
Development and Validation of an Ultra-High Performance Liquid Chromatography-Tandem Mass Spectrometry Method for Simultaneous Determination of Four Type B Trichothecenes and Masked Deoxynivalenol in Various Feed Products


Abstract
:1. Introduction
2. Results and Discussion
2.1. Optimization of Sample Preparation
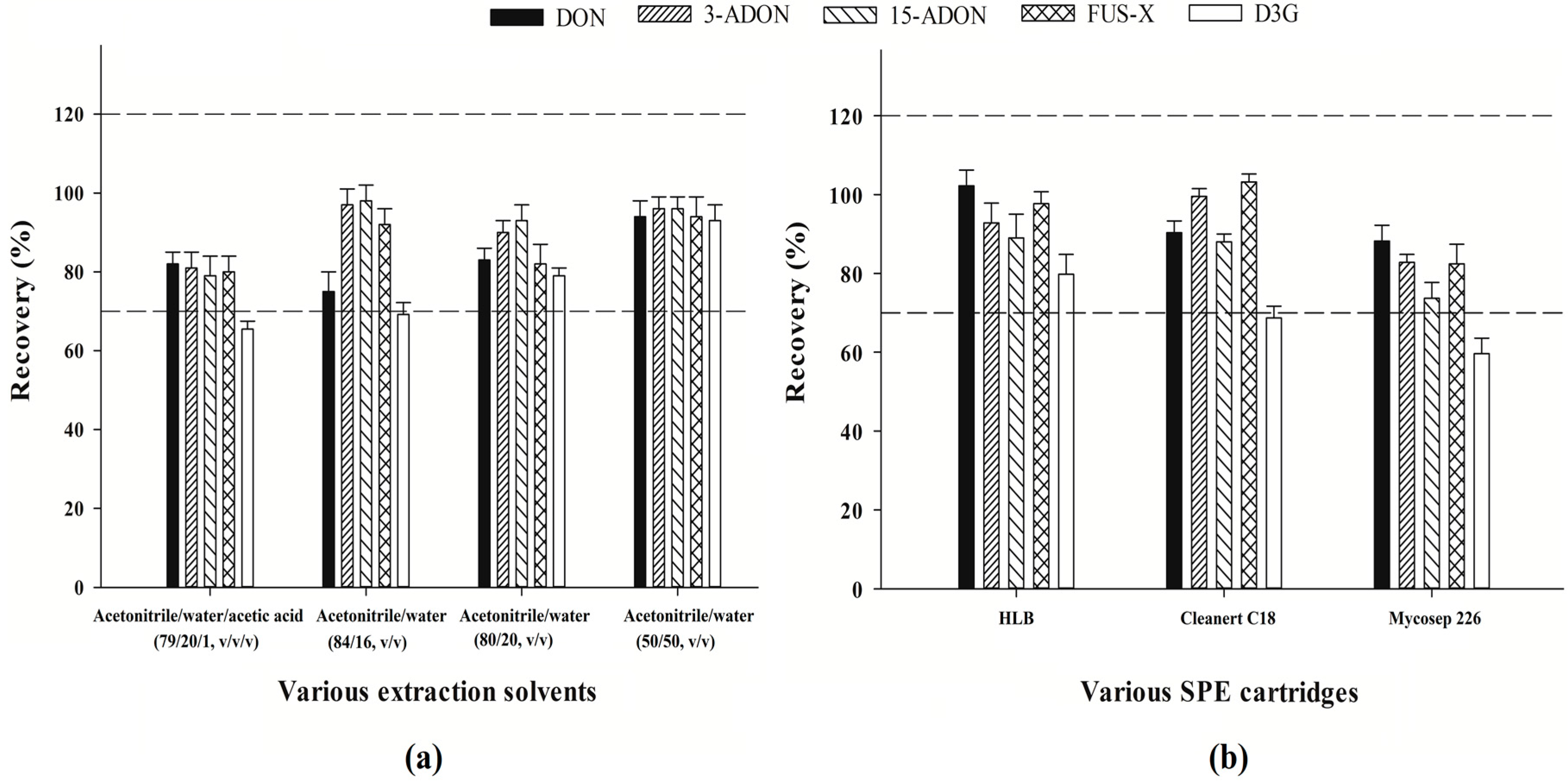
2.1.1. Selection of Extraction Method
2.1.2. Optimization of SPE Clean-Up Procedure
2.1.3. Evaluation of the HLB SPE Clean-up Method
2.2. Optimization of UHPLC-MS/MS Conditions
2.3. Method Validation
2.4. Method Application
3. Materials and Methods
3.1. Chemicals and Regents
3.2. Preparation of Standard Solutions
3.3. Samples
3.4. Sample Preparation
3.5. UHPLC-MS/MS Analysis
3.6. Method Validation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DON | deoxynivalenol |
| 3-ADON | 3-acetyldeoxynivalenol |
| 15-ADON | 15-acetyldeoxynivalenol |
| FUS-X | fusarenon X |
| D3G | deoxynivalenol-3-glucoside |
| UHPLC-MS/MS | ultra-high performance liquid chromatography-tandem mass spectrometry |
| TLC | thin-layer chromatography |
| ELISA | enzyme-linked immune-sorbent assay |
| HPLC | high-performance liquid chromatography |
| ESI | electrospray ionization |
| APCI | atmospheric pressure chemical ionization |
| LLE | liquid liquid extraction |
| DSPE | dispersive solid-phase extraction |
| SPE | solid phase extraction |
| LOD | limit of detection |
| LOQ | limit of quantification |
| MRM | multiple reaction monitoring |
| S/N | signal-to-noise ratio |
| SSE | signal suppression/enhancement |
| RSD | relative standard deviation |
References
- Urban, M.; Daniels, S.; Mott, E.; Hammond-Kosack, K. Arabidopsis is susceptible to the cereal ear blight fungal pathogens Fusarium graminearum and Fusarium culmorum. Plant J. 2002, 32, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Kokkonen, M.; Ojala, L.; Parikka, P.; Jestoi, M. Mycotoxin production of selected Fusarium species at different culture conditions. Int. J. Food Microbiol. 2010, 143, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Jiao-Jie, M.; Chuan-Chuan, Y.; Xiao-Hui, L.; Hong-Ru, J.; Bing, S.; Feng-Qin, L. Simultaneous determination of masked deoxynivalenol and some important type B trichothecenes in Chinese corn kernels and corn-based products by ultra-performance liquid chromatography-tandem mass spectrometry. J. Agric. Food Chem. 2012, 60, 11638–11646. [Google Scholar] [CrossRef] [PubMed]
- Ward, T.J.; Bielawski, J.P.; Kistler, H.C.; Sullivan, E.; O’Donnell, K. Ancestral polymorphism and adaptive evolution in the trichothecene mycotoxin gene cluster of phytopathogenic Fusarium. Proc. Natl. Acad. Sci. USA 2002, 99, 9278–9283. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhang, Y.; Shao, S.; Cai, Z.; Feng, L.; Pan, H.; Wang, Z. Simultaneous determination of multi-component mycotoxin contaminants in foods and feeds by ultra-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2007, 1143, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Li, F.Q.; Yu, C.C.; Shao, B.; Wang, W.; Yu, H.X. Natural occurrence of masked deoxynivalenol and multi-mycotoxins in cereals from China harvested in 2007 and 2008. Zhonghua Yu Fang Yi Xue Za Zhi 2011, 45, 57–63. [Google Scholar] [PubMed]
- Jin, P.G.; Han, Z.; Cai, Z.X.; Wu, Y.J.; Ren, Y.P. Simultaneous determination of 10 mycotoxins in grain by ultra-high-performance liquid chromatography-tandem mass spectrometry using 13C15-deoxynivalenol as internal standard. Food Addit. Contam. Part A Chem. Anal. Control Expo Risk Assess. 2010, 27, 1701–1713. [Google Scholar] [CrossRef] [PubMed]
- Marin, S.; Ramos, A.J.; Cano-Sancho, G.; Sanchis, V. Mycotoxins: Occurrence, toxicology, and exposure assessment. Food Chem. Toxicol. 2013, 60, 218–237. [Google Scholar] [CrossRef] [PubMed]
- Spanjer, M.C.; Rensen, P.M.; Scholten, J.M. LC-MS/MS multi-method for mycotoxins after single extraction, with validation data for peanut, pistachio, wheat, maize, cornflakes, raisins and figs. Food Addit. Contam. Part A Chem. Anal. Control Expo Risk Assess. 2008, 25, 472–489. [Google Scholar] [CrossRef] [PubMed]
- Juan, C.; Ritieni, A.; Manes, J. Occurrence of Fusarium mycotoxins in Italian cereal and cereal products from organic farming. Food Chem. 2013, 141, 1747–1755. [Google Scholar] [CrossRef] [PubMed]
- Ediage, E.N.; di Mavungu, J.D.; Monbaliu, S.; van Peteghem, C.; de Saeger, S. A validated multianalyte LC-MS/MS method for quantification of 25 mycotoxins in cassava flour, peanut cake and maize samples. J. Agric. Food Chem. 2011, 59, 5173–5180. [Google Scholar] [CrossRef] [PubMed]
- Eriksen, G.S.; Pettersson, H. Toxicological evaluation of trichothecenes in animal feed. Anim. Feed Sci. Technol. 2004, 114, 205–239. [Google Scholar] [CrossRef]
- Miura, K.; Aminova, L.; Murayama, Y. Fusarenon-X induced apoptosis in HL-60 cells depends on caspase activation and cytochrome c release. Toxicology 2002, 172, 103–112. [Google Scholar] [CrossRef]
- Alkadri, D.; Nipoti, P.; Doll, K.; Karlovsky, P.; Prodi, A.; Pisi, A. Study of fungal colonization of wheat kernels in Syria with a focus on Fusarium species. Int. J. Mol. Sci. 2013, 14, 5938–5951. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Rao, Q.; Song, S.; Liu, N.; Han, Z.; Hou, J.; Wu, A. Simultaneous determination of major type B trichothecenes and deoxynivalenol-3-glucoside in animal feed and raw materials using improved DSPE combined with LC-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 963, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Uegaki, R.; Tohno, M.; Yamamura, K.; Tsukiboshi, T.; Uozumi, S. Natural occurrence of mycotoxins in forage maize during crop growth in Japan: Case study. Mycotoxin Res. 2015, 31, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Rychlik, M.; Humpf, H.U.; Marko, D.; Danicke, S.; Mally, A.; Berthiller, F.; Klaffke, H.; Lorenz, N. Proposal of a comprehensive definition of modified and other forms of mycotoxins including “masked” mycotoxins. Mycotoxin Res. 2014, 30, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Nathanail, A.V.; Sarikaya, E.; Jestoi, M.; Godula, M.; Peltonen, K. Determination of deoxynivalenol and deoxynivalenol-3-glucoside in wheat and barley using liquid chromatography coupled to mass spectrometry: On-line clean-up versus conventional sample preparation techniques. J. Chromatogr. A 2014, 1374, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Suman, M.; Bergamini, E.; Catellani, D.; Manzitti, A. Development and validation of a liquid chromatography/linear ion trap mass spectrometry method for the quantitative determination of deoxynivalenol-3-glucoside in processed cereal-derived products. Food Chem. 2013, 136, 1568–1576. [Google Scholar] [CrossRef] [PubMed]
- Simsek, S.; Ovando-Martinez, M.; Ozsisli, B.; Whitney, K.; Ohm, J.B. Occurrence of deoxynivalenol and deoxynivalenol-3-glucoside in hard red spring wheat grown in the USA. Toxins 2013, 5, 2656–2670. [Google Scholar] [CrossRef] [PubMed]
- Varga, E.; Malachova, A.; Schwartz, H.; Krska, R.; Berthiller, F. Survey of deoxynivalenol and its conjugates deoxynivalenol-3-glucoside and 3-acetyl-deoxynivalenol in 374 beer samples. Food Addit. Contam. Part A Chem. Anal. Control Expo Risk Assess. 2013, 30, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Zachariasova, M.; Dzuman, Z.; Veprikova, Z.; Hajkova, K.; Jiru, M.; Vaclavikova, M.; Zachariasova, A.; Pospichalova, M.; Florian, M.; Hajslova, J. Occurrence of multiple mycotoxins in European feedingstuffs, assessment of dietary intake by farm animals. Anim. Feed Sci. Technol. 2014, 193, 124–140. [Google Scholar] [CrossRef]
- Nagl, V.; Woechtl, B.; Schwartz-Zimmermann, H.E.; Hennig-Pauka, I.; Moll, W.D.; Adam, G.; Berthiller, F. Metabolism of the masked mycotoxin deoxynivalenol-3-glucoside in pigs. Toxicol. Lett. 2014, 229, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, F.; Krska, R.; Domig, K.J.; Kneifel, W.; Juge, N.; Schuhmacher, R.; Adam, G. Hydrolytic fate of deoxynivalenol-3-glucoside during digestion. Toxicol. Lett. 2011, 206, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Shephard, G.S. Determination of mycotoxins in human foods. Chem. Soc. Rev. 2008, 37, 2468–2477. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Luo, X.; Yang, S.; Cao, X.; Wang, Z.; Shi, W.; Zhang, S. High specific monoclonal antibody production and development of an ELISA method for monitoring T-2 toxin in rice. J. Agric. Food Chem. 2014, 62, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Almeida, I.; Martins, H.M.; Santos, S.; Costa, J.M.; Bernardo, F. Co-occurrence of mycotoxins in swine feed produced in Portugal. Mycotoxin Res. 2011, 27, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Han, Z.; Aerts, J.; Nie, D.; Jin, M.; Shi, W.; Zhao, Z.; de Saeger, S.; Zhao, Y.; Wu, A. A reliable liquid chromatography-tandem mass spectrometry method for simultaneous determination of multiple mycotoxins in fresh fish and dried seafoods. J. Chromatogr. A 2015, 1387, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Malachova, A.; Dzuman, Z.; Veprikova, Z.; Vaclavikova, M.; Zachariasova, M.; Hajslova, J. Deoxynivalenol, deoxynivalenol-3-glucoside, and enniatins: The major mycotoxins found in cereal-based products on the Czech market. J. Agric. Food Chem. 2011, 59, 12990–12997. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, F.; Dall’Asta, C.; Schuhmacher, R.; Lemmens, M.; Adam, G.; Krska, R. Masked mycotoxins: Determination of a deoxynivalenol glucoside in artificially and naturally contaminated wheat by liquid chromatography-tandem mass spectrometry. J. Agric. Food Chem. 2005, 53, 3421–3425. [Google Scholar] [CrossRef] [PubMed]
- Commission Recommendation of 17 August 2006 on the Presence of Deoxynivalenol, Zearalenone, Ochratoxin A, T-2 and HT-2 and Fumonisins in Products Intended for Animal Feeding. Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/?qid=1464705395792&uri=CELEX:32006H0576 (accessed on 9 March 2016).
- Long, G.; Winefordner, J. Linearization of electron capture detector response to strongly responding compounds. J. Anal. Chem. 1983, 55, 713–724. [Google Scholar]
- Cavaliere, C.; Foglia, P.; Guarino, C.; Nazzari, M.; Samperi, R.; Lagana, A. Determination of aflatoxins in olive oil by liquid chromatography-tandem mass spectrometry. Anal. Chim. Acta 2007, 596, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Zheng, Y.; Luan, L.; Ren, Y.; Wu, Y. Analysis of ochratoxin A and ochratoxin B in traditional Chinese medicines by ultra-high-performance liquid chromatography-tandem mass spectrometry using [13C20]-ochratoxin A as an internal standard. J. Chromatogr. A 2010, 1217, 4365–4374. [Google Scholar] [CrossRef] [PubMed]
- Sulyok, M.; Berthiller, F.; Krska, R.; Schuhmacher, R. Development and validation of a liquid chromatography/tandem mass spectrometric method for the determination of 39 mycotoxins in wheat and maize. J. Rapid Commun. Mass Spectrom. 2006, 20, 2649–2659. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Si, W.; Jiang, K.; Nie, D.; Wu, Y.; Zhao, Z.; de Saeger, S.; Han, Z. Multi-walled carbon nanotubes as solid-phase extraction sorbents for simultaneous determination of type A trichothecenes in maize, wheat and rice by ultra-high performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2015, 1423, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: All feed samples are available from the authors.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrix | Mycotoxins | Linear Range (μg/kg) | Slope | Intercept | R2 | LOD a | LOQ b |
|---|---|---|---|---|---|---|---|
| (X ± SD) | (X ± SD) | (μg/kg) | (μg/kg) | ||||
| Neat solvents | DON | 1–200 | 519 ± 57 | 3153 ± 125 | 0.999 | ||
| 3-ADON | 2–200 | 90 ± 7 | −108 ± 14 | 0.999 | |||
| 15-ADON | 1–200 | 238 ± 14 | 140 ± 13 | 0.997 | |||
| FUS-X | 1–200 | 447 ± 25 | −82 ± 7 | 0.999 | |||
| D3G | 1–200 | 98 ± 8 | 45 ± 4 | 0.999 | |||
| Formula feed | DON | 1–200 | 485 ± 18 | 1197 ± 57 | 0.999 | 0.08 | 0.10 |
| 3-ADON | 5–200 | 70 ± 7 | 26 ± 4 | 0.996 | 2.09 | 4.17 | |
| 15-ADON | 2–200 | 152 ± 13 | 972 ± 48 | 0.991 | 0.57 | 1.21 | |
| FUS-X | 2–200 | 335 ± 14 | 294 ± 11 | 0.998 | 0.44 | 1.93 | |
| D3G | 1–200 | 75 ± 4 | 729 ± 54 | 0.990 | 0.46 | 0.93 | |
| Concentrated feed | DON | 1–200 | 414 ± 19 | 45368 ± 234 | 0.995 | 0.23 | 0.52 |
| 3-ADON | 5–200 | 59 ± 8 | 357 ± 24 | 0.997 | 2.31 | 4.85 | |
| 15-ADON | 2–200 | 100 ± 9 | 3472 ± 98 | 0.995 | 0.98 | 1.86 | |
| FUS-X | 2–200 | 154 ± 8 | 272 ± 18 | 0.999 | 0.68 | 1.57 | |
| D3G | 1–200 | 70 ± 5 | 438 ± 21 | 0.993 | 0.42 | 0.98 | |
| Premixed feed | DON | 1–200 | 462 ± 17 | 3404 ± 68 | 0.990 | 0.12 | 0.24 |
| 3-ADON | 5–200 | 84 ± 4 | −108 ± 5 | 0.999 | 1.32 | 2.98 | |
| 15-ADON | 2–200 | 202 ± 18 | 3020 ± 152 | 0.997 | 0.74 | 1.86 | |
| FUS-X | 2–200 | 381 ± 21 | −440 ± 26 | 0.998 | 0.58 | 1.24 | |
| D3G | 1–200 | 89 ± 5 | 36 ± 8 | 0.994 | 0.29 | 0.7 |
| Mycotoxins | Spiked Levels (μg/kg) | Formula Feed | Concentrated Feed | Premixed Feed | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Recovery | Intra-RSD | Inter-RSD | Recovery | Intra-RSD | Inter-RSD | Recovery | Intra-RSD | Inter-RSD | ||
| (X ± SD) | (X ± SD) | (X ± SD) | ||||||||
| DON | 10 | 102.3 ± 11.8 | 11.6 | 11.9 | 91.9 ± 7.8 | 8.4 | 7.9 | 95.6 ± 2.7 | 2.8 | 10.2 |
| 50 | 95.7 ± 3.9 | 4.8 | 2.6 | 78.0 ± 5.5 | 7.1 | 7.7 | 98.5 ± 5.3 | 5.4 | 10.7 | |
| 200 | 91.5 ± 5.0 | 5.5 | 6.1 | 94 ± 1.9 | 2.1 | 6.1 | 94.2 ± 10.8 | 11.5 | 13.2 | |
| 3-ADON | 10 | 89.5 ± 10.1 | 3.6 | 11.3 | 92.1 ± 8.7 | 11.5 | 9.5 | 86.4 ± 9.6 | 10.3 | 11.2 |
| 50 | 92.3 ± 9.6 | 11.5 | 10.4 | 93.0 ± 10.1 | 3.5 | 10.9 | 85.6 ± 8.5 | 8.5 | 9.9 | |
| 200 | 92.9 ± 8.1 | 6.1 | 8.7 | 88.9 ± 9.9 | 5.9 | 10.2 | 88.3 ± 9.1 | 7.2 | 10.3 | |
| 15-ADON | 10 | 96.1 ± 9.5 | 2.7 | 9.8 | 94.7 ± 8.6 | 8.4 | 9.1 | 89.5 ± 9.9 | 11.9 | 11.1 |
| 50 | 90.5 ± 7.7 | 8.6 | 8.5 | 94.4 ± 9.8 | 4.8 | 10.4 | 84.1 ± 5.7 | 2.9 | 6.8 | |
| 200 | 92.6 ± 9.7 | 11.2 | 10.5 | 88.0 ± 8.9 | 9.0 | 10.1 | 96.2 ± 10.7 | 9.6 | 9.9 | |
| FUS-X | 10 | 95.1 ± 7.3 | 7.6 | 9.7 | 95.1 ± 12.7 | 13.3 | 8.9 | 82.4 ± 4.8 | 5.9 | 11.8 |
| 50 | 101.1 ± 7.5 | 7.4 | 7.5 | 81.0 ± 6.6 | 8.2 | 11.5 | 100.1 ± 5.3 | 5.3 | 11.5 | |
| 200 | 97.6 ± 1.1 | 1.1 | 4.2 | 93.6 ± 3.5 | 3.7 | 10.7 | 78.5 ± 6.5 | 8.3 | 11.6 | |
| D3G | 10 | 99.1 ± 10.5 | 10.6 | 11.2 | 101.8 ± 13.5 | 13.2 | 8.4 | 80.1 ± 4.6 | 5.7 | 12.9 |
| 50 | 85.7 ± 4.0 | 4.6 | 5.6 | 82.5 ± 11.2 | 13.5 | 14.9 | 108.1 ± 8.5 | 7.8 | 14.6 | |
| 200 | 89.2 ± 1.9 | 2.1 | 4.1 | 83.9 ± 9.9 | 11.7 | 13.4 | 79.3 ± 2.3 | 2.9 | 13.2 | |
| Mycotoxin | Formula Feed | Concentrated Feed | Premixed Feed | |||
|---|---|---|---|---|---|---|
| Positive/Total Samples | Range (μg/kg) | Positive/Total Samples | Range (μg/kg) | Positive/Total Samples | Range (μg/kg) | |
| DON | 9/11 | 47.1–864.5 | 6/8 | 11.6–277.6 | 8/12 | 97.4–776.3 |
| 3-ADON | 8/11 | 5.1–221.8 | 5/8 | 5.6–56.4 | 5/12 | 26.5–135.1 |
| 15-ADON | 6/11 | 5.0–350.4 | 5/8 | 5.7–160.2 | 2/12 | 99.5–332.8 |
| FUS-X | 0/11 | ND | 2/8 | 11.9–14.6 | 0/12 | ND |
| D3G | 8/11 | 2.1–21.6 | 3/8 | 3.5–34.8 | 6/12 | 2.1–30.1 |
| Mycotoxin | Retention Time (min) | Precursor Ion (m/z) | Products Ion (m/z) | Collision Energy (eV) |
|---|---|---|---|---|
| DON | 3.1 | 297.1 [M + H]+ | 249.1 * | 10 |
| 231.1 | 13 | |||
| 3-ADON | 5.6 | 396.9 [M + CH3COO]− | 337.1 * | 14 |
| 307.1 | 8 | |||
| 15-ADON | 5.5 | 356.1 [M + NH4]+ | 339.1 * | 12 |
| 136.9 | 6 | |||
| FUS-X | 3.8 | 355.2 [M + H]+ | 247.1 * | 13 |
| 229.1 | 15 | |||
| D3G | 2.8 | 517.2 [M − H]− | 457.1 * | 14 |
| 427.1 | 22 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Z.; Bai, B.; Jin, P.; Fan, K.; Guo, W.; Zhao, Z.; Han, Z. Development and Validation of an Ultra-High Performance Liquid Chromatography-Tandem Mass Spectrometry Method for Simultaneous Determination of Four Type B Trichothecenes and Masked Deoxynivalenol in Various Feed Products. Molecules 2016, 21, 747. https://doi.org/10.3390/molecules21060747
Fan Z, Bai B, Jin P, Fan K, Guo W, Zhao Z, Han Z. Development and Validation of an Ultra-High Performance Liquid Chromatography-Tandem Mass Spectrometry Method for Simultaneous Determination of Four Type B Trichothecenes and Masked Deoxynivalenol in Various Feed Products. Molecules. 2016; 21(6):747. https://doi.org/10.3390/molecules21060747
Chicago/Turabian StyleFan, Zhichen, Bing Bai, Peng Jin, Kai Fan, Wenbo Guo, Zhihui Zhao, and Zheng Han. 2016. "Development and Validation of an Ultra-High Performance Liquid Chromatography-Tandem Mass Spectrometry Method for Simultaneous Determination of Four Type B Trichothecenes and Masked Deoxynivalenol in Various Feed Products" Molecules 21, no. 6: 747. https://doi.org/10.3390/molecules21060747
APA StyleFan, Z., Bai, B., Jin, P., Fan, K., Guo, W., Zhao, Z., & Han, Z. (2016). Development and Validation of an Ultra-High Performance Liquid Chromatography-Tandem Mass Spectrometry Method for Simultaneous Determination of Four Type B Trichothecenes and Masked Deoxynivalenol in Various Feed Products. Molecules, 21(6), 747. https://doi.org/10.3390/molecules21060747