
In Vitro Assessment of CYP-Mediated Drug Interactions for Kinsenoside, an Antihyperlipidemic Candidate


 , and
, and 
Abstract
:1. Introduction
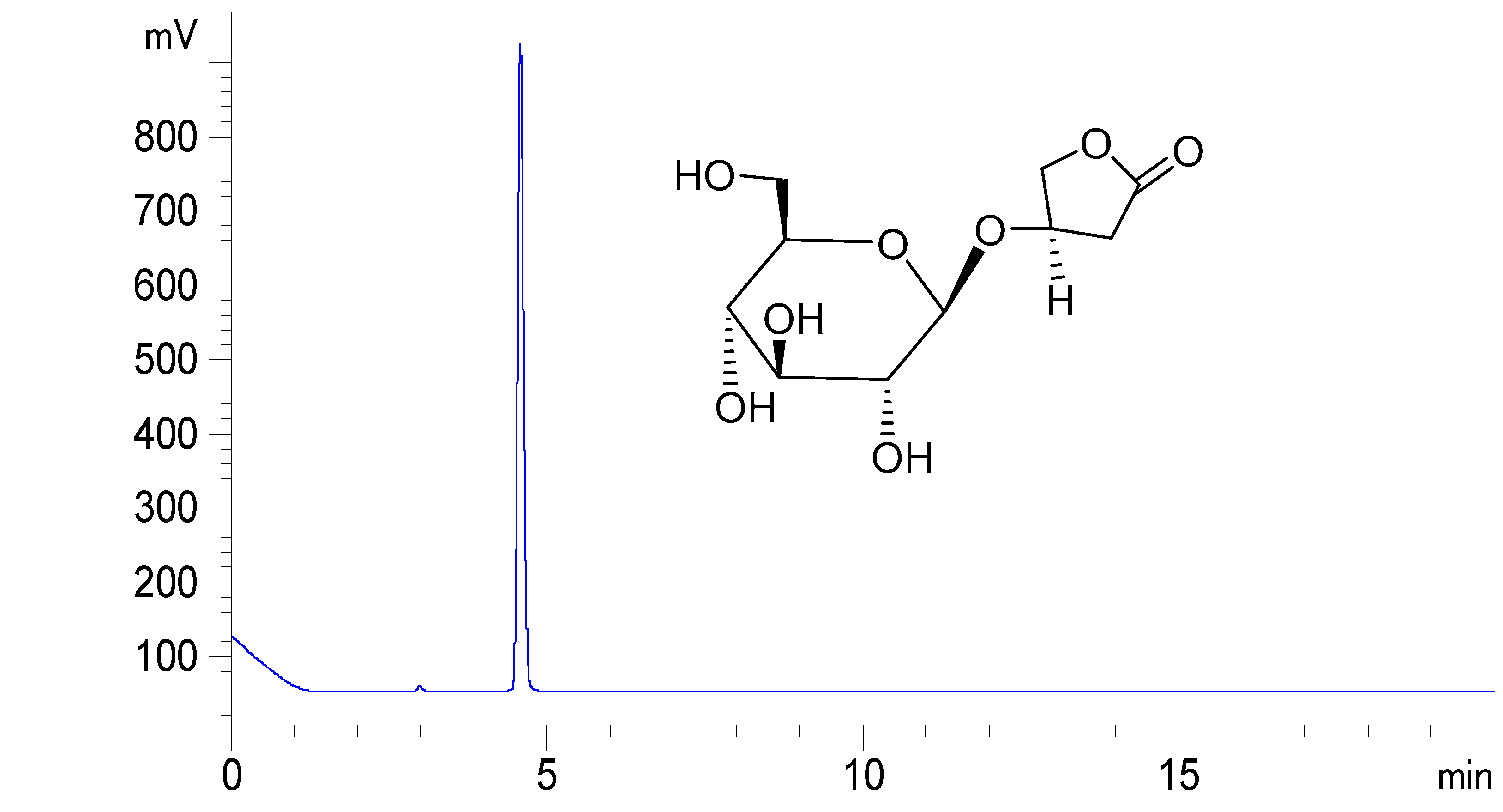
2. Results
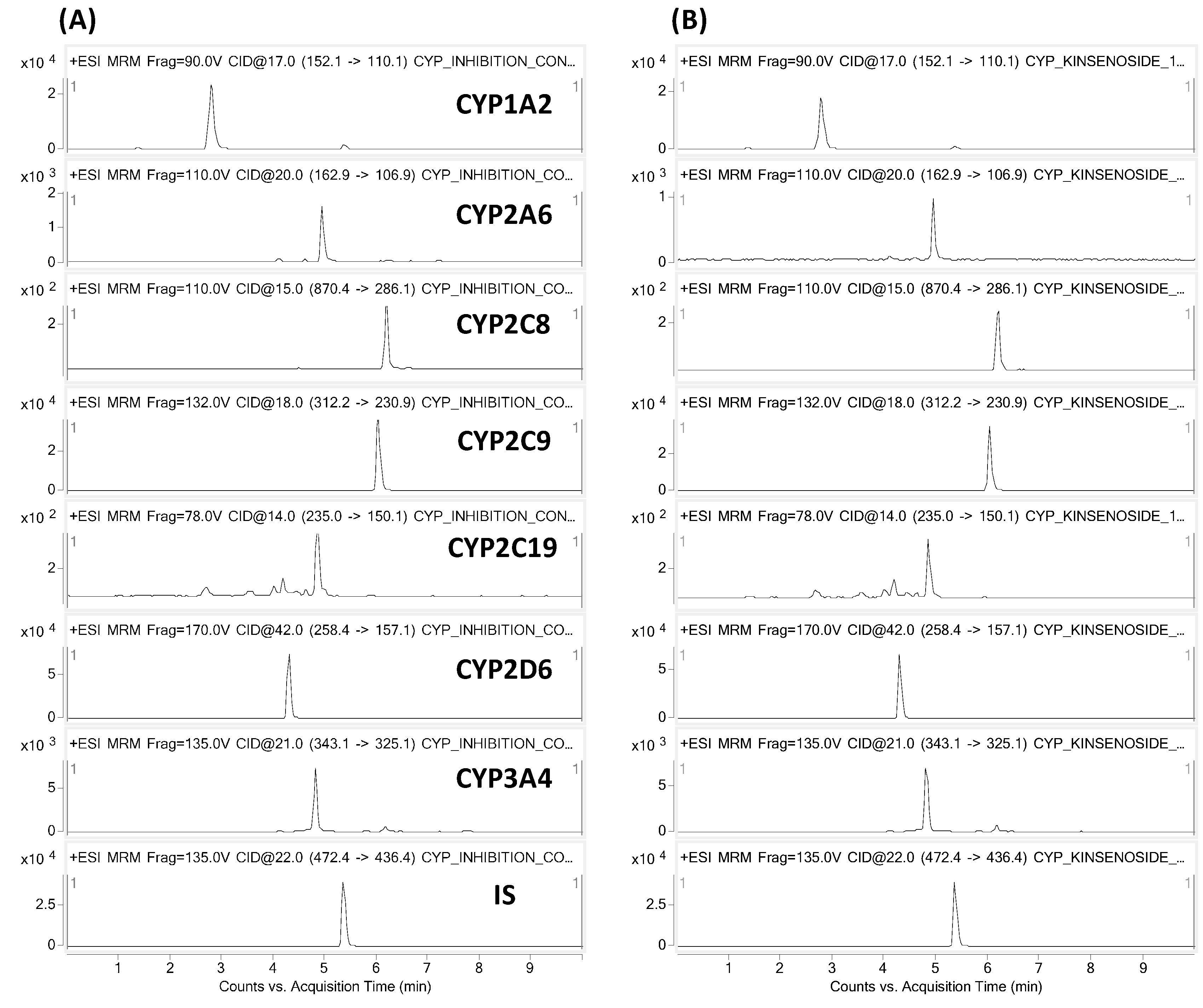
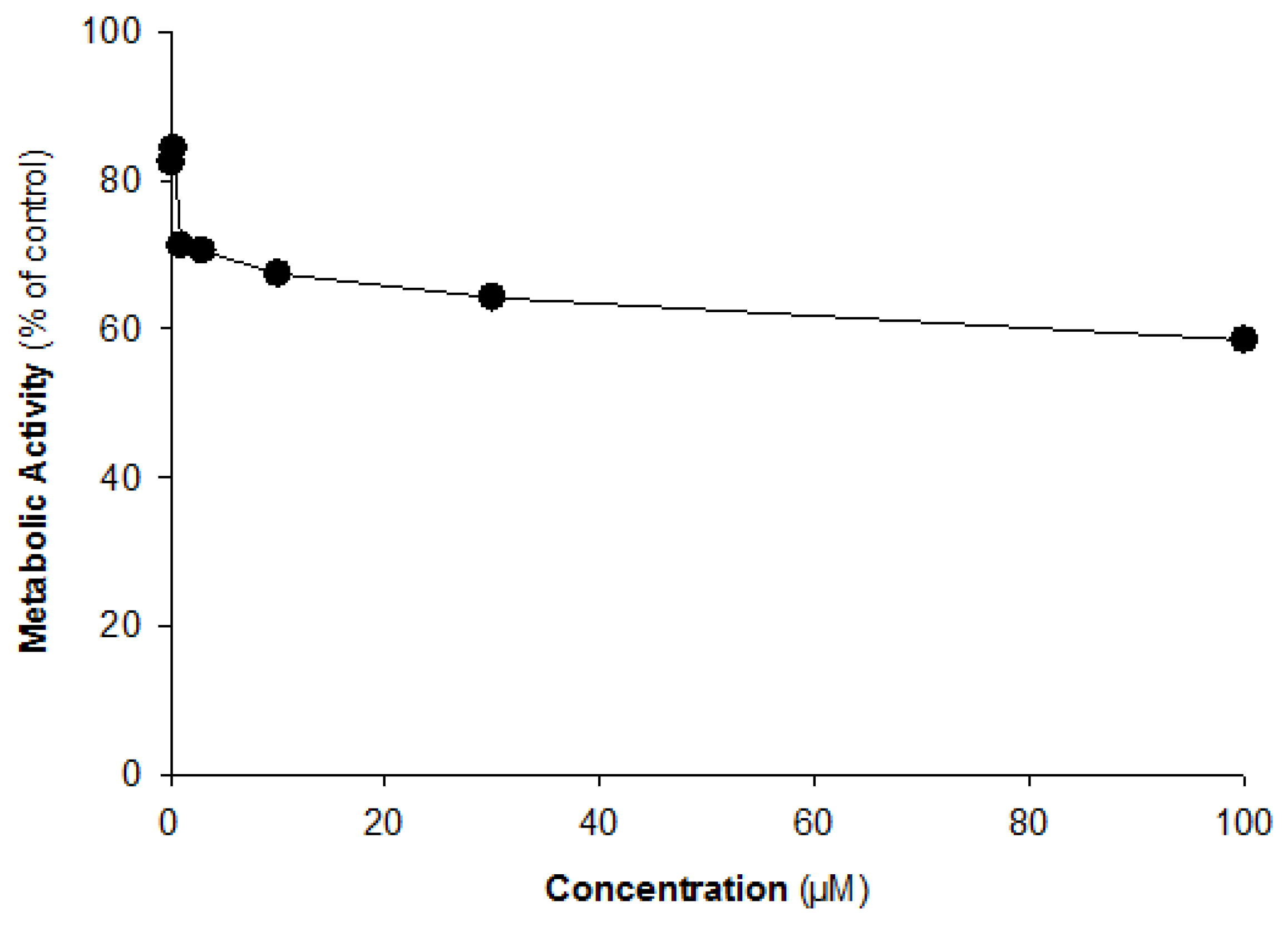
2.1. CYP Inhibition Assay
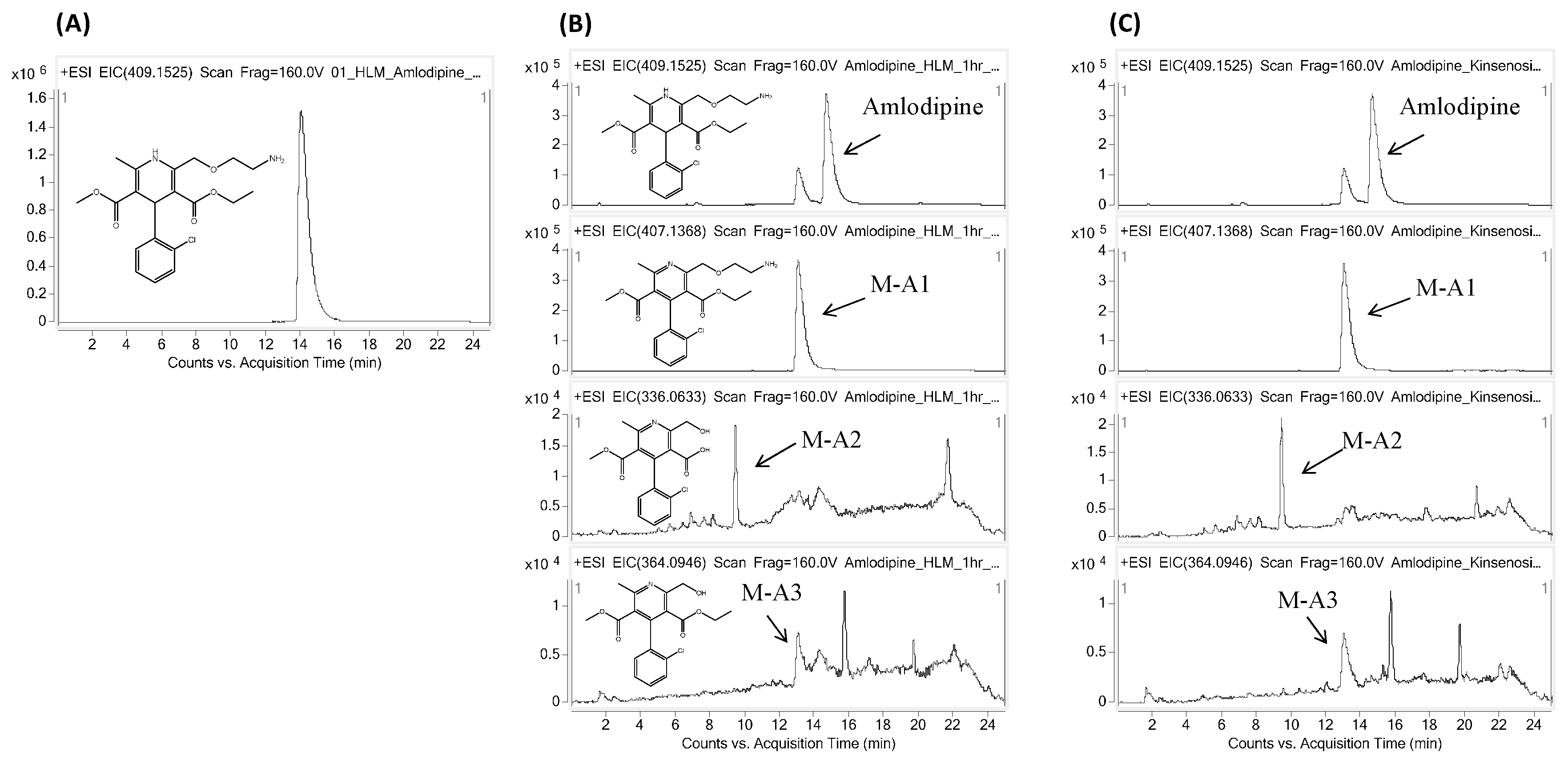
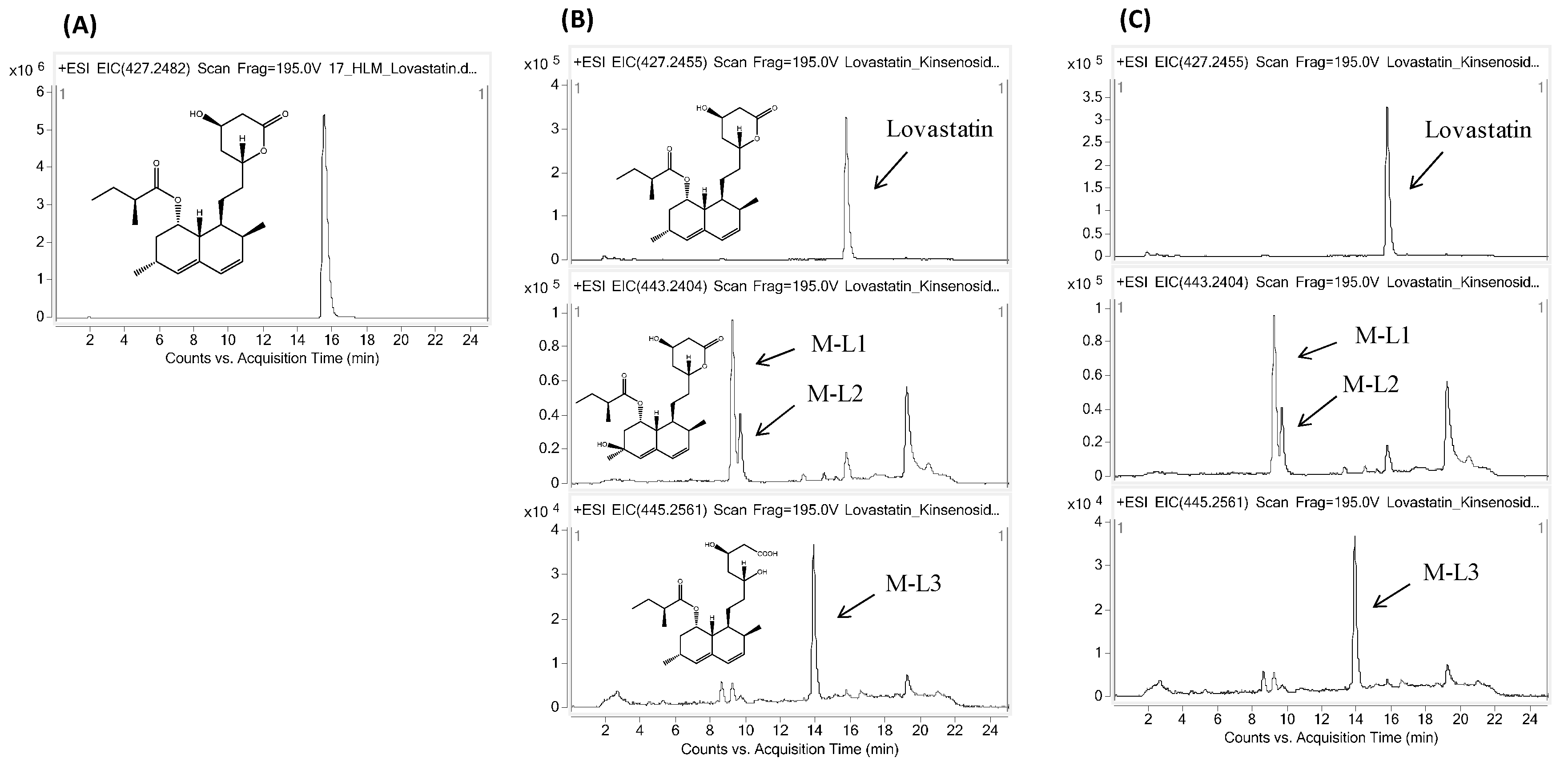
2.2. Effects of Kinsenoside on the Metabolism of Amlodipine and Lovastatin
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. CYP Inhibition Assay
4.3. Kinsenoside CYP Interaction Assay for Amlodipine and Lovastatin
4.4. Sample Preparation
4.5. LC-MS/MS Analysis
4.6. Liquid Chromatography/Quadrupole Time-of-Flight Mass Spectrometry (LC-QTOF MS)
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| cDNA | complementary deoxyribonucleic acid |
| CVD | cardiovascular disease |
| DW | distilled water |
| EIC | extracted ion chromatograms |
| ELSD | evaporative light scattering detector |
| HMG-CoA | 3-hydroxy-3-methylglutaryl-coenzyme A |
| HPLC | high-performance liquid chromatography |
| LC/MS/MS | liquid chromatography/tandem mass spectrometry |
| M-A | amlodipine’s metabolite |
| M-L | lovastatin’s metabolite |
| NADP+ | nicotinamide adenine dinucleotide phosphate |
| NGS | NADPH-generating system |
| STD | standard |
References
- Du, X.M.; Irino, N.; Furusho, N.; Hayashi, J.; Shoyama, Y. Pharmacologically active compounds in the Anoect Chilus and Goodyera species. J. Nat. Med. 2008, 62, 132–148. [Google Scholar] [CrossRef] [PubMed]
- Du, X.M.; Sun, N.Y.; Tamura, T.; Mohri, A.; Sugiura, M.; Yoshizawa, T.; Irino, N.; Hayashi, J.; Shoyama, Y. Higher yielding isolation of kinsenoside in Anoect Chilus and its antihyperliposis effect. Biol. Pharm. Bull. 2001, 24, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cai, J.; Ruan, H.; Pi, H.; Wu, J. Antihyperglycemic activity of kinsenoside, a high yielding constituent from Anoect Chilus roxburghii in streptozot Cin diabetic rats. J. Ethnopharmacol. 2007, 114, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.B.; Lin, H.; Wu, J.B.; Lin, W.C. Kinsenoside prevents ovariectomy-induced bone loss and suppresses oste Clastogenesis by regulating classical NF-κB pathways. Osteoporos. Int. 2013, 24, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.B.; Wu, J.B.; Lin, H.; Lin, W.C. Kinsenoside isolated from Anoect Chilus formosanus suppresses LPS-stimulated inflammatory reactions in macrophages and endotoxin sh Ck in mice. Shock 2011, 35, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, Z.L.; Tian, J.; Shi, W.; Liu, Y.Q. The semisynthetic spin-labelled derivatives of 3-hydroxybutanolide as potential oxidative stress inhibitors. Nat. Prod. Res. 2014, 28, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.L.; Liu, Q.; Xiao, B.; Zhou, J.; Zhang, J.G.; Li, Y. The vascular protective properties of kinsenoside isolated from Anoect Chilus roxburghii under high glucose condition. Fitoterapia 2013, 86, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.C.; Wu, Y.W.; Lin, W.C. Antihyperglycaemic And Anti-Oxidant Properties of Anoect Chilus Formosanus in Diabetic Rats. Clin. Exp. Pharmacol. Physiol. 2002, 29, 684–688. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.B.; Lin, W.L.; Hsieh, C.C.; Ho, H.Y.; Tsay, H.S.; Lin, W.C. The hepatoprotective activity of kinsenoside from Anoect Chilus formosanus. Phytother. Res. 2007, 21, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Solimene, U. WHO Guidelines for Assessing Quality of Herbal Medicines with Reference to Contaminants and Residues; World Health Organization: Geneva, Switzerland, 2007; p. 105. [Google Scholar]
- Walker, P.S.; Donovan, J.A. Herbal remedies: Natural caveats. Int. J. Dermatol. 1999, 38, 746–748. [Google Scholar] [CrossRef] [PubMed]
- Bent, S.; Goldberg, H.; Padula, A.; Avins, A.L. Spontaneous bleeding ass Ciated with Ginkgo biloba. J. Gen. Intern. Med. 2005, 20, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, M. In vitro assessment of the allelic variants of cyt Chrome P450. Drug Metab. Pharmacokinet. 2012, 27, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Russell, D.W. Clinical importance of the cyt Chromes P450. Lancet 2002, 360, 1155–1162. [Google Scholar] [CrossRef]
- Peters, W.H.; Kremers, P.G. Cyt Chromes P-450 in the intestinal mucosa of man. Biochem. Pharmacol. 1989, 38, 1535–1538. [Google Scholar] [CrossRef]
- Shimada, T.; Yamazaki, H.; Mimura, M.; Inui, Y.; Guengerich, F.P. Interindividual variations in human liver cyt Chrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: Studies with liver microsomes of 30 Japanese and 30 Caucasians. J. Pharmacol. Exp. Ther. 1994, 270, 414–423. [Google Scholar] [PubMed]
- Ono, S.; Hatanaka, T.; Hotta, H.; Satoh, T.; Gonzalez, F.; Tsutsui, M. Specificity of substrate and inhibitor probes for cyt Chrome P450s: Evaluation of in vitro metabolism using cDNA-expressed human P450s and human liver microsomes. Xenobiotica 1996, 26, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Boullata, J. Natural health product interactions with medication. Nutr. Clin. Pract. 2005, 20, 33–51. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.F.; Chou, K.C. Molecular modeling of cyt Chrome P450 and drug metabolism. Curr. Drug Metab. 2010, 11, 342–346. [Google Scholar] [CrossRef] [PubMed]
- National Institutes of Health, Office of Dietary Supplements. Botanical Dietary Supplements: Background Information. Available online: http://ods.od.nih.gov/factsheets/botanicalbackground.asp (accessed on 14 June 2016).
- Tom, L.; Amy, P. The effect of cyt Chrome P450 metabolism on drug response, interactions, and adverse effects. Eastern Virginia Medical School, Norfolk, Virginia. Am. Fam. Phys. 2007, 76, 391–396. [Google Scholar]
- Jacobsen, W.; Kirchner, G.; Hallensleben, K.; Mancinelli, L.; Deters, M.; Hackbarth, I.; Benet, L.Z.; Sewing, K.F.; Christians, U. Comparison of cyt Chrome P-450-dependent metabolism and drug interactions of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors lovastatin and pravastatin in the liver. Drug Metab. Dispos. 1999, 27, 173–179. [Google Scholar] [PubMed]
- Davis, B.; Cutler, J.A.; Gordon, D. Major outcomes in high risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel bl Cker vs. diuretic: The Antihypertensive and Lipid Lowering treatment to prevent Heart Attack Trial (ALLHAT). Jama 2002, 288, 2981–2997. [Google Scholar]
- Sever, P.S.; Dahlof, B.; Poulter, N.R.; Wedel, H.; Beevers, G.; Caulfield, M.; Collins, R.; Kjeldsen, S.E.; Kristinsson, A.; McInnes, G.T. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial-Lipid Lowering Arm (ASCOT-LLA): A multicentre randomised controlled trial. Lancet 2003, 361, 1149–1158. [Google Scholar]
- Wald, N.J.; Law, M.R. A strategy to reduce cardiovascular disease by more than 80%. BMJ 2003, 326, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Gæde, P.; Lund-Andersen, H.; Parving, H.H.; Pedersen, O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N. Engl. J. Med. 2008, 358, 580–591. [Google Scholar] [CrossRef] [PubMed]
- FDA USA. Guidance for Industry Drug Interaction Studies-Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations, Februry 2012. Available online: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm292362.pdf (accessed on 14 June 2016).
- Davidson, M.H.; Palmisano, J.; Wilson, H.; Liss, C.; Dicklin, M.R. A multicenter, randomized, double-blind clinical trial comparing the low-density lipoprotein cholesterol-lowering ability of lovastatin 10, 20, and 40 mg·d−1 with fluvastatin 20 and 40 mg·d–1. Clin. Ther. 2003, 25, 2738–2753. [Google Scholar] [CrossRef]
- Downs, J.R.; Clearfield, M.; Weis, S.; Whitney, E.; Shapiro, D.R.; Beere, P.A.; Langendorfer, A.; Stein, E.A.; Kruyer, W.; Gotto, A.M., Jr. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Jama 1998, 279, 1615–1622. [Google Scholar] [CrossRef] [PubMed]
- Melin, J.M.; Struble, W.E.; Tipping, R.W.; Reynolds, J.M.; Vassil, T.C.; Levy, S.J.; Petrohoy, T.M.; Midgette, P.; Hemwall, E.L.; Levine, J.G. A consumer use study of over-the-counter lovastatin (CUSTOM). Am. J. Cardiol. 2004, 94, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Dietschy, J.M.; Wilson, J.D. Regulation of cholesterol metabolism. N. Engl. J. Med. 1970, 282, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Halpin, R.; Ulm, E.; Till, A.; Kari, P.; Vyas, K.; Hunninghake, D.; Duggan, D. Biotransformation of lovastatin. V. Species differences in in vivo metabolite profiles of mouse, rat, dog, and human. Drug Metab. Dispos. 1993, 21, 1003–1011. [Google Scholar] [PubMed]
- Vyas, K.; Kari, P.; Pitzenberger, S.; Halpin, R.; Ramjit, H.; Arison, B.; Murphy, J.; Hoffman, W.; Schwartz, M.; Ulm, E. Biotransformation of lovastatin. I. Structure elucidation of in vitro and in vivo metabolites in the rat and mouse. Drug Metab. Dispos. 1990, 18, 203–211. [Google Scholar] [PubMed]
- Grabarkiewicz, T.; Grobelny, P.; Hoffmann, M.; Mielcarek, J. DFT study on hydroxy acid-lactone interconversion of statins: The case of fluvastatin. Org. Biomol. Chem. 2006, 4, 4299–4306. [Google Scholar] [CrossRef] [PubMed]
- Haria, M.; Wagstaff, A. Erratum to: Amlodipine. A reappraisal of its pharmacological properties and therapeutic use in cardiovascular disease. Drugs 1995, 50, 896. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Brian, W.R.; Iwasaki, M.; Sari, M.A.; Baeaernhielm, C.; Berntsson, P. Oxidation of dihydropyridine calcium channel bl Ckers and analogs by human liver cyt Chrome P-450 IIIA4. J. Med. Chem. 1991, 34, 1838–1844. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.U.; Kim, I.S.; Choi, M.S.; Luo, Z.; Yao, G.; Xue, Y.; Zhang, Y.; Yoo, H.H. Development of a hydrophilic interaction liquid chromatography-tandem mass spectrometric method for the determination of kinsenoside, an antihyperlipidemic candidate, in rat plasma and its application to pharmacokinetic studies. J. Pharm. Biomed. Anal. 2016, 120, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not Available.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P450 Specific Metabolite | Metabolite Formation (% of Control) | ||||||
|---|---|---|---|---|---|---|---|
| Kinsenoside Concentrations (µM) | |||||||
| 0.1 | 0.3 | 1 | 3 | 10 | 30 | 100 | |
| Acetaminophen (1A2) | 84.3 | 78.8 | 76.4 | 80.1 | 77.5 | 78.3 | 75.6 |
| 7-OH-coumarin (2A6) | 82.4 | 84.3 | 71.2 | 70.5 | 67.4 | 64.2 | 58.3 |
| 6-OH-paclitaxel (2C8) | 91.8 | 84.2 | 94.4 | 92.6 | 80.5 | 82.9 | 81.6 |
| 4-OH-diclofenac (2C9) | 92.2 | 94.5 | 91.7 | 89.2 | 92.5 | 92.3 | 88.5 |
| 4-OH-mephenytoin (2C19) | 88.6 | 95.2 | 100.2 | 99.4 | 99.3 | 95.5 | 89.2 |
| Dextromethorphan (2D6) | 92.1 | 101.7 | 100.5 | 98.9 | 96.2 | 95.3 | 92.8 |
| 1-OH-midazolam (3A4) | 88.4 | 91.0 | 94.8 | 89.2 | 85.7 | 86.4 | 82.9 |
| Drug | Metabolites | RT | Chemical Formula * | Mass | Error | |
|---|---|---|---|---|---|---|
| Theoretical | Experimental | |||||
| Amlodipine | Parent | 14.8 | C20H26ClN2O5 | 409.1525 | 409.1511 | 3.4 |
| M-A1 | 13.1 | C20H24ClN2O5 | 407.1368 | 407.1349 | 4.7 | |
| M-A2 | 9.5 | C16H15ClNO5 | 336.0633 | 336.0614 | 5.6 | |
| M-A3 | 13.0 | C18H19ClNO5 | 364.0946 | 364.0923 | 6.3 | |
| Lovastatin | Parent | 15.8 | C24H36O5Na | 427.2455 | 427.2429 | 6.1 |
| M-L1 | 9.3 | C24H36O6Na | 443.2404 | 443.2403 | 0.4 | |
| M-L2 | 9.8 | C24H36O6Na | 443.2404 | 443.2400 | −5.6 | |
| M-L3 | 13.9 | C24H38O6Na | 445.2561 | 445.2545 | 3.6 | |
| P450-Isozyme | Tested Metabolites | Precursor Ion | Product Ion |
|---|---|---|---|
| CYP 1A2 | Acetaminophen | 152.1 | 110.1 |
| CYP 2A6 | 7-OH-coumarin | 162.9 | 106.9 |
| CYP 2C8 | 6-OH-paclitaxel | 870.4 | 286.1 |
| CYP 2C9 | 4-OH-diclofenac | 312.2 | 230.9 |
| CYP 2C19 | 4-OH-mephenytoin | 235.0 | 150.1 |
| CYP 2D6 | Dextrorphan | 258.3 | 157.1 |
| CYP 3A4 | 1-OH-midazolam | 343.1 | 325.1 |
| Internal Standard | Terfenadine | 472.4 | 436.4 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehman, S.U.; Choi, M.S.; Kim, I.S.; Luo, Z.; Xue, Y.; Yao, G.; Zhang, Y.; Yoo, H.H. In Vitro Assessment of CYP-Mediated Drug Interactions for Kinsenoside, an Antihyperlipidemic Candidate. Molecules 2016, 21, 800. https://doi.org/10.3390/molecules21060800
Rehman SU, Choi MS, Kim IS, Luo Z, Xue Y, Yao G, Zhang Y, Yoo HH. In Vitro Assessment of CYP-Mediated Drug Interactions for Kinsenoside, an Antihyperlipidemic Candidate. Molecules. 2016; 21(6):800. https://doi.org/10.3390/molecules21060800
Chicago/Turabian StyleRehman, Shaheed Ur, Min Sun Choi, In Sook Kim, Zengwei Luo, Yongbo Xue, Guangming Yao, Yonghui Zhang, and Hye Hyun Yoo. 2016. "In Vitro Assessment of CYP-Mediated Drug Interactions for Kinsenoside, an Antihyperlipidemic Candidate" Molecules 21, no. 6: 800. https://doi.org/10.3390/molecules21060800
APA StyleRehman, S. U., Choi, M. S., Kim, I. S., Luo, Z., Xue, Y., Yao, G., Zhang, Y., & Yoo, H. H. (2016). In Vitro Assessment of CYP-Mediated Drug Interactions for Kinsenoside, an Antihyperlipidemic Candidate. Molecules, 21(6), 800. https://doi.org/10.3390/molecules21060800