, V.; Gonzalez-Alvarez, M.; Gonzalez-Alvarez, I.; Garcia-Gimenez, J.L.; Diaz Cabanas, M.J.; Bermejo, M.; Corma, A. A promising camptothecin derivative: Semisynthesis, antitumor activity and intestinal permeability. Eur. J. Med. Chem. 2014, 83, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Jayasooriya, R.G.; Choi, Y.H.; Hyun, J.W.; Kim, G.Y. Camptothecin sensitizes human hepatoma Hep3B cells to TRAIL-mediated apoptosis via ROS-dependent death receptor 5 upregulation with the involvement of MAPKs. Environ. Toxicol. Pharmacol. 2014, 38, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Warrell, R.P., Jr.; Coonley, C.J.; Gee, T.S. Homoharringtonine: An effective new drug for remission induction in refractory nonlymphoblastic leukemia. J. Clin. Oncol. 1985, 3, 617–621. [Google Scholar] [PubMed]
- Ling, Y.H.; Tseng, M.T.; Harty, J.I. Effects of homoharringtonine on protein glycosylation in human bladder carcinoma cell T-24. Cancer Res. 1989, 49, 76–80. [Google Scholar] [PubMed]
- Ko, Y.J.; Canil, C.M.; Mukherjee, S.D.; Winquist, E.; Elser, C.; Eisen, A.; Reaume, M.N.; Zhang, L.; Sridhar, S.S. Nanoparticle albumin-bound paclitaxel for second-line treatment of metastatic urothelial carcinoma: A single group, multicentre, phase 2 study. Lancet Oncol. 2013, 14, 769–776. [Google Scholar] [CrossRef]
- Einzig, A.I.; Hochster, H.; Wiernik, P.H.; Trump, D.L.; Dutcher, J.P.; Garowski, E.; Sasloff, J.; Smith, T.J. A phase II study of taxol in patients with malignant melanoma. Investig. New Drugs 1991, 9, 59–64. [Google Scholar] [CrossRef]
- Hostanska, K.; Jurgenliemk, G.; Abel, G.; Nahrstedt, A.; Saller, R. Willow bark extract (BNO1455) and its fractions suppress growth and induce apoptosis in human colon and lung cancer cells. Cancer Detect. Prev. 2007, 31, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.; Lee, J.Y.; Hung, H.Y.; Shi, Q.; Lin, L.; Zhao, Y.; Goto, M.; Yang, P.C.; Kuo, S.C.; Chen, H.W.; et al. Novel curcumin analogs to overcome EGFR-TKI lung adenocarcinoma drug resistance and reduce EGFR-TKI-induced GI adverse effects. Bioorg. Med. Chem. 2015, 23, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.X.; Li, N.; Wu, J.L.; Zhou, Y.L.; He, J.X.; Liu, L.; Leung, E.L. Celastrol induces apoptosis in gefitinib-resistant non-small cell lung cancer cells via caspases-dependent pathways and Hsp90 client protein degradation. Molecules 2014, 19, 3508–3522. [Google Scholar] [CrossRef] [PubMed]
- Yoda, A.; Hokin, L.E. On the reversibility of binding of cardiotonic steroids to a partially purified (Na+K)-activated adenosinetriphosphatase from beef brain. Biochem. Biophys. Res. Commun. 1970, 40, 880–886. [Google Scholar] [CrossRef]
- De Pover, A.; Godfraind, T. Influence of 16 beta formylation on Na, K-ATPase inhibition by cardiac glycosides. Naunyn Schmiedebergs Arch. Pharmacol. 1982, 321, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar] [CrossRef]
- Gu, Y.; Turck, C.W.; Morgan, D.O. Inhibition of CDK2 activity in vivo by an associated 20K regulatory subunit. Nature 1993, 366, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ma, X.; Jones, H.M.; Chan, L.L.; Song, F.; Zhang, W.; Bae-Jump, V.L.; Zhou, C. Evaluation of the antitumor effects of c-Myc-Max heterodimerization inhibitor 100258-F4 in ovarian cancer cells. J. Transl. Med. 2014, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.L.; Sui, Y.; Qiao, C.F.; Yip, K.Y.; Leung, R.K.; Tsui, S.K.; Lee, H.M.; Wong, H.K.; Zhu, X.; Siu, J.J.; et al. Sustained antidiabetic effects of a berberine-containing Chinese herbal medicine through regulation of hepatic gene expression. Diabetes 2012, 61, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wang, Q.; Zhang, Y. A systems pharmacology approach to decipher the mechanism of danggui-shaoyao-san decoction for the treatment of neurodegenerative diseases. J. Ethnopharmacol. 2016, 3, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Lengfelder, E.; Hofmann, W.K.; Nowak, D. Impact of arsenic trioxide in the treatment of acute promyelocytic leukemia. Leukemia 2012, 26, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Li, P.C.; Konkimalla, V.S.; Kaina, B. From traditional Chinese medicine to rational cancer therapy. Trends Mol. Med. 2007, 13, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Gheorghiade, M.; van Veldhuisen, D.J.; Colucci, W.S. Contemporary use of digoxin in the management of cardiovascular disorders. Circulation 2006, 113, 2556–2564. [Google Scholar] [CrossRef] [PubMed]
- Watabe, M.; Masuda, Y.; Nakajo, S.; Yoshida, T.; Kuroiwa, Y.; Nakaya, K. The cooperative interaction of two different signaling pathways in response to bufalin induces apoptosis in human leukemia U937 cells. J. Biol. Chem. 1996, 271, 14067–14072. [Google Scholar] [PubMed]
- Yeh, J.Y.; Huang, W.J.; Kan, S.F.; Wang, P.S. Effects of bufalin and cinobufagin on the proliferation of androgen dependent and independent prostate cancer cells. Prostate 2003, 54, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Han, K.Q.; Huang, G.; Gu, W.; Su, Y.H.; Huang, X.Q.; Ling, C.Q. Anti-tumor activities and apoptosis-regulated mechanisms of bufalin on the orthotopic transplantation tumor model of human hepatocellular carcinoma in nude mice. World J. Gastroenterol. 2007, 13, 3374–3379. [Google Scholar] [CrossRef] [PubMed]
- Stenkvist, B.; Bengtsson, E.; Dahlqvist, B.; Eriksson, O.; Jarkrans, T.; Nordin, B. Cardiac glycosides and breast cancer, revisited. New Engl. J. Med. 1982, 306, 484. [Google Scholar] [PubMed]
- Trenti, A.; Grumati, P.; Cusinato, F.; Orso, G.; Bonaldo, P.; Trevisi, L. Cardiac glycoside ouabain induces autophagic cell death in non-small cell lung cancer cells via a JNK-dependent decrease of Bcl-2. Biochem. Pharmacol. 2014, 89, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Wolle, D.; Lee, S.J.; Li, Z.; Litan, A.; Barwe, S.P.; Langhans, S.A. Inhibition of epidermal growth factor signaling by the cardiac glycoside ouabain in medulloblastoma. Cancer Med. 2014, 3, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Kometiani, P.; Liu, L.; Askari, A. Digitalis-induced signaling by Na+/K+-ATPase in human breast cancer cells. Mol. Pharmacol. 2005, 67, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Chang, H.H.; Lai, Y.H.; Lin, C.H.; Chen, M.H.; Chang, G.C.; Tsai, M.F.; Chen, J.J. Digoxin Suppresses Tumor Malignancy through Inhibiting Multiple Src-Related Signaling Pathways in Non-Small Cell Lung Cancer. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- McConkey, D.J.; Lin, Y.; Nutt, L.K.; Ozel, H.Z.; Newman, R.A. Cardiac glycosides stimulate Ca2+ increases and apoptosis in androgen-independent, metastatic human prostate adenocarcinoma cells. Cancer Res. 2000, 60, 3807–3812. [Google Scholar] [PubMed]
- Elbaz, H.A.; Stueckle, T.A.; Wang, H.Y.; O’Doherty, G.A.; Lowry, D.T.; Sargent, L.M.; Wang, L.; Dinu, C.Z.; Rojanasakul, Y. Digitoxin and a synthetic monosaccharide analog inhibit cell viability in lung cancer cells. Toxicol. Appl. Pharmacol. 2012, 258, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Okina, Y.; Takeuchi, F.; Yokomichi, T.; Takada, Y.; Kataoka, T. Cardenolide aglycones inhibit tumor necrosis factor alpha-induced expression of intercellular adhesion molecule-1 at the translation step by blocking Na(+)/K(+)-ATPase. Biol. Pharm. Bull. 2015, 38, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Peng, J.J.; Gao, H.; Li, H.; Li, D.; Tan, Y.; Zhang, T. Digoxin downregulates NDRG1 and VEGF through the inhibition of HIF-1alpha under hypoxic conditions in human lung adenocarcinoma A549 cells. Int. J. Mol. Sci. 2013, 14, 7273–7285. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhan, Y.; Xu, R.; Shao, R.; Jiang, J.; Wang, Z. Src mediates extracellular signal-regulated kinase 1/2 activation and autophagic cell death induced by cardiac glycosides in human non-small cell lung cancer cell lines. Mol. Carcinog. 2015, 54, E26–E34. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.K.; Ryu, B.J.; Choi, S.W.; Kim, S.H.; Lee, K. Inactivation of Src-to-ezrin pathway: A possible mechanism in the ouabain-mediated inhibition of A549 cell migration. Biomed. Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Li, Y.; Su, S.; Wang, N.; Wang, H.; Li, J. Inhibition of cell migration by ouabain in the A549 human lung cancer cell line. Oncol. Lett. 2013, 6, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Matsuo, K.; Ogura, H.; Bai, L.; Toki, A.; Wang, L.; Ando, M.; Kataoka, T. Odoroside A and ouabain inhibit Na+/K+-ATPase and prevent NF-kappaB-inducible protein expression by blocking Na+-dependent amino acid transport. Biochem.Pharmacol. 2009, 78, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qiu, Q.; Shen, J.J.; Li, D.D.; Jiang, X.J.; Si, S.Y.; Shao, R.G.; Wang, Z. Cardiac glycosides induce autophagy in human non-small cell lung cancer cells through regulation of dual signaling pathways. Int. J. Biochem. Cell Biol. 2012, 44, 1813–1824. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.S. Reduction of doxorubicin cytotoxicity by ouabain: Correlation with topoisomerase-induced DNA strand breakage in human and hamster cells. Cancer Res. 1988, 48, 725–730. [Google Scholar] [PubMed]
- Verheye-Dua, F.A.; Bohm, L. Influence of ouabain on cell inactivation by irradiation. Strahlenther. Onkol. 1996, 172, 156–161. [Google Scholar] [PubMed]
- Yakisich, J.S.; Azad, N.; Venkatadri, R.; Kulkarni, Y.; Wright, C.; Kaushik, V.; O’Doherty, G.A.; Iyer, A.K. Digitoxin and its synthetic analog MonoD have potent antiproliferative effects on lung cancer cells and potentiate the effects of hydroxyurea and paclitaxel. Oncol. Rep. 2015, 35, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Ninsontia, C.; Chanvorachote, P. Ouabain mediates integrin switch in human lung cancer cells. Anticancer Res. 2014, 34, 5495–5502. [Google Scholar] [PubMed]
- Owa, T.; Yoshino, H.; Yoshimatsu, K.; Nagasu, T. Cell cycle regulation in the G1 phase: A promising target for the development of new chemotherapeutic anticancer agents. Curr. Med. Chem. 2001, 8, 1487–1503. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Trovesi, C.; Manfrini, N.; Falcettoni, M.; Longhese, M.P. Regulation of the DNA damage response by cyclin-dependent kinases. J. Mol. Biol. 2013, 425, 4756–4766. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W. Cyclin dependent kinase inhibitors. Cancer Surv. 1997, 29, 91–107. [Google Scholar] [PubMed]
- Deng, C.X. BRCA1: Cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006, 34, 1416–1426. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.; Pabla, N.; Dong, Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell. Mol. Life Sci. 2013, 70, 4009–4021. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Yu, H.; Liang, X.; Xu, J.; Cai, X. Chk1-induced CCNB1 overexpression promotes cell proliferation and tumor growth in human colorectal cancer. Cancer Biol. Ther. 2014, 15, 1268–1279. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, J.M.; Kothari, A.; Chambers, T.C. Cyclin B1 overexpression induces cell death independent of mitotic arrest. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Yam, C.H.; Fung, T.K.; Poon, R.Y. Cyclin A in cell cycle control and cancer. Cell. Mol. Life Sci. 2002, 59, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Sanli, T.; Steinberg, G.R.; Singh, G.; Tsakiridis, T. AMP-activated protein kinase (AMPK) beyond metabolism: A novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol. Ther. 2014, 15, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol. 2009, 196, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Bensimon, A.; Aebersold, R.; Shiloh, Y. Beyond ATM: The protein kinase landscape of the DNA damage response. FEBS Lett. 2011, 585, 1625–1639. [Google Scholar] [CrossRef] [PubMed]
- Storozhuk, Y.; Hopmans, S.N.; Sanli, T.; Barron, C.; Tsiani, E.; Cutz, J.C.; Pond, G.; Wright, J.; Singh, G.; Tsakiridis, T. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br. J. Cancer 2013, 108, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Koh, H.; Kim, M.; Kim, Y.; Lee, S.Y.; Karess, R.E.; Lee, S.H.; Shong, M.; Kim, J.M.; Kim, J.; et al. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature 2007, 447, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalving, M.; Gietema, J.A.; Lefrandt, J.D.; de Jong, S.; Reyners, A.K.; Gans, R.O.; de Vries, E.G. Metformin: Taking away the candy for cancer? Eur. J. Cancer 2010, 46, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Fassett, J.T.; Hu, X.; Xu, X.; Lu, Z.; Zhang, P.; Chen, Y.; Bache, R.J. AMPK attenuates microtubule proliferation in cardiac hypertrophy. Am. J. Physiol. 2013, 304, H749–H758. [Google Scholar] [CrossRef] [PubMed]
- Weisenberg, R.C. Microtubule formation in vitro in solutions containing low calcium concentrations. Science 1972, 177, 1104–1105. [Google Scholar] [CrossRef] [PubMed]
- Marx, A.; Nugoor, C.; Panneerselvam, S.; Mandelkow, E. Structure and function of polarity-inducing kinase family MARK/Par-1 within the branch of AMPK/Snf1-related kinases. FASEB J. 2010, 24, 1637–1648. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Evans, A.; Pham, J.; Plosky, B. Cellular stress responses: A balancing act. Mol. Cell 2010, 40, 175. [Google Scholar] [CrossRef] [PubMed]
- Mitchison, T.; Kirschner, M. Dynamic instability of microtubule growth. Nature 1984, 312, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Cueva, J.G.; Hsin, J.; Huang, K.C.; Goodman, M.B. Posttranslational acetylation of alpha-tubulin constrains protofilament number in native microtubules. Curr. Biol. 2012, 22, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Peris, L.; Thery, M.; Faure, J.; Saoudi, Y.; Lafanechere, L.; Chilton, J.K.; Gordon-Weeks, P.; Galjart, N.; Bornens, M.; Wordeman, L.; et al. Tubulin tyrosination is a major factor affecting the recruitment of CAP-Gly proteins at microtubule plus ends. J. Cell Biol. 2006, 174, 839–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peris, L.; Wagenbach, M.; Lafanechere, L.; Brocard, J.; Moore, A.T.; Kozielski, F.; Job, D.; Wordeman, L.; Andrieux, A. Motor-dependent microtubule disassembly driven by tubulin tyrosination. J. Cell Biol. 2009, 185, 1159–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackeh, R.; Lorin, S.; Ratier, A.; Mejdoubi-Charef, N.; Baillet, A.; Bruneel, A.; Hamai, A.; Codogno, P.; Pous, C.; Perdiz, D. Reactive oxygen species, AMP-activated protein kinase, and the transcription cofactor p300 regulate α-tubulin acetyltransferase-1 (αTAT-1/MEC-17)-dependent microtubule hyperacetylation during cell stress. J. Biol. Chem. 2014, 289, 11816–11828. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Ishihara, K.; Cooper, G.T. Cytoskeletal role in the contractile dysfunction of hypertrophied myocardium. Science 1993, 260, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Medellin, D.C.; Zhou, Q.; Scott, R.; Hill, R.M.; Frail, S.K.; Dasari, R.; Ontiveros, S.J.; Pelly, S.C.; van Otterlo, W.A.; Betancourt, T.; et al. Novel Microtubule-Targeting 7-Deazahypoxanthines Derived from Marine Alkaloid Rigidins with Potent in Vitro and in Vivo Anticancer Activities. J. Med. Chem. 2015, 59, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Denning, D.P.; Hirose, T. Anti-tubulins DEPendably induce apoptosis. Nat. Cell Biol. 2014, 16, 741–743. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the 800 compounds are available from the authors.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CC50 Value (μM) | ||
|---|---|---|
| Candidate Compounds | H1975 (EGFR L858R+T790M) | A549 (EGFR WT) |
| Digitoxin | 0.19 ± 0.06 | 0.18 ± 0.02 |
| Daunorubicin | 0.24 ± 0.11 | 0.23 ± 0.03 |
| Proscillaridin | 0.32 ± 0.21 | 0.042 ± 0.02 |
| Pyrromycin | 0.33 ± 0.13 | 0.031 ± 0.003 |
| Strophanthidinic acid lactone acetate | 0.36 ± 0.17 | 0.08 ± 0.03 |
| Plumbagin | 0.39 ± 0.13 | 2.52 ± 1.84 |
| Camptothecin | 0.41 ± 0.17 | 0.11 ± 0.02 |
| Digydrocelastryl Diacetate | 0.52 ± 0.14 | 2.16 ± 1.16 |
| Celastrol | 0.53 ± 0.15 | 1.49 ± 0.79 |
| Valinomycin | 0.64 ± 0.26 | 0.43 ± 0.20 |
| Antimucin | 0.64 ± 0.38 | 4.23 ± 2.30 |
| Picropodophyllin | 0.75 ± 0.33 | 1.02 ± 0.54 |
| Anthothecol | 0.79 ± 0.22 | 2.46 ± 1.31 |
| 5alpha-cholestan-3beta-ol-6-one | 0.81 ± 0.39 | 1.02 ± 0.58 |
| Patulin | 1.03 ± 0.29 | 1.62 ± 0.44 |
| Rotenone | 1.06 ± 0.46 | 0.82 ± 0.36 |
| Gambofic acid | 1.15 ± 0.69 | 3.85 ± 1.81 |
| Strophanthidin | 1.19 ± 0.43 | 0.16 ± 0.03 |
| Estrone Benzoate | 1.40 ± 0.39 | 0.40 ± 0.14 |
| Picropodophyllin Acentate | 1.57 ± 0.69 | 0.67 ± 0.25 |
| Paclitaxel | 1.67 ± 0.80 | 0.79 ± 0.49 |
| Monensin Sodium | 1.71 ± 0.76 | 1.16 ± 0.35 |
| Dihydrorotenone | 2.27 ± 1.10 | 2.21 ± 1.36 |
| Isorotenone | 2.49 ± 1.2 | 1.35 ± 0.796 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.-Z.; Chen, X.; Fan, X.-X.; He, J.-X.; Huang, J.; Xiao, D.-K.; Zhou, Y.-L.; Zheng, S.-Y.; Xu, J.-H.; Yao, X.-J.; et al. Compound Library Screening Identified Cardiac Glycoside Digitoxin as an Effective Growth Inhibitor of Gefitinib-Resistant Non-Small Cell Lung Cancer via Downregulation of α-Tubulin and Inhibition of Microtubule Formation. Molecules 2016, 21, 374. https://doi.org/10.3390/molecules21030374
Zhang Y-Z, Chen X, Fan X-X, He J-X, Huang J, Xiao D-K, Zhou Y-L, Zheng S-Y, Xu J-H, Yao X-J, et al. Compound Library Screening Identified Cardiac Glycoside Digitoxin as an Effective Growth Inhibitor of Gefitinib-Resistant Non-Small Cell Lung Cancer via Downregulation of α-Tubulin and Inhibition of Microtubule Formation. Molecules. 2016; 21(3):374. https://doi.org/10.3390/molecules21030374
Chicago/Turabian StyleZhang, Yi-Ze, Xi Chen, Xing-Xing Fan, Jian-Xing He, Jun Huang, Da-Kai Xiao, Yan-Ling Zhou, Sen-You Zheng, Jia-Hui Xu, Xiao-Jun Yao, and et al. 2016. "Compound Library Screening Identified Cardiac Glycoside Digitoxin as an Effective Growth Inhibitor of Gefitinib-Resistant Non-Small Cell Lung Cancer via Downregulation of α-Tubulin and Inhibition of Microtubule Formation" Molecules 21, no. 3: 374. https://doi.org/10.3390/molecules21030374
APA StyleZhang, Y. -Z., Chen, X., Fan, X. -X., He, J. -X., Huang, J., Xiao, D. -K., Zhou, Y. -L., Zheng, S. -Y., Xu, J. -H., Yao, X. -J., Liu, L., & Leung, E. L. -H. (2016). Compound Library Screening Identified Cardiac Glycoside Digitoxin as an Effective Growth Inhibitor of Gefitinib-Resistant Non-Small Cell Lung Cancer via Downregulation of α-Tubulin and Inhibition of Microtubule Formation. Molecules, 21(3), 374. https://doi.org/10.3390/molecules21030374