
Chemical Synthesis of Deoxynivalenol-3-β-d-[13C6]-glucoside and Application in Stable Isotope Dilution Assays


Abstract
:1. Introduction
2. Results
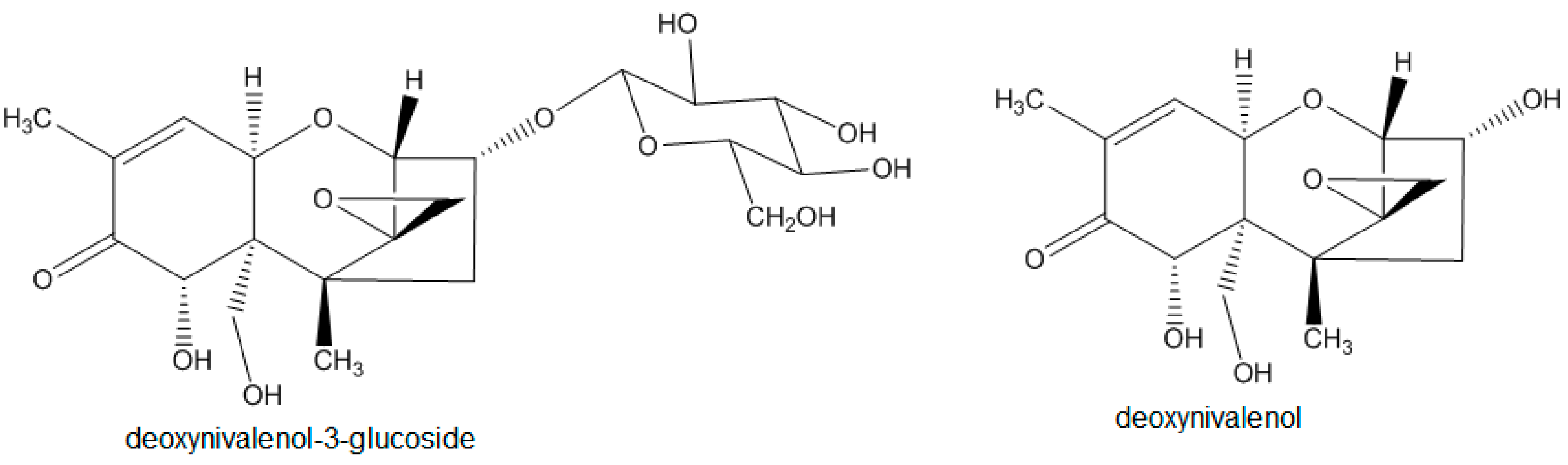
2.1. Syntheses
2.1.1. Synthesis of 2,3,4,6-Tetraacetyl-1-bromo-α-d-[13C6]-glucopyranoside
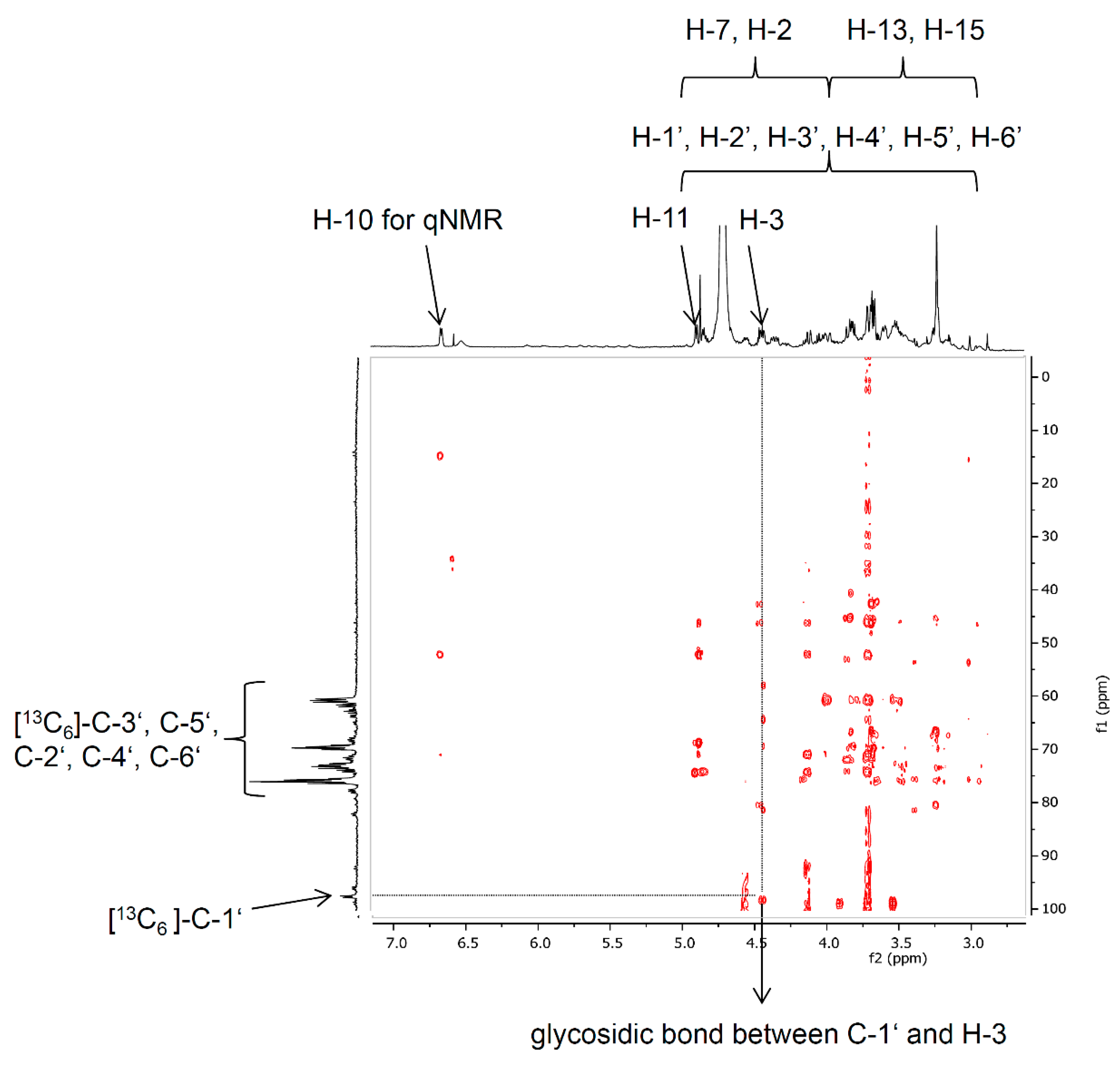
2.1.2. Synthesis of Deoxynivalenol-3-β-d-[13C6]-glucoside
2.2. Method Development
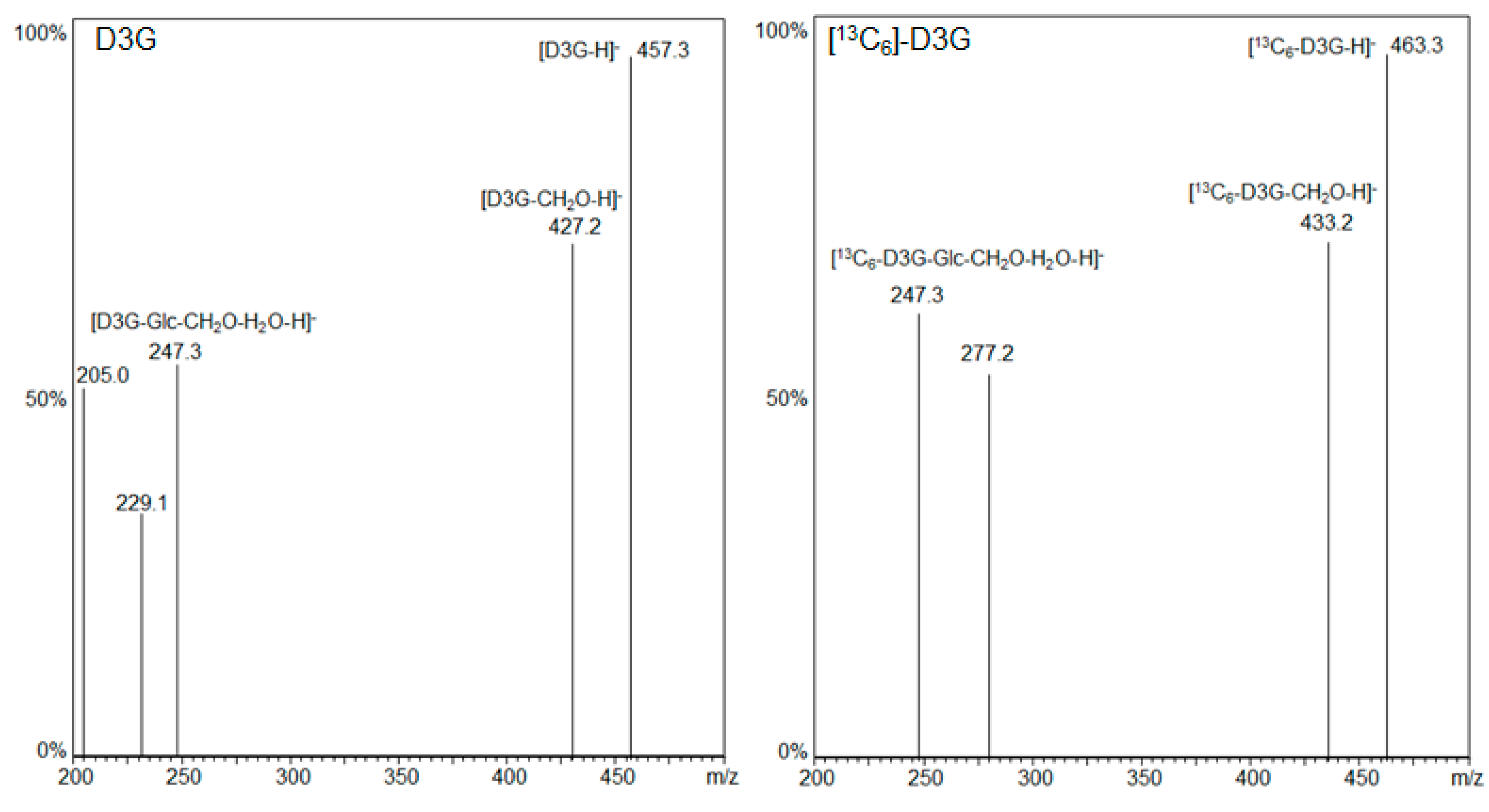
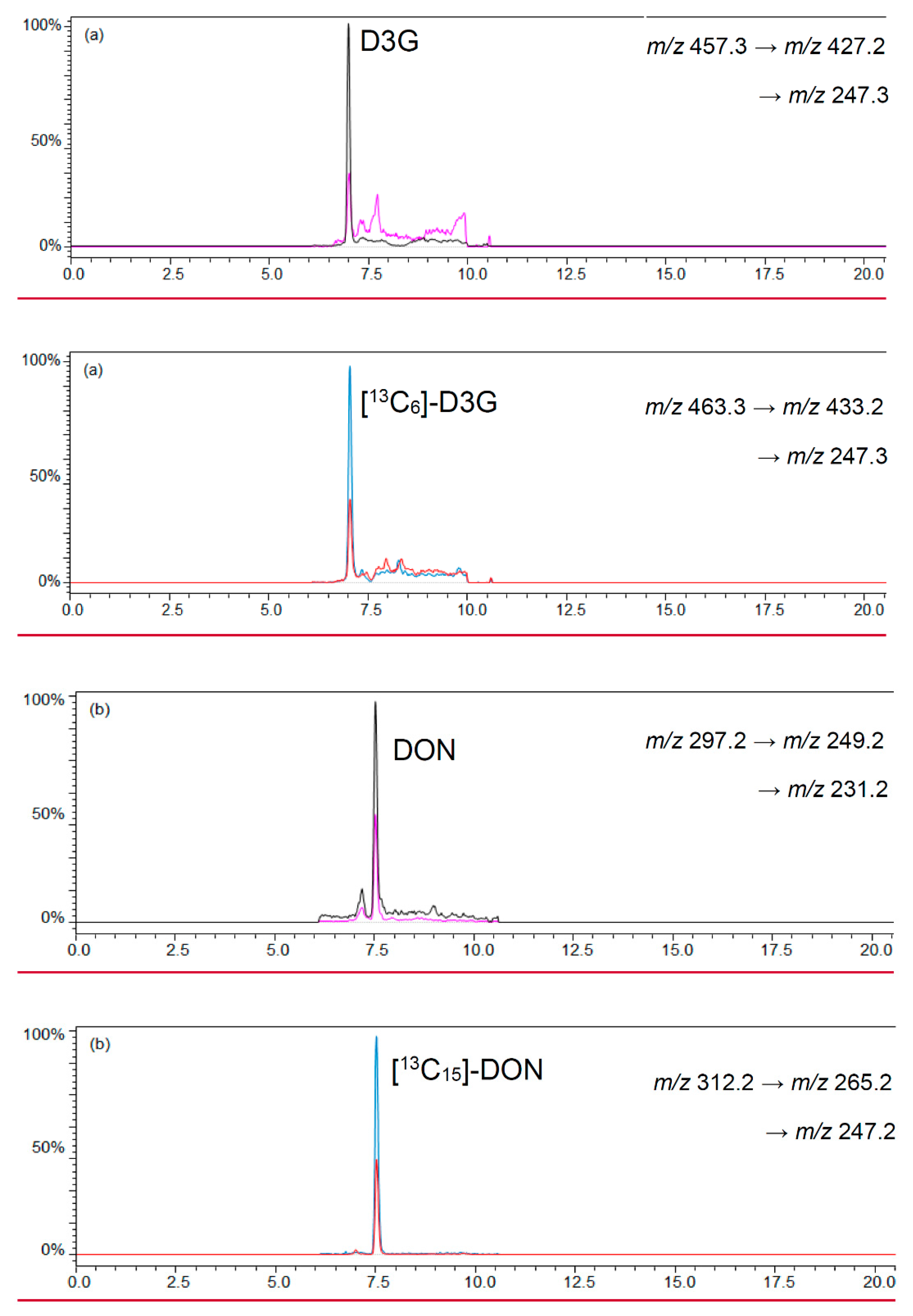
2.2.1. LC-MS/MS
2.2.2. Calibration and Quantitation
2.2.3. Sample Purification
2.3. Method Validation
2.3.1. Limits of Detection and Quantitation
2.3.2. Recovery
2.3.3. Precision
2.4. Analysis of D3G and DON in Beer Samples
3. Discussion
3.1. Syntheses
3.2. Method Development
3.3. Method Validation
3.4. Analysis of D3G and DON in Beer Samples
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Syntheses
4.2.1. Synthesis of 2,3,4,6-Tetraacetyl-1-bromo-α-d-[13C6]-glucopyranoside
4.2.2. Synthesis of Deoxynivalenol-3-β-d-[13C6]-glucoside
4.3. Preparative HPLC
4.4. NMR and qNMR
4.5. Preparation of Standard Solution
4.6. LC-MS/MS
4.7. Calibration and Quantitation
4.8. Sample Preparation
4.9. Method Validation
4.9.1. Limits of Detection and Quantitation
4.9.2. Recovery
4.9.3. Precision
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| D3G | Deoxynivalenol-3-glucoside |
| DON | Deoxynivalenol |
| ESI | Electrospray Ionization |
| HMBC | Heteronuclear Multiple Bond Correlation |
| LC-MS/MS | Liquid chromatography combined with tandem mass spectrometry |
| LOD | Limit of Detection |
| LOQ | Limit of Quantitation |
| NMR | Nuclear Magnetic Resonance |
| qNMR | quantitative NMR |
| SIDA | stable isotope dilution assay |
| SPE | solid phase extraction |
References
- Parry, D.W.; Jenkinson, P.; McLeed, L. Fusarium ear blight (scab) in small grain cereals—A review. Plant Pathol. 1995, 44, 207–218. [Google Scholar] [CrossRef]
- Desjardins, A.E. Fusarium Mycotoxins: Chemistry, Genetics and Biology; APS Press: Saint Paul, MN, USA, 2006. [Google Scholar]
- The Commission of the European Communities. Commission Regulation (EC) No. 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs; Official Journal of the European Union, European Union: Brussels, Belgium, 2006. [Google Scholar]
- Food and Agricultural Organization of the United Nations/World Health Organization (FAO/WHO). Safety evaluation of certain contaminants in food/prepared by the Seventy-second meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA). In WHO Food Additives Series No. 63/FAO JECFA Monographs 8; World Health Organization: Geneva, Switzerland and Rome, Italy, 2011. [Google Scholar]
- Sewald, N.; von Gleissenthall, J.L.; Schuster, M.; Müller, G.; Aplin, R.T. Structural Elucidation of a Plant Metabolite of 4-Deosoxynivalenol. Tetrahedron 1992, 3, 953–960. [Google Scholar] [CrossRef]
- Berthiller, F.; Dall’Asta, C.; Schuhmacher, R.; Lemmens, M.; Adam, G.; Krska, R. Masked Mycotoxins: Determination of a Deoxynivalenol Glucoside in Artificially and Naturally Contaminated Wheat by Liquid Chromatography-Tandem Mass Spectrometry. J. Agric. Food Chem. 2005, 53, 3421–3425. [Google Scholar] [CrossRef] [PubMed]
- Nagl, V.; Schwartz, H.; Krska, R.; Moll, W.-D.; Knasmüller, S.; Ritzmann, M.; Adam, G.; Berthiller, F. Metabolism of the masked mycotoxin deoxynivalenol-3-glucoside in rats. Toxicol. Lett. 2012, 213, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Nagl, V.; Woechtl, B.; Schwartz-Zimmermann, H.E.; Hennig-Pauka, I.; Moll, W.-D.; Adam, G.; Berthiller, F. Metabolism of the masked mycotoxin deoxynivalenol-3-glucoside in pigs. Toxicol. Lett. 2014, 229, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, F.; Krska, R.; Domig, K.J.; Kneifel, W.; Juge, N.; Schuhmacher, R.; Adam, G. Hydrolytic fate of deoxynivalenol-3-glucoside during digestion. Toxicol. Lett. 2011, 206, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Rychlik, M.; Humpf, H.-U.; Marko, D.; Dänicke, S.; Mally, A.; Berthiller, F.; Klaffke, H.; Lorenz, N. Proposal of a comprehensive definition of modified and other forms of mycotoxins including “masked” mycotoxins. Mycotoxin Res. 2014, 30, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, F.; Crews, C.; Dall’Asta, C.; Saeger, S.D.; Haesaert, G.; Karlovsky, P.; Oswald, I.P.; Seefelder, W.; Speijers, G.; Stroka, J. Masked mycotoxins: A review. Mol. Nutr. Food Res. 2013, 57, 165–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zachariasova, M.; Vaclavikova, M.; Lacina, O.; Vaclavik, L.; Hajslova, J. Deoxynivalenol Oligoglycosides: New “Masked” Fusarium Toxins Occurring in Malt, Beer, and Breadstuff. J. Agric. Food Chem. 2012, 60, 9280–9291. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, S.A.; Boddu, J.; Berthiller, F.; Hametner, C.; Stupar, R.M.; Adam, G.; Muehlbauer, G.J. Transcriptome Analysis of the Barley-Deoxynivalenol Interaction: Evidence for a Role of Glutathione in Deoxynivalenol Detoxification. Am. Phytopathol. Soc. 2010, 23, 962–976. [Google Scholar] [CrossRef] [PubMed]
- Kluger, B.; Bueschl, C.; Lemmens, M.; Michlmayr, H.; Malachova, A.; Koutnik, A.; Maloku, I.; Berthiller, F.; Adam, G.; Krska, R.; et al. Biotransformation of the Mycotoxin Deoxynivalenol in Fusarium Resistant and Susceptible Near Isogenic Wheat Lines. PLoS ONE 2015, 10, e0119656. [Google Scholar] [CrossRef] [PubMed]
- Warth, B.; Fruhmann, P.; Wiesenberger, G.; Kluger, B.; Sarkanj, B.; Lemmens, M.; Hametner, C.; Fröhlich, J.; Adam, G.; Krska, R.; et al. Deoxynivalenol-sulfates: Identification and quantification of novel conjugated (masked) mycotoxins in wheat. Anal. Bioanal. Chem. 2015, 407, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Yoshinari, T.; Sakuda, S.; Furihata, K.; Furusawa, H.; Ohnishi, T.; Sugita-Konishi, Y.; Ishizaki, N.; Terajima, J. Structural determination of a nivalenol glucoside and development of an analytical method for the simultaneous determination of nivalenol and deoxynivalenol, and their glucosides, in wheat. J. Agric. Food Chem. 2014, 62, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Stanic, A.; Uhlig, S.; Solhaug, A.; Rise, F.; Wilkins, A.L.; Miles, C.O. Preparation and Characterization of Cysteine Adducts of Deoxynivalenol. J. Agric. Food Chem. 2016, in press. [Google Scholar]
- Habler, K.; Rychlik, M. Multi-mycotoxin stable isotope dilution LC-MS/MS method for Fusarium toxins in cereals. Anal. Bioanal. Chem. 2016, 408, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Habler, K.; Hofer, K.; Geißinger, C.; Schüler, J.; Hückelhoven, R.; Hess, M.; Gastl, M.; Rychlik, M. Fate of Fusarium Toxins during the Malting Process. J. Agric. Food Chem. 2016, 64, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Nathanail, A.V.; Syvähuoko, J.; Malachová, A.; Jestoi, M.; Varga, E.; Michlmayr, H.; Adam, G.; Sieviläinen, E.; Berthiller, F.; Peltonen, K. Simultaneous determination of major type A and B trichothecenes, zearalenone and certain modified metabolites in Finnish cereal grains with a novel liquid chromatography-tandem mass spectrometric method. Anal. Bioanal. Chem. 2015, 407, 4745–4755. [Google Scholar] [CrossRef] [PubMed]
- Kostelanska, M.; Zachariasova, M.; Dzuman, Z.; Hajslova, J.; Ehrenbergerova, J.; Cerkal, R.; Vaculova, K.; Mikyska, A.; Psota, V. Fusarium Mycotoxins in Spring Barley and Their Occurrence Within the Technological Chain Barley-Malt-Beer. Kvas. Prumysl 2011, 57, 209–214. [Google Scholar]
- Malachova, A.; Dzuman, Z.; Veprikova, Z.; Vaclavikova, M.; Zachariasova, M.; Hajslova, J. Deoxynivalenol, Deoxynivalenol-3-glucoside, and Enniatins: The Major Mycotoxins Found in Cereal-Based Products on the Czech Market. J. Agric. Food Chem. 2011, 59, 12990–12997. [Google Scholar] [CrossRef] [PubMed]
- Lancova, K.; Hajslova, J.; Poustka, J.; Krplova, A.; Zachariasova, M.; Dostalek, P.; Sachambula, L. Transfer of Fusarium mycotoxins and ‘masked’ deoxynivalenol (deoxynivalenol-3-glucoside) from field barley through malt to beer. Food Addit. Contam. 2008, 25, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Varga, E.; Malachova, A.; Schwartz, H.; Krska, R.; Berthiller, F. Survey of deoxynivalenol and its conjugates deoxynivalenol-3-glucoside and 3-acetyl-deoxynivalenol in 374 beer samples. Food Addit. Contam. Part A 2013, 30, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Klötzel, M.; Gutsche, B.; Lauber, U.; Humpf, H.-U. Determination of 12 type A and B trichothecenes in cereals by liquid chromatography-electrospray ionization tandem mass spectrometry. J. Agric. Food Chem. 2005, 53, 8904–8910. [Google Scholar] [CrossRef] [PubMed]
- Asam, S.; Rychlik, M. Synthesis of Four Carbon-13-Labeled Type A Trichothecene Mycotoxins and Their Application as Internal Standards in Stable Isotope Dilution Assays. J. Agric. Food Chem. 2006, 54, 6535–6546. [Google Scholar] [CrossRef] [PubMed]
- Ravindranathan Kartha, K.P.; Jennings, H.J. A Simplified, One-Pot Preparation of Acetobromosugars from Reducing Sugars. J. Carbohydr. Chem. 1990, 9, 777–781. [Google Scholar] [CrossRef]
- Redemann, E.C.; Niemann, C. Acetobromoglucose: [2,3,4,6-Tetraacetyl-α-d-glucopyranosyl bromide]. Org. Synth. 1955, 3, 11. [Google Scholar]
- Koschella, A.; Dorn, S.; Heinze, T.; Togola, A.; Paulsen, B.S. Synthesis and characterization of branched polysaccharides by reaction of cellulose with 2,3,4,6-tetraacetyl-1-bromo-α-Dglucopyranoside. Arkivoc 2012, 3, 76–89. [Google Scholar]
- Savard, M.E. Deoxynivalenol Fatty Acid and Glucoside Conjugates. J. Agric. Food Chem. 1991, 39, 570–574. [Google Scholar] [CrossRef]
- Mikula, H.; Hametner, C.; Berthiller, F.; Warth, B.; Krska, R.; Adam, G.; Fröhlich, J. Fast and reproducible chemical synthesis of zeralenone-14-beta, d-glucuronide. World Mykotoxin J. 2012, 5, 289–296. [Google Scholar] [CrossRef]
- Mikula, H.; Weber, J.; Lexmüller, S.; Bichl, G.; Schwartz, H.; Varga, E.; Berthiller, F.; Hametner, C.; Krska, R.; Fröhlich, J. Simultaneous preparation of α/β-zearalenol glucosides and glucuronides. Carbohydr. Res. 2013, 373, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Malachová, A.; Štočková, L.; Wakker, A.; Varga, E.; Krska, R.; Michlmayr, H.; Adam, G.; Berthiller, F. Critical evaluation of indirect methods for the determination of deoxynivalenol and its conjugated forms in cereals. Anal. Bioanal. Chem. 2015, 407, 6009–6020. [Google Scholar] [CrossRef] [PubMed]
- Korn, M.; Frank, O.; Hofmann, T.; Rychlik, M. Development of stable isotope dilution assays for ochratoxin A in blood samples. Anal. Biochem. 2011, 419, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, F.; Schuhmacher, R.; Buttinger, G.; Freudenschuss, M.; Adam, G.; Krska, R. Synthesis of Deoxynivalenol-Glucosides and their Characerization using a QTrap LC-MS/MS. Mycotoxin Res. 2003, 19, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Agilent Technologies. New SPE Sorbent for Clean-up of Fusarium Toxin-Contaminated Cereals & Cereal-based Foods, Bond Elut Mycotoxin. 2010. Available online: https://www.agilent.com/cs/library/applications/SI-00295.pdf (accessed on 3 May 2016).
- Agilent Technologies. LC/MS/MS of Trichothecenes and Zearalenone in Wheat Using Different Sample Prep Methods. 2012. Available online: https://www.agilent.com/cs/library/applications/5990-9107EN.pdf (accessed on 3 May 2016).
- Vogelgesang, J.; Hädrich, J. Limits of detection, identification and determination: A statistical approach for practitioners. Accredit. Qual. Assur. 1998, 3, 242–255. [Google Scholar] [CrossRef]
- Koenigs, W.; Knorr, E. Ueber einige Derivate des Traubenzuckers und der Galactose. Ber. Dtsch. Chem. Ges. 1901, 34, 957–981. [Google Scholar] [CrossRef]
- Schmidt, R.R.; Michel, J. Facile Synthesis of α- and β-O-Glycosyl Imidates; Preparation of Glycosides and Disaccharides. Angew. Chem. Int. Ed. Engl. 1980, 19, 731–732. [Google Scholar] [CrossRef]
- Van Beyersbergen Henegouwen, W.G.; Hiemstra, H. Studies toward the Total Synthesis of the Oxindole Alkaloid Gelsedine: An Efficient Allene-Terminated N-Acyliminium Ion Cyclization. J. Org. Chem. 1997, 62, 8862–8867. [Google Scholar] [CrossRef]
- Blackwell, B.A.; Greenhalgh, R.; Bain, A.D. Carbon-13 and proton nuclear magnetic resonance spectral assignments of deoxynivalenol and other mycotoxins from Fusarium graminearum. J. Agric. Food Chem. 1984, 32, 1078–1083. [Google Scholar] [CrossRef]
- Kostelanska, M.; Hajslova, J.; Zachariasova, M.; Malachova, A.; Kalachova, K.; Poustka, J.; Fiala, J.; Scott, P.M.; Berthiller, F.; Krska, R. Occurrence of Deoxynivalenol and Its Major Conjugate, Deoxynivalenol-3-Glucoside, in Beer and Some Brewing Intermediates. J. Agric. Food Chem. 2009, 57, 3187–3194. [Google Scholar] [CrossRef] [PubMed]
- Zachariasova, M.; Cajka, T.; Godula, M.; Malachova, A.; Veprikova, Z.; Hajslova, J. Analysis of multiple mycotoxins in beer employing (ultra)-high-resolution mass spectrometry. Rapid Commun. Mass Spectrom. 2010, 24, 3357–3367. [Google Scholar] [CrossRef] [PubMed]
- Vaclavikova, M.; Malachova, A.; Veprikova, Z.; Dzuman, Z.; Zachariasova, M.; Hajslova, J. ‘Emerging’ mycotoxins in cereals processing chains: Changes of enniatins during beer and bread making. Food Chem. 2013, 136, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Al-Taher, F.; Banaszewski, K.; Jackson, L.; Zweigenbaum, J.; Ryu, D.; Cappozzo, J. Rapid Method for the Determination of Multiple Mycotoxins in Wines and Beers by LC-MS/MS Using a Stable Isotope Dilution Assay. J. Agric. Food Chem. 2013, 61, 2378–2384. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Gastl, M.; Linkmeyer, A.; Hess, M.; Rychlik, M. Fate of enniatins and beauvericin during the malting and brewing process determined by stable isotope dilution assays. LWT Food Sci. Technol. 2014, 56, 469–477. [Google Scholar] [CrossRef]
- Malachova, A.; Varga, E.; Schwartz, H.; Krska, R.; Berthiller, F. Development, validation and application of an LC-MS/MS based method for the determination of deoxynivalenol and its conjugates in different types of beer. World Mycotoxin J. 2012, 5, 261–270. [Google Scholar] [CrossRef]
- Harcz, P.; Tangni, E.K.; Wilmart, O.; Moons, E.; van Peteghem, C.; de Saeger, S.; Schneider, Y.-J.; Larondelle, Y.; Pussemier, L. Intake of ochratoxin A and deoxynivalenol through beer consumption in Belgium. Food Addit. Contam. 2007, 24, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Zachariasova, M.; Hajslova, J.; Kostelanska, M.; Poustka, J.; Krplova, A.; Cuhra, P.; Hochel, I. Deoxynivalenol and its conjugates in beer: A critical assessment of data obtained by enzyme-linked immunosorbent assay and liquid chromatography coupled to tandem mass spectrometry. Anal. Chim. Acta 2008, 625, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Precursor Ion m/z | Product Ion m/z | Q1 Pre Bias (V) | Q3 Pre Bias (V) | CE (V) |
|---|---|---|---|---|---|
| D3G | 457.3 | 427.2 | 17 | 13 | 16 |
| 247.3 | 22 | 15 | 22 | ||
| [13C6]-D3G | 463.3 | 433.2 | 17 | 13 | 16 |
| 247.3 | 22 | 15 | 22 | ||
| DON | 297.2 | 249.2 | −21 | −18 | −12 |
| 231.2 | −20 | −17 | −12 | ||
| [13C15]-DON | 312.2 | 265.2 | −21 | −18 | −12 |
| 247.2 | −20 | −17 | −12 |
| Analyte | LOD (μg/L) | LOQ (μg/L) | Precision (n = 3) (RSD %) | Recoveries (%) at Spiking Levels of | |||
|---|---|---|---|---|---|---|---|
| Intraday | Interday | 10 μg/L | 20 μg/L | 30 μg/L | |||
| D3G | 2.99 | 8.84 | 0.5 | 5 | 112 ± 3 | 110 ± 3 | 105 ± 1 |
| DON | 1.49 | 4.44 | 0.3 | 7 | 97 ± 1 | 107 ± 2 | 108 ± 2 |
| Beer Type | Country | Alcohol Content (vol %) | D3G (μg/L) | DON (μg/L) | Molar Ratio n(D3G)/n(DON) |
|---|---|---|---|---|---|
| Lager | Germany | 5.4 | - | - | - |
| Wheat beer b | Germany | 5.1 | 10.6 | 20.3 | 0.34 |
| Lager | Taiwan | 5.0 | 15.3 | 6.43 | 1.54 |
| Lager | China | 4.7 | 8.31 a | 5.11 | 1.05 |
| Lager | USA | 5.0 | 20.1 | 14.1 | 0.92 |
| IPA | USA | 6.5 | - | 9.77 | - |
| Triple Golden Ale | USA | 8.0 | 18.1 | 9.40 | 1.25 |
| Porter | USA | 5.3 | - | - | - |
| Belgian Style IPA | USA | 8.3 | 63.3 | 28.3 | 1.45 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habler, K.; Frank, O.; Rychlik, M. Chemical Synthesis of Deoxynivalenol-3-β-d-[13C6]-glucoside and Application in Stable Isotope Dilution Assays. Molecules 2016, 21, 838. https://doi.org/10.3390/molecules21070838
Habler K, Frank O, Rychlik M. Chemical Synthesis of Deoxynivalenol-3-β-d-[13C6]-glucoside and Application in Stable Isotope Dilution Assays. Molecules. 2016; 21(7):838. https://doi.org/10.3390/molecules21070838
Chicago/Turabian StyleHabler, Katharina, Oliver Frank, and Michael Rychlik. 2016. "Chemical Synthesis of Deoxynivalenol-3-β-d-[13C6]-glucoside and Application in Stable Isotope Dilution Assays" Molecules 21, no. 7: 838. https://doi.org/10.3390/molecules21070838
APA StyleHabler, K., Frank, O., & Rychlik, M. (2016). Chemical Synthesis of Deoxynivalenol-3-β-d-[13C6]-glucoside and Application in Stable Isotope Dilution Assays. Molecules, 21(7), 838. https://doi.org/10.3390/molecules21070838