
Recent Advances in Metal-Free Quinoline Synthesis


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
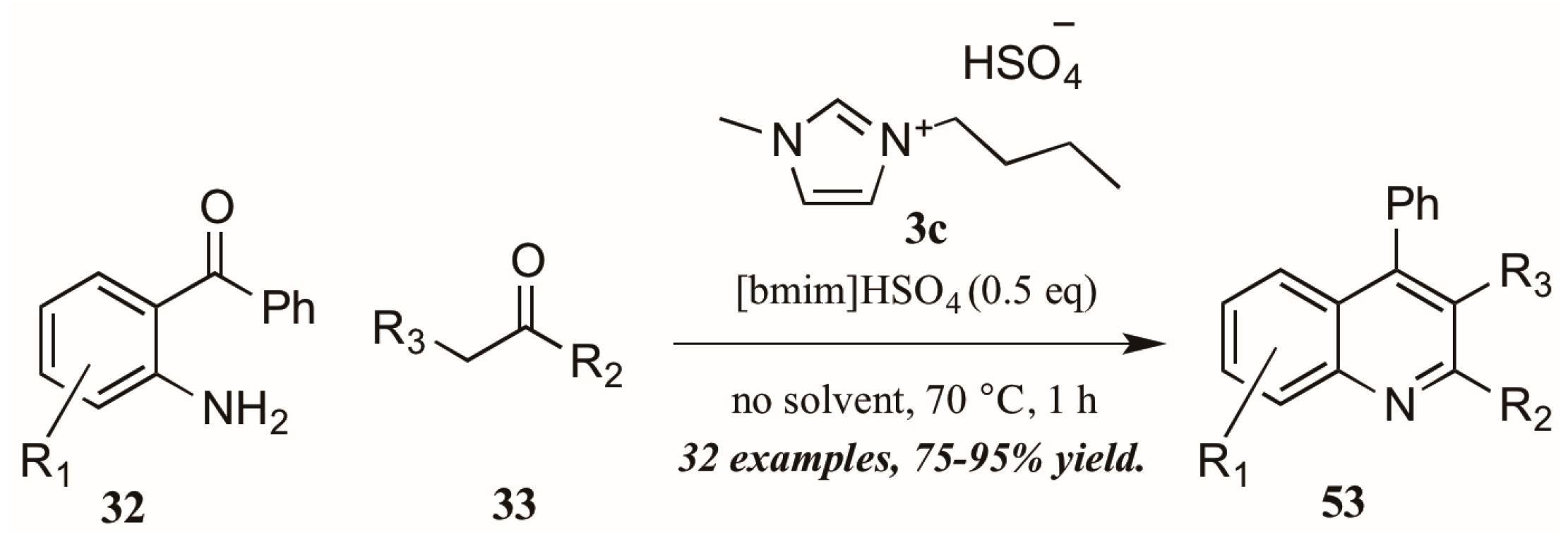{kind=link}
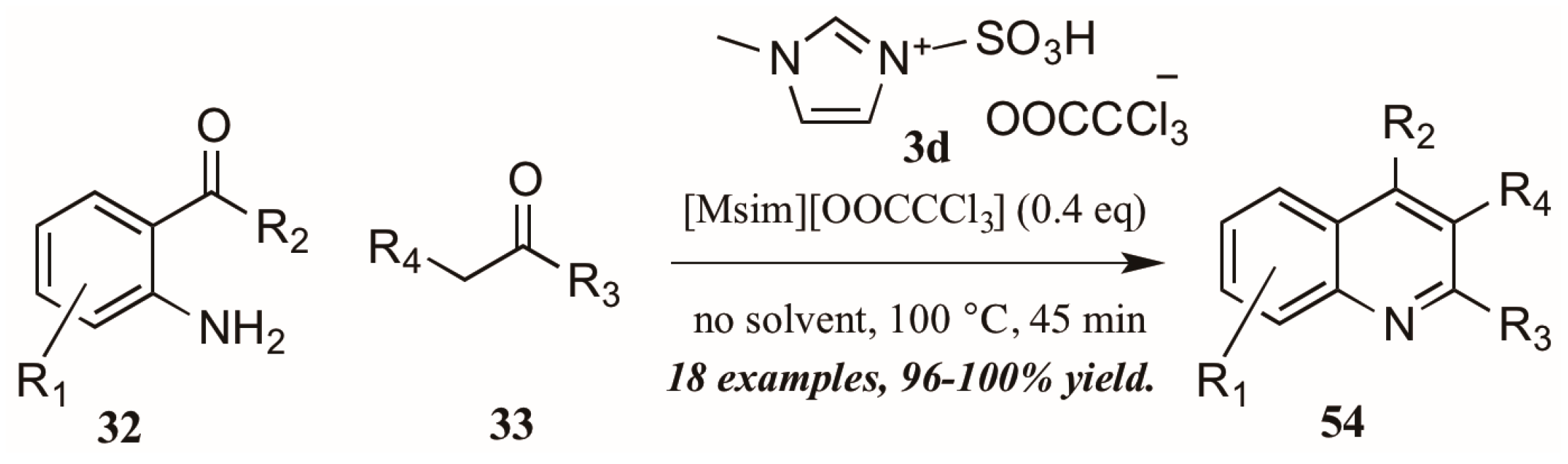{kind=link}
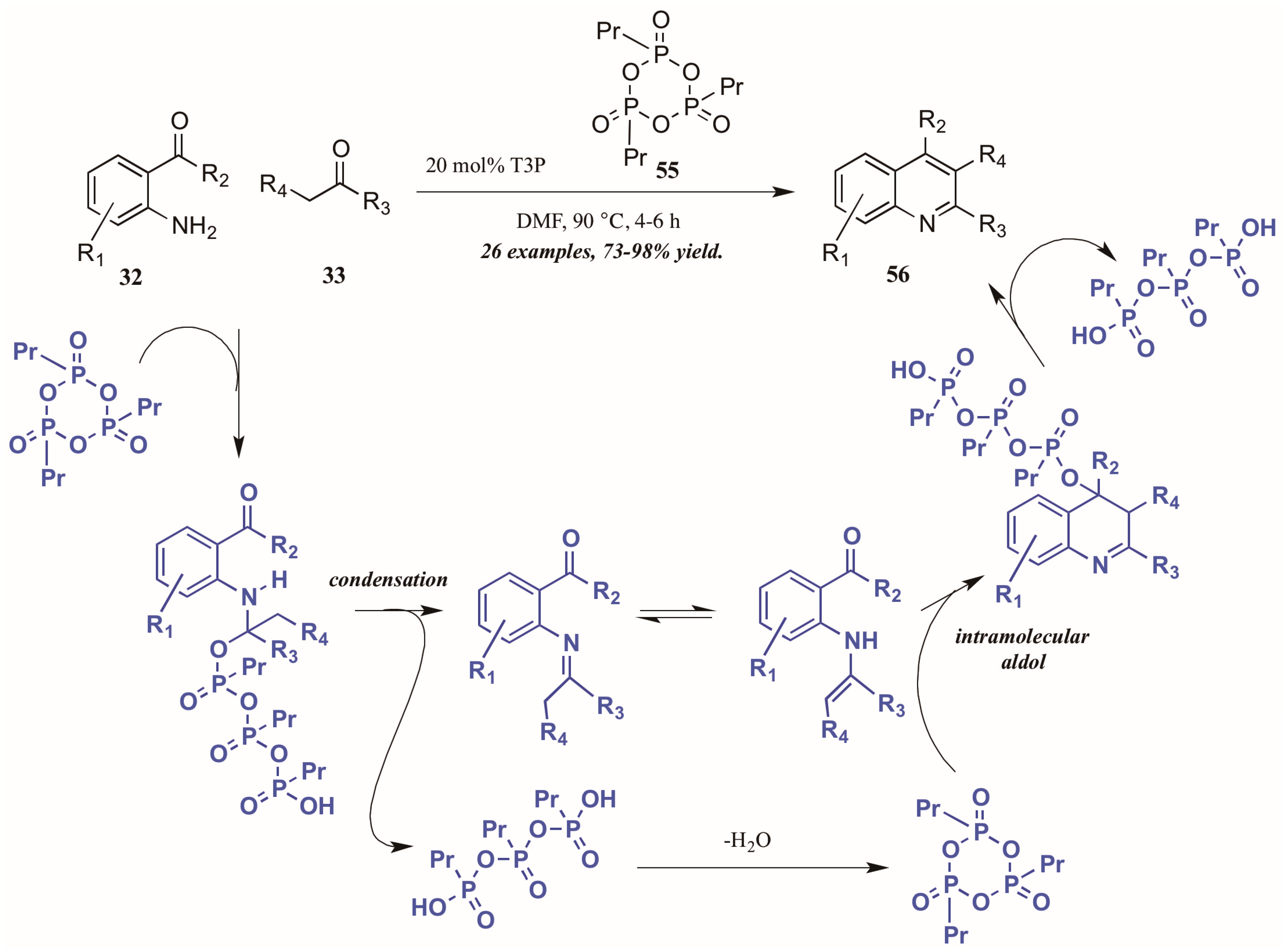{kind=link}
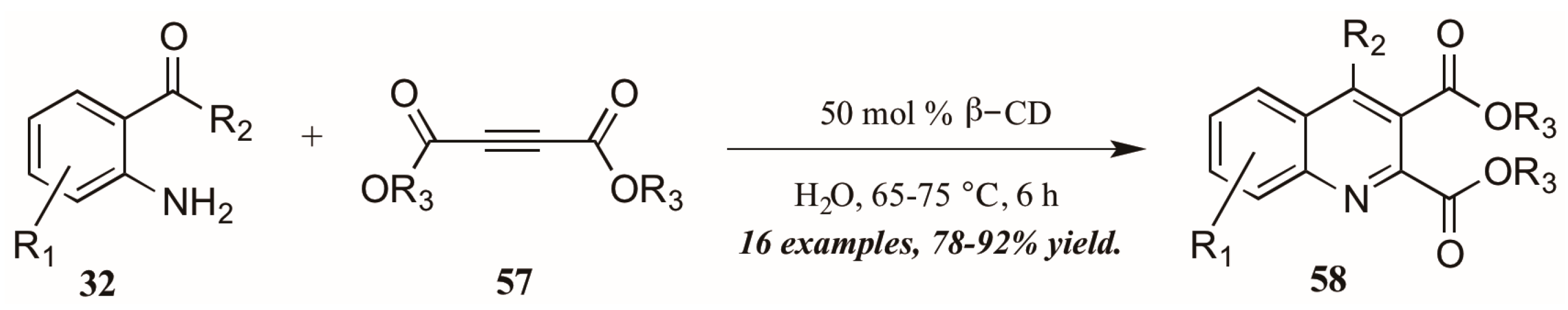{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
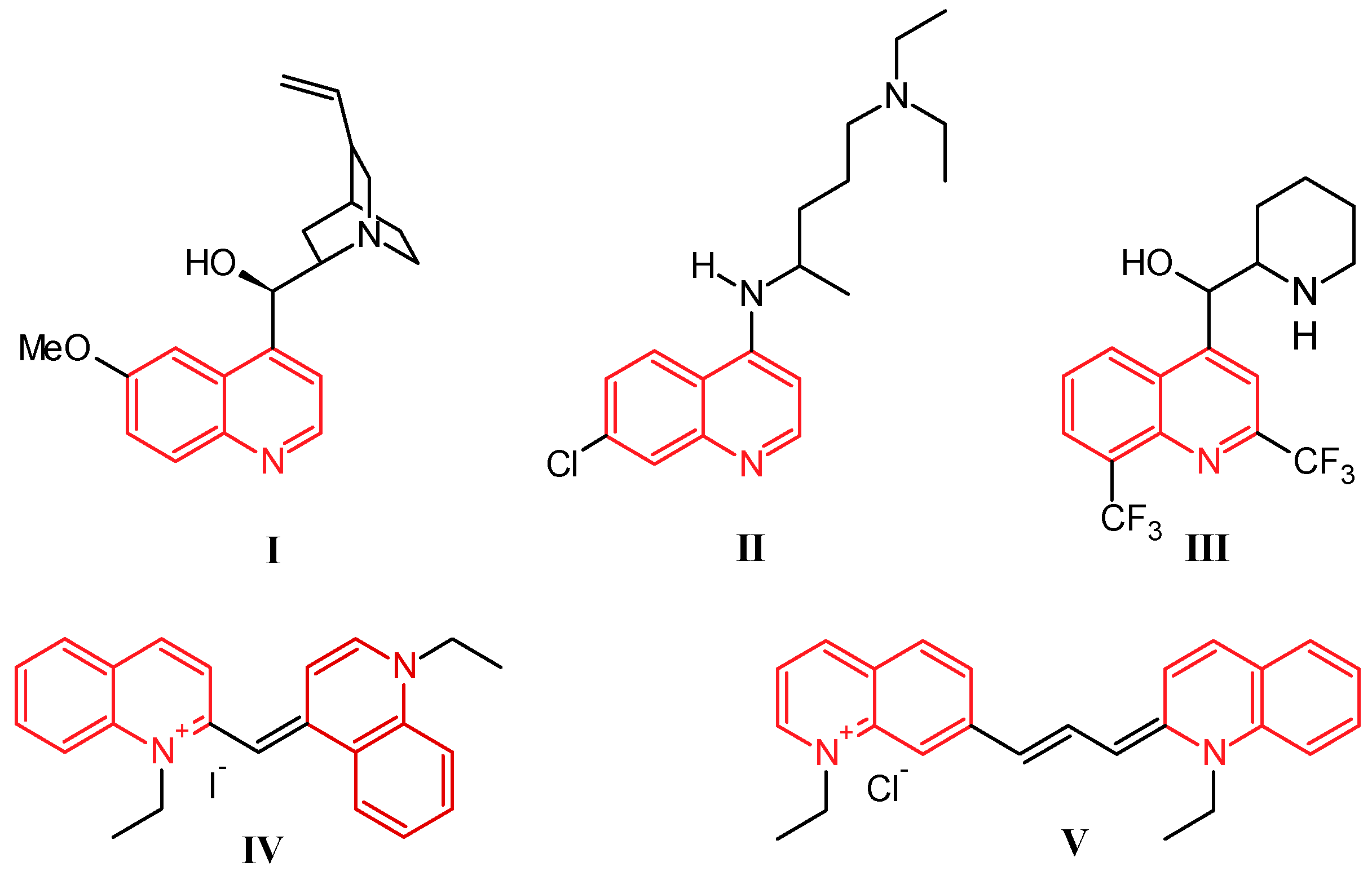
1. Introduction
2. Established Methods of Quinoline Synthesis
3. Quinoline Synthesis via Modifications to Aniline-Based Established Methods
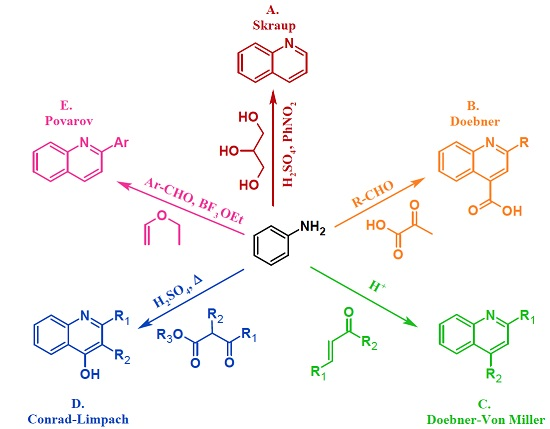
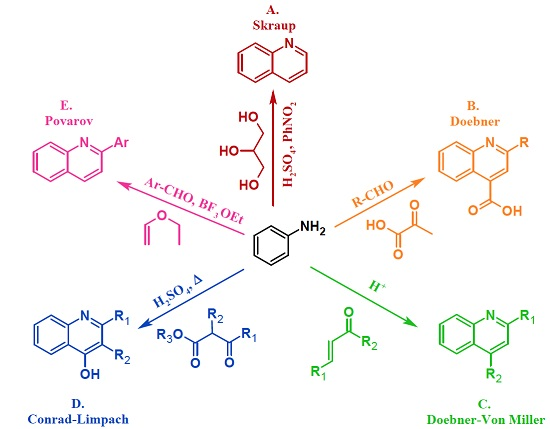
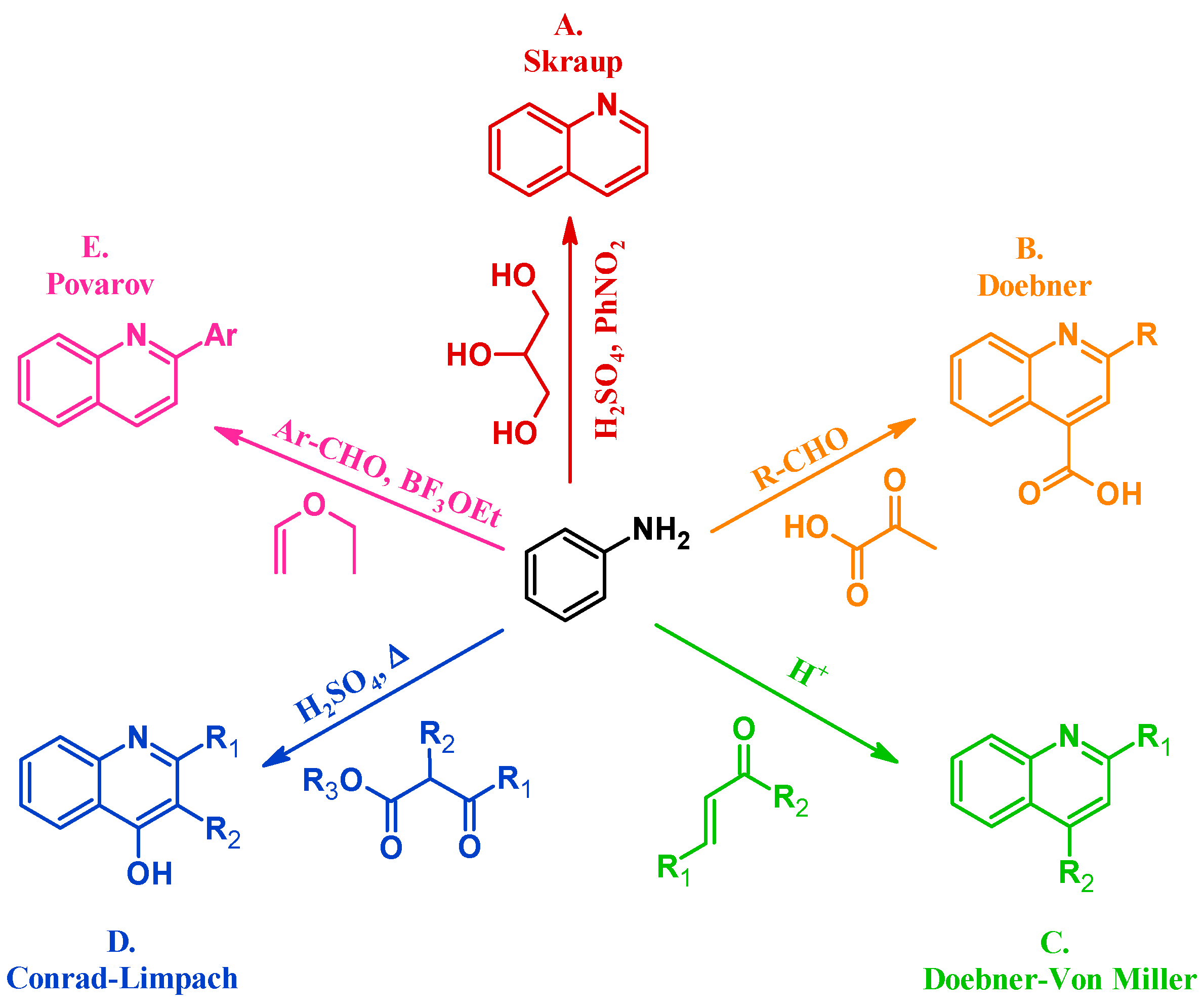
3.1. Skraup Reaction
3.2. Doebner Reaction
3.3. Doebner–Von Miller Reaction
3.4. Conrad–Limpach Reaction
3.5. Povarov Reaction
4. Quinoline Synthesis via Modifications to Non-Aniline-Based Established Methods
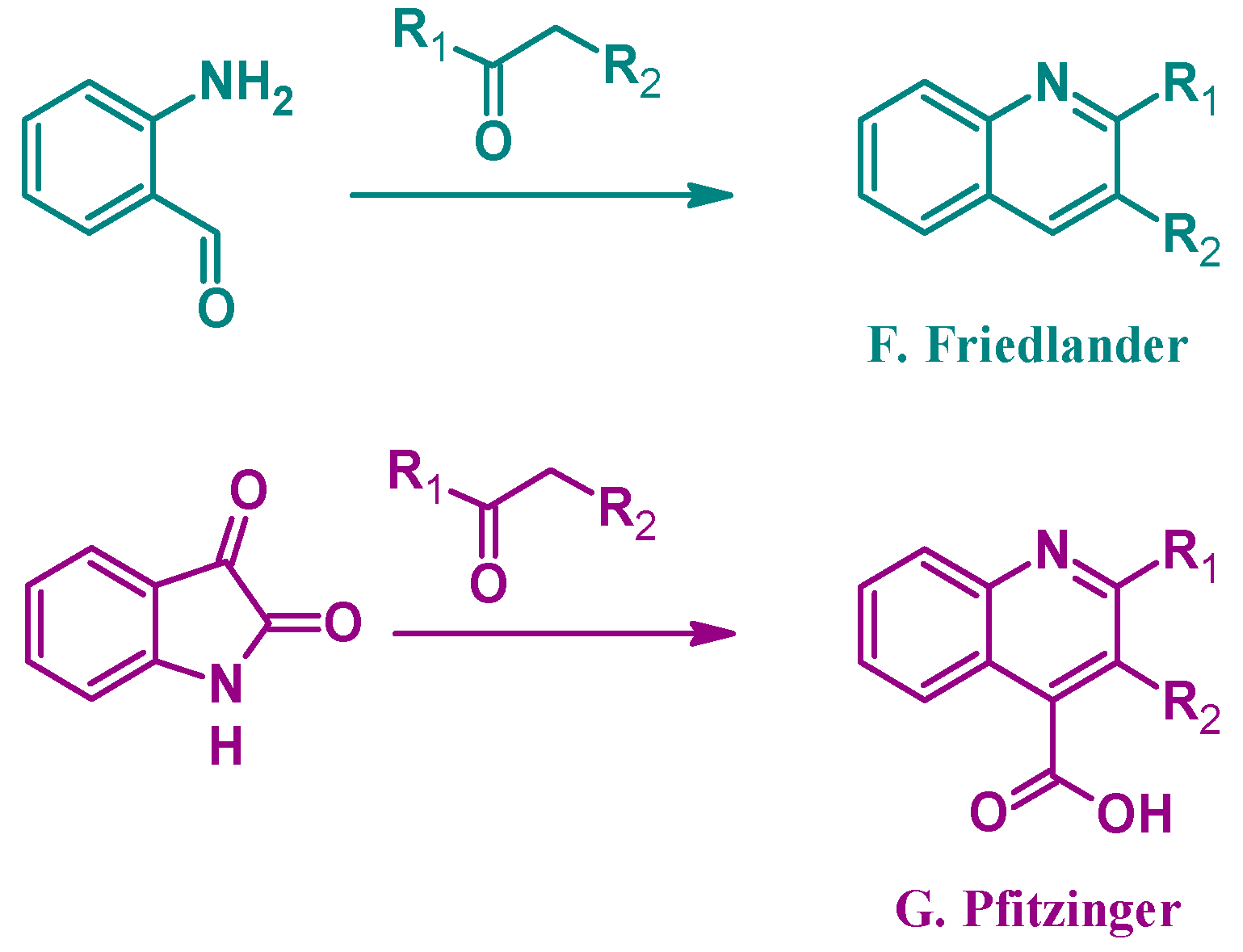
4.1. Pfitzinger Reaction
4.2. Friedländer Reaction
5. Quinoline Synthesis via Novel Synthetic Routes
5.1. Electrophilic Cyclization
5.2. Oxidative Cyclization
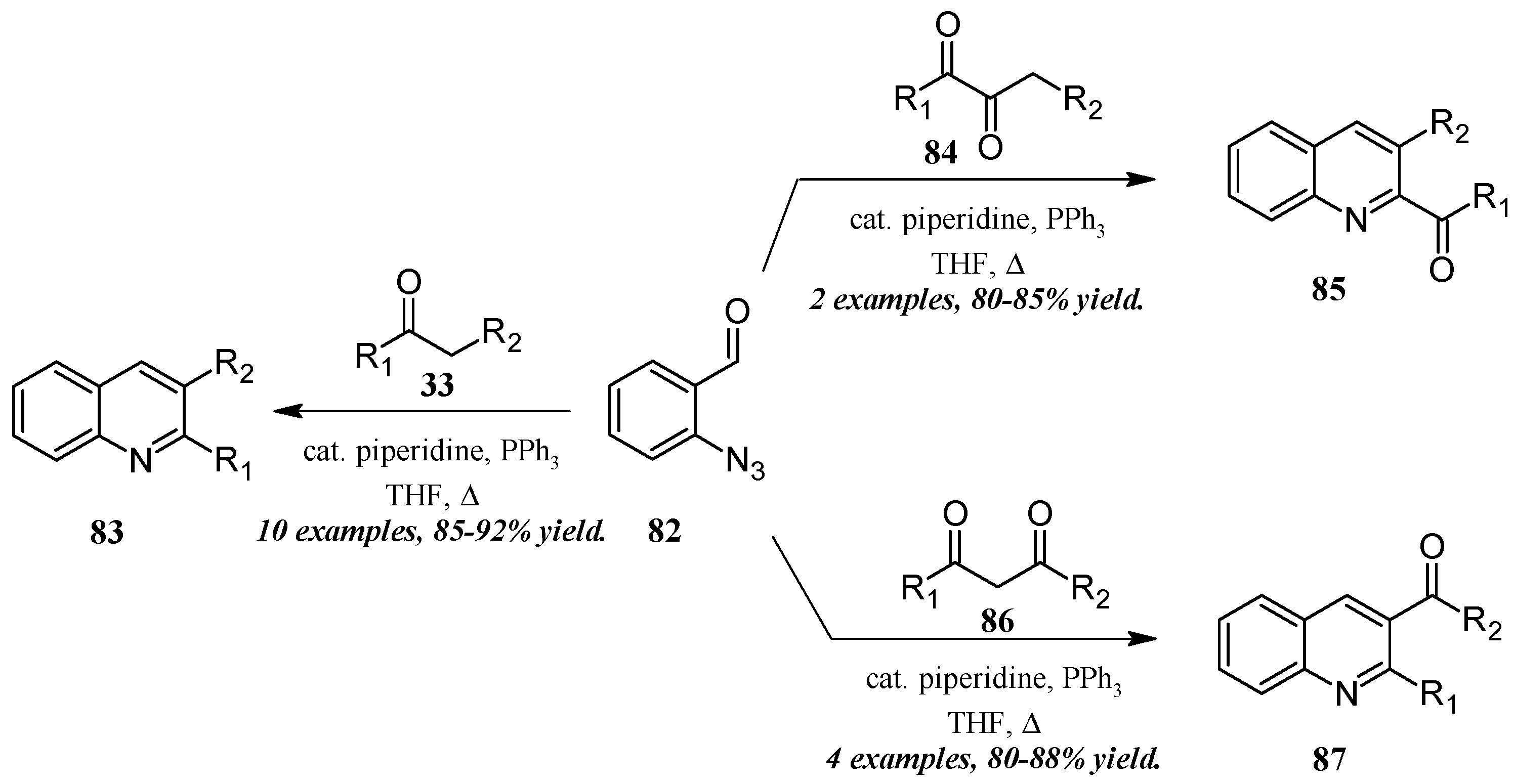
5.3. Aza-Wittig Cascade Reactions
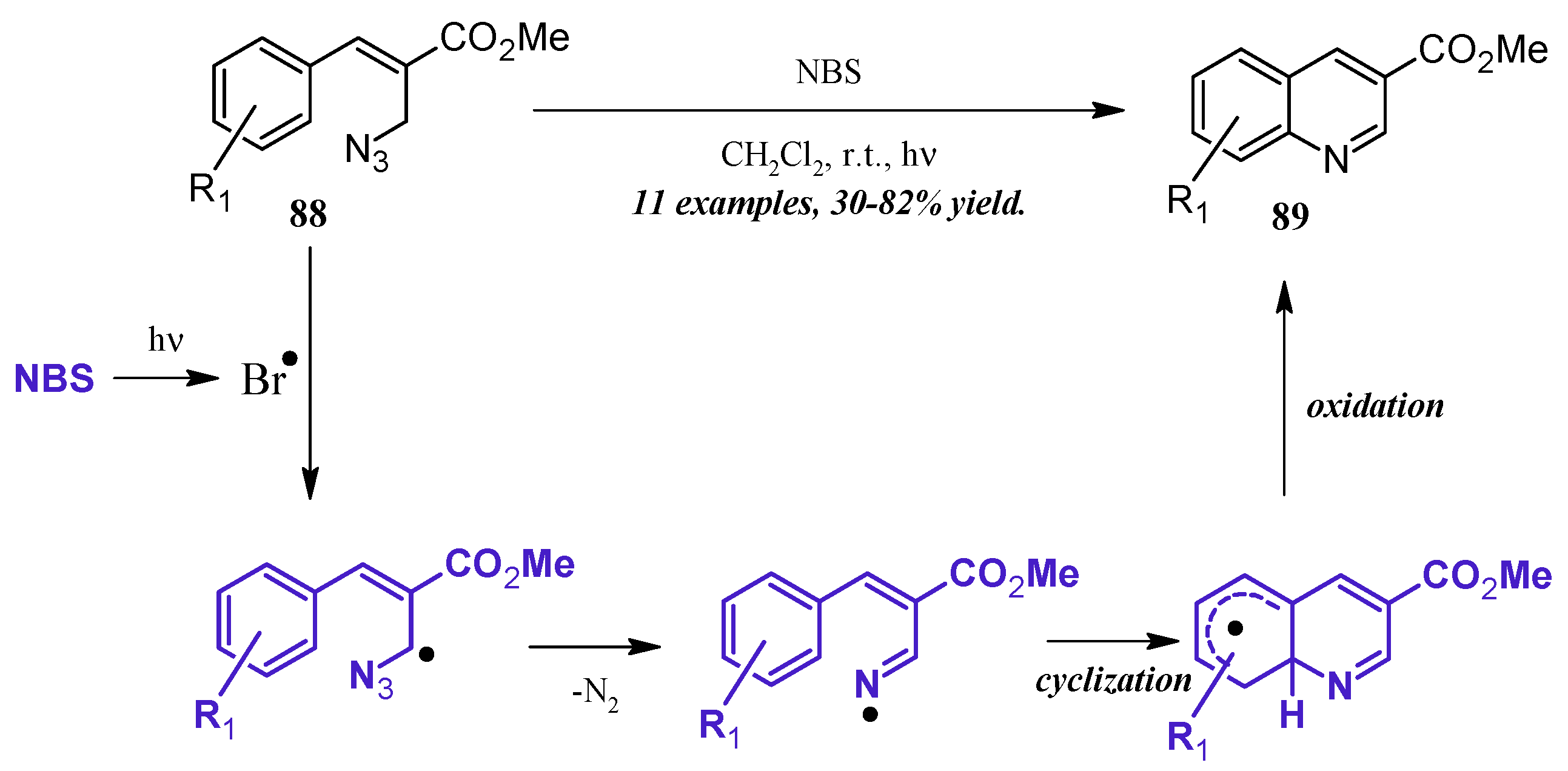
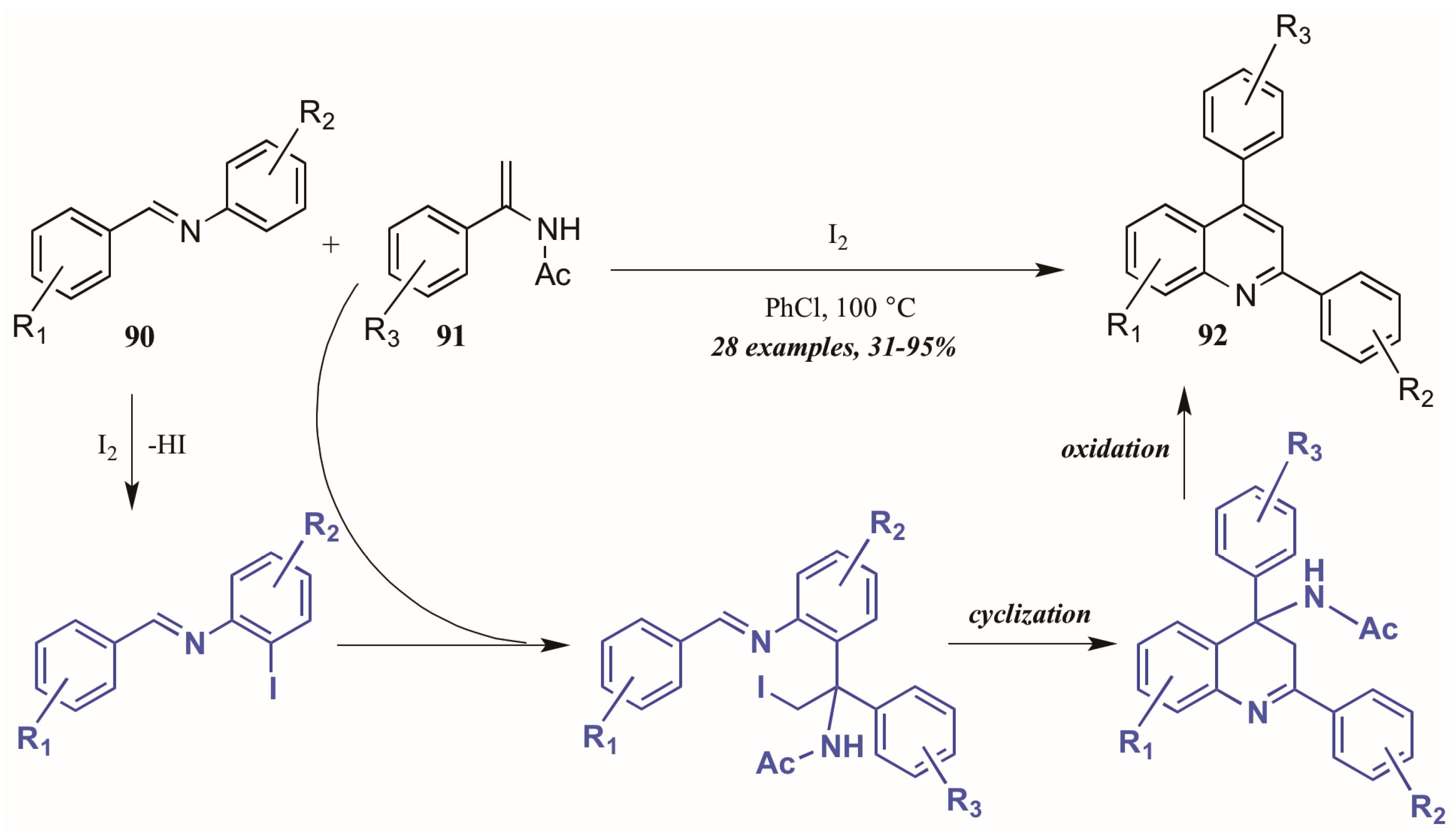
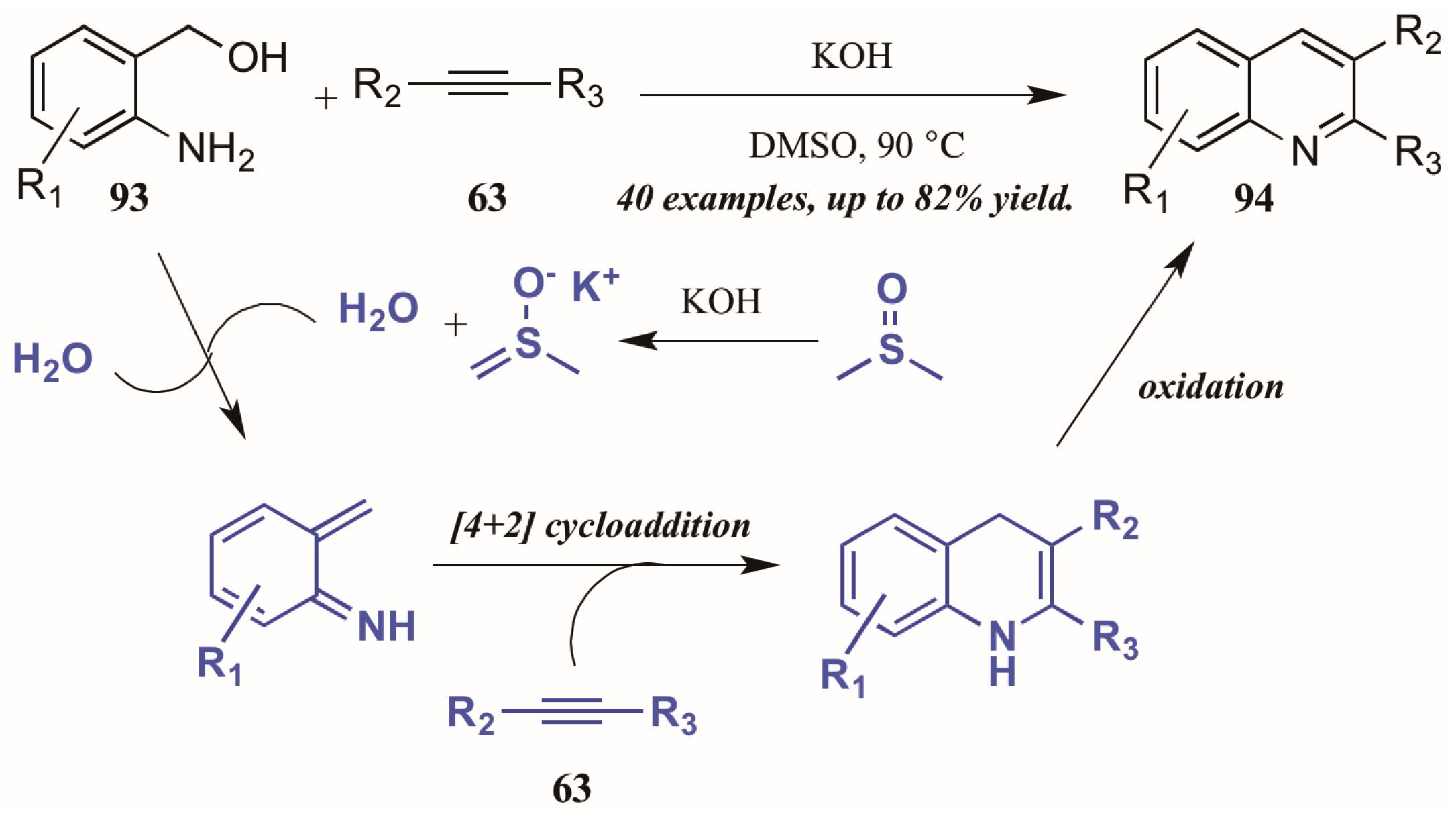
5.4. Other (Radical-Promoted, Cycloaddition, I2 Catalyzed)
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Meldola, R. Coal and What We Get from It: A Romance of Applied Science; Society for Promoting Christian Knowledge: London, UK, 1913. [Google Scholar]
- Achan, J.; Talisuna, A.O.; Erhart, A.; Yeka, A.; Tibenderana, J.K.; Baliraine, F.N.; Rosenthal, P.J.; D’Alessandro, U. Quinine, an old anti-malarial drug in a modern world: Role in the treatment of malaria. Malar J. 2011, 10, 144. [Google Scholar] [CrossRef] [PubMed]
- Chanda, T.; Verma, R.K.; Singh, M.S. InCl3-driven regioselective synthesis of functionalized/annulated quinolines: Scope and limitations. Chem. Asian J. 2012, 7, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.E.; Rittle, K.E.; Bock, M.G.; DiPardo, R.M.; Freidinger, R.M.; Whitter, W.L.; Lundell, G.F.; Veber, D.F.; Anderson, P.S.; Chang, R.S.L.; et al. Methods for drug discovery: development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem. 1988, 31, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- Bräse, S. Privileged Scaffolds in Medicinal Chemistry: Design, Synthesis, Evaluation; Royal Society of Chemistry: London, UK, 2015. [Google Scholar]
- Denmark, S.E.; Venkatraman, S. On the Mechanism of the Skraup–Doebner–Von Miller Quinoline Synthesis. J. Org. Chem. 2006, 71, 1668–1676. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J. Name Reactions: A Collection of Detailed Mechanisms and Synthetic Applications; Springer International Publishing: Cham, Switzerland, 2014. [Google Scholar]
- Povarov, L.S.; Grigos, V.I.; Mikhailov, B.M. Reaction of benzylideneaniline with some unsaturated compounds. Bull. Acad. Sci. USSR 1963, 12, 1878–1880. [Google Scholar] [CrossRef]
- Bharate, J.B.; Vishwakarma, R.A.; Bharate, S.B. Metal-free domino one-pot protocols for quinoline synthesis. RSC Adv. 2015, 5, 42020–42053. [Google Scholar] [CrossRef]
- Gaina, L.; Cristea, C.; Moldovan, C.; Porumb, D.; Surducan, E.; Deleanu, C.; Mahamoud, A.; Barbe, J.; Silberg, I.A. Microwave-assisted synthesis of phenothiazine and quinoline derivatives. Int. J. Mol. Sci. 2007, 8, 70–80. [Google Scholar] [CrossRef]
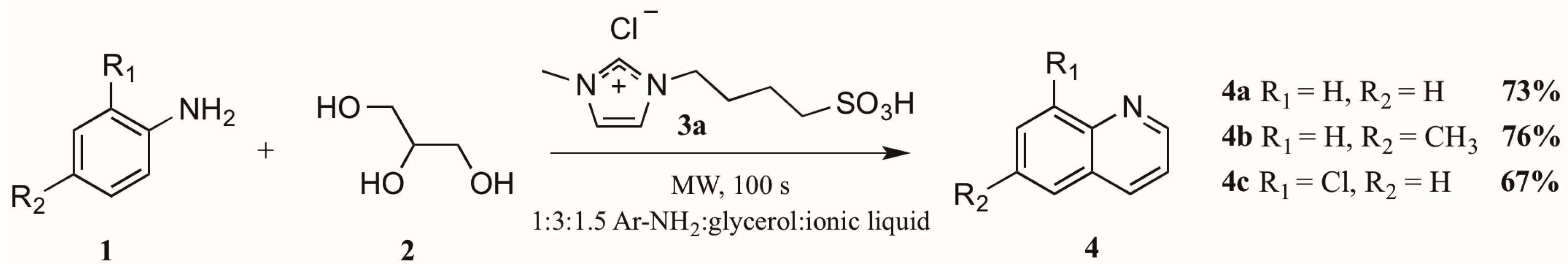
- Amarasekara, A.S.; Hasan, M.A. 1-(1-Alkylsulfonic)-3-methylimidazolium chloride Bronsted acidic ionic liquid catalyzed Skraup synthesis of quinolines under microwave heating. Tetrahedron Lett. 2014, 55, 3319–3321. [Google Scholar] [CrossRef]
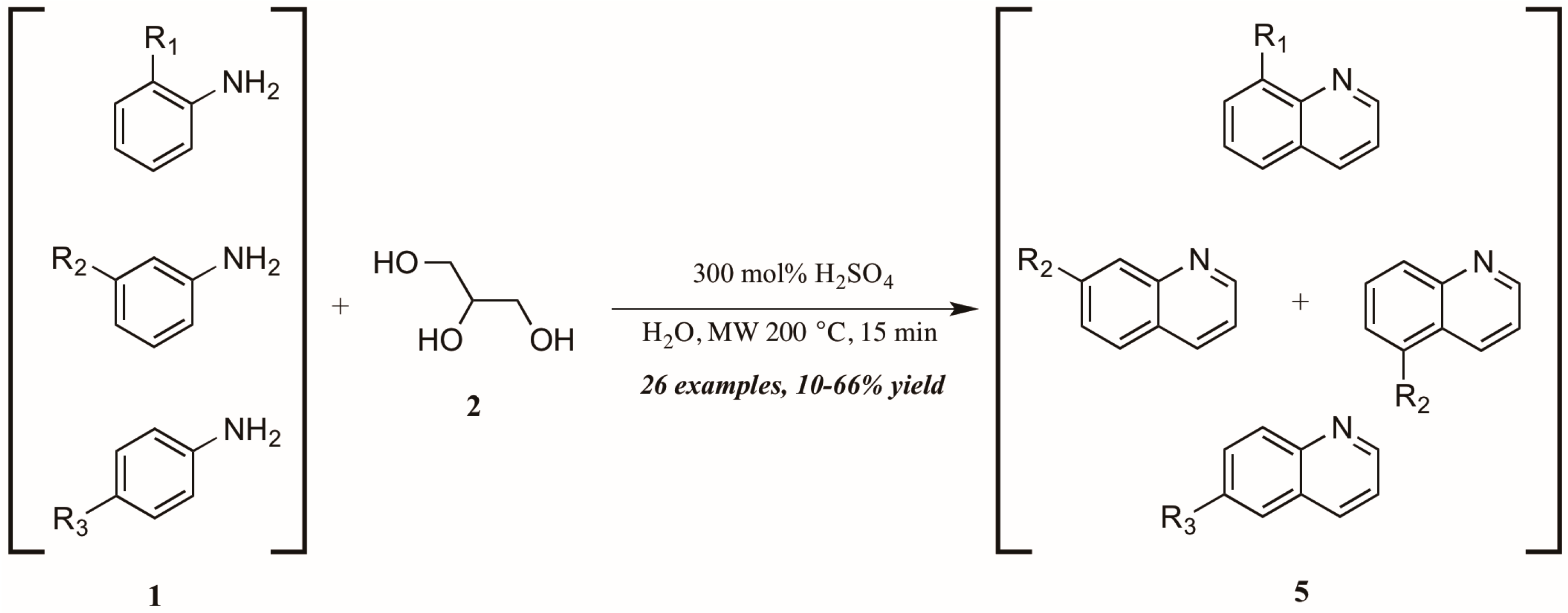
- Saggadi, H.; Luart, D.; Thiebault, N.; Polaert, I.; Estel, L.; Len, C. Quinoline and phenanthroline preparation starting from glycerol via improved microwave-assisted modified Skraup reaction. RSC Adv. 2014, 4, 21456–21464. [Google Scholar] [CrossRef]
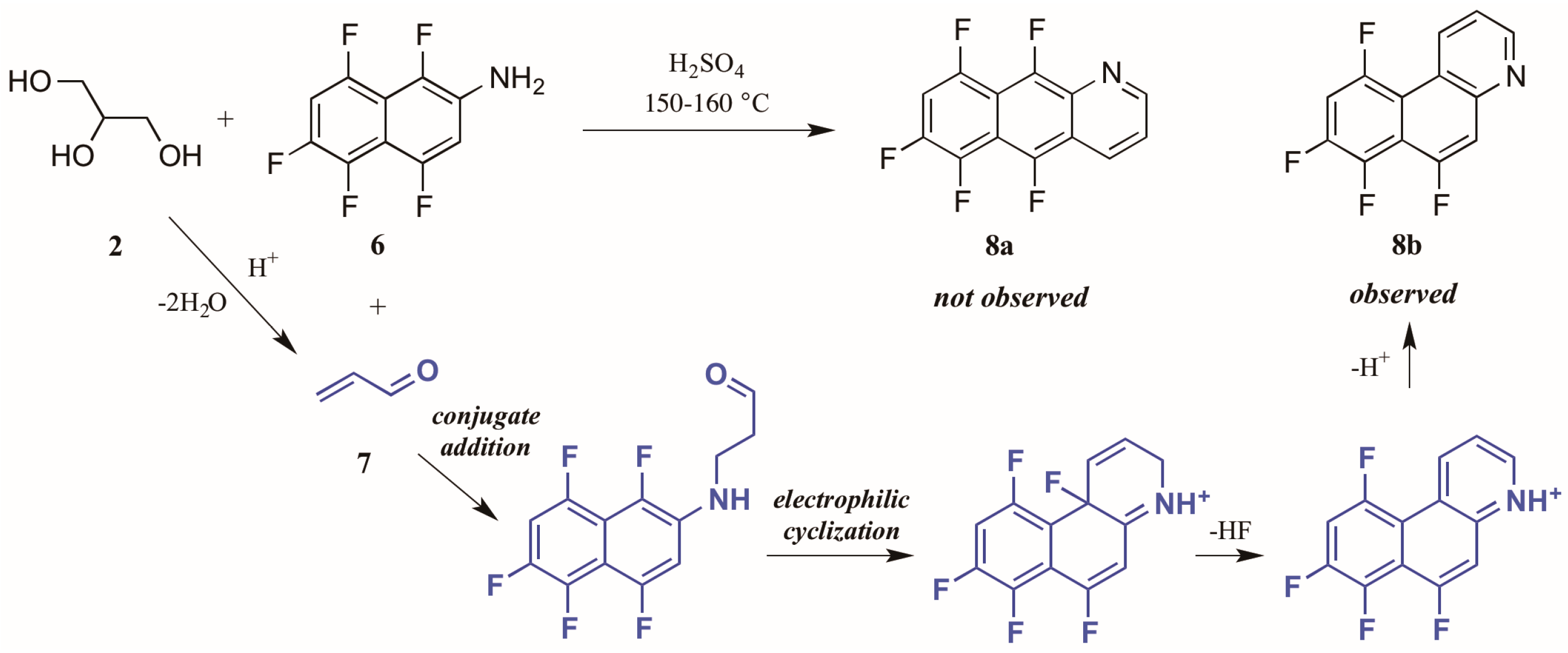
- Selivanova, G.A.; Reshetov, A.V.; Bagryanskaya, I.Y.; Shteingarts, V.D. Skraup-like cyclization of polyfluoro-2-naphthylamines: Vicarious electrophilic substitution of fluorine. J. Fluorine Chem. 2012, 137, 113–116. [Google Scholar] [CrossRef]
- Wang, L.-M.; Hu, L.; Chen, H.-J.; Sui, Y.-Y.; Shen, W. One-pot synthesis of quinoline-4-carboxylic acid derivatives in water: Ytterbium perfluorooctanoate catalyzed Doebner reaction. J. Fluorine Chem. 2009, 130, 406–409. [Google Scholar] [CrossRef]
- Guo, Q.; Wang, W.; Teng, W.; Chen, L.; Wang, Y.; Shen, B. Oxidant effect of H2O2 for the syntheses of quinoline derivatives via one-pot reaction of aniline and aldehyde. Synth. Commun. 2012, 42, 2574–2584. [Google Scholar] [CrossRef]
- Bharate, J.B.; Bharate, S.B.; Vishwakarma, R.A. Metal-Free, ionic liquid-mediated synthesis of functionalized quinolines. ACS Comb. Sci. 2014, 16, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-G.; Cheng, X.; Zhou, Q.-L. A convenient synthesis of 2-alkyl-8-quinoline carboxylic acids. Synth. Commun. 2002, 32, 2477–2481. [Google Scholar] [CrossRef]
- Ramann, G.A.; Cowen, B.J. Quinoline synthesis by improved Skraup–Doebner–Von Miller reactions utilizing acrolein diethyl acetal. Tetrahedon Lett. 2015, 56, 6436–6439. [Google Scholar] [CrossRef]
- Eisch, J.J.; Dluzniewski, T. Mechanism of the Skraup and Doebner-von Miller quinoline syntheses: Cyclization of alpha,beta-unsaturated N-Aryliminium salts via 1,3-diazetidinium ion intermediates. J. Org. Chem. 1988, 54, 1269–1274. [Google Scholar] [CrossRef]
- Reitsema, R.H. The chemistry of 4-hydroxyquinolines. Chem. Rev. 1948, 1, 43–68. [Google Scholar] [CrossRef]
- Brouet, J.-C.; Gu, S.; Peet, N.P.; Williams, J.D. A survey of solvents for the Conrad-Limpach synthesis of 4-hydroxyquinolones. Synth. Commun. 2009, 39, 5193–5196. [Google Scholar] [CrossRef] [PubMed]
- Gattu, R.; Basha, R.S.; Bagdi, P.R.; Khan, A.T. One-pot three-component regioselective synthesis of C1-functionalised 3-arylbenzo[f]quinoline. RSC Adv. 2016, 6, 11675–11682. [Google Scholar] [CrossRef]
- Ganem, B. Strategies for innovation in multicomponent reaction design. Acc. Chem. Res. 2009, 42, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Mao, Z.; Wang, Y.; Chen, W.; Lin, X. Molecular iodine-catalyzed and air-mediated tandem synthesis of quinolines via three-component reaction of amines, aldehydes, and alkynes. Tetrahedron 2011, 67, 3858–3862. [Google Scholar] [CrossRef]
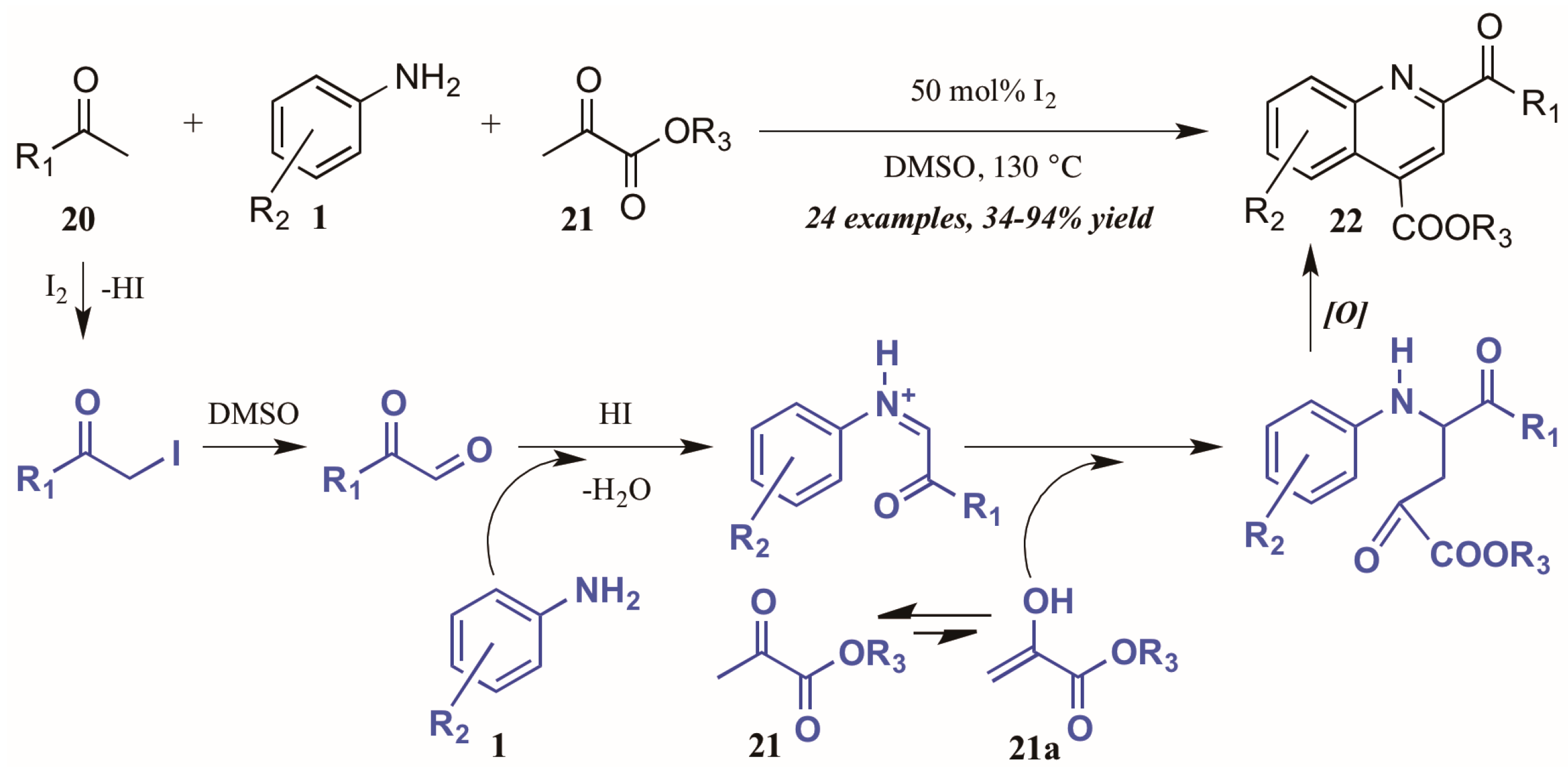
- Gao, Q.; Liu, S.; Wu, X.; Zhang, J.; Wu, A. Coproduct promoted Povarov reaction: Synthesis of substituted quinolines from methyl ketones, arylamines, and α-ketoesters. J. Org. Chem. 2015, 80, 5984–5991. [Google Scholar] [CrossRef] [PubMed]
- Shvekhgeimer, M. The Pfitzinger reaction. (review). Chem. Heterocycl. Compd. 2004, 40, 257–294. [Google Scholar] [CrossRef]
- Ivachtchenko, A.V.; Kobak, V.V.; Il’yin, A.P.; Trifilenkov, A.S.; Busel, A.A. New scaffolds for combinatorial synthesis. II. 6-sulfamoylquinolinecarboxylic acids. J. Comb. Chem. 2003, 5, 645–652. [Google Scholar] [CrossRef]
- Zhu, H.; Yang, R.F.; Yun, L.H.; Li, J. Facile and efficient synthesis of quinoline-4-carboxylic acids under microwave irradiation. Chin. Chem. Lett. 2010, 21, 35–38. [Google Scholar] [CrossRef]
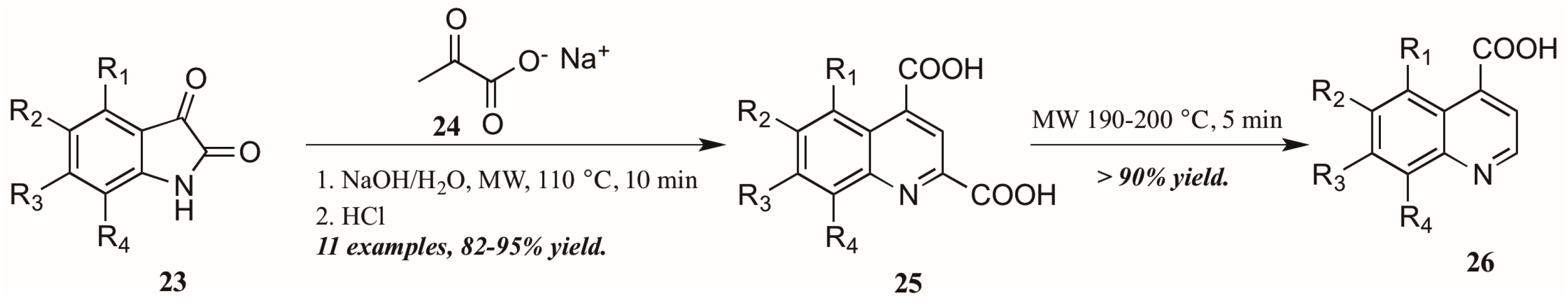
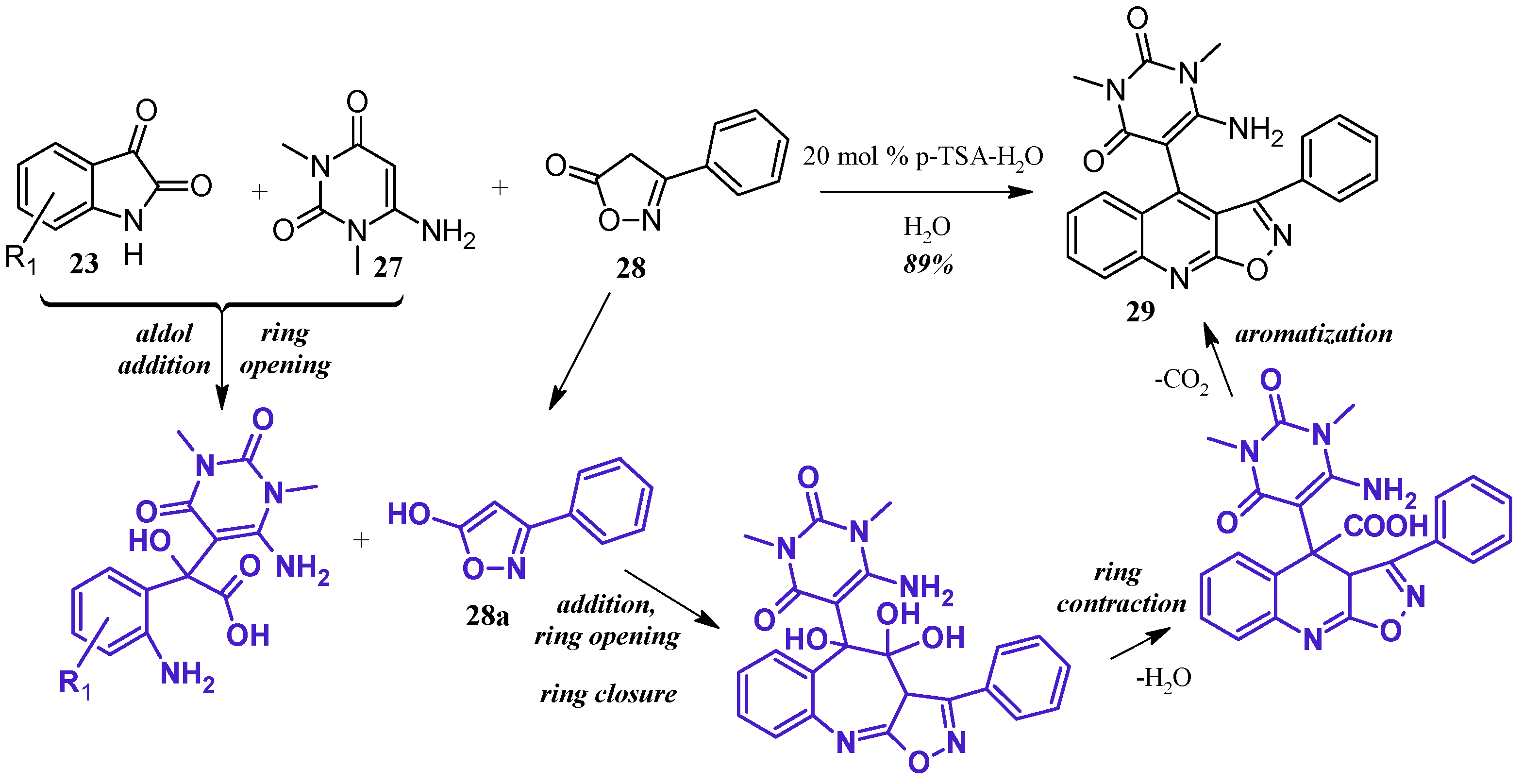
- Poomathi, N.; Mayakrishnan, S.; Muralidharan, D.; Srinivasan, R.; Perumal, P.T. Reaction of isatins with 6-amino uracils and isoxazoles: Isatin ring-opening vs. annulations and regioselective synthesis of isoxazole fused quinoline scaffolds in water. Green Chem. 2015, 17, 3362–3372. [Google Scholar] [CrossRef]
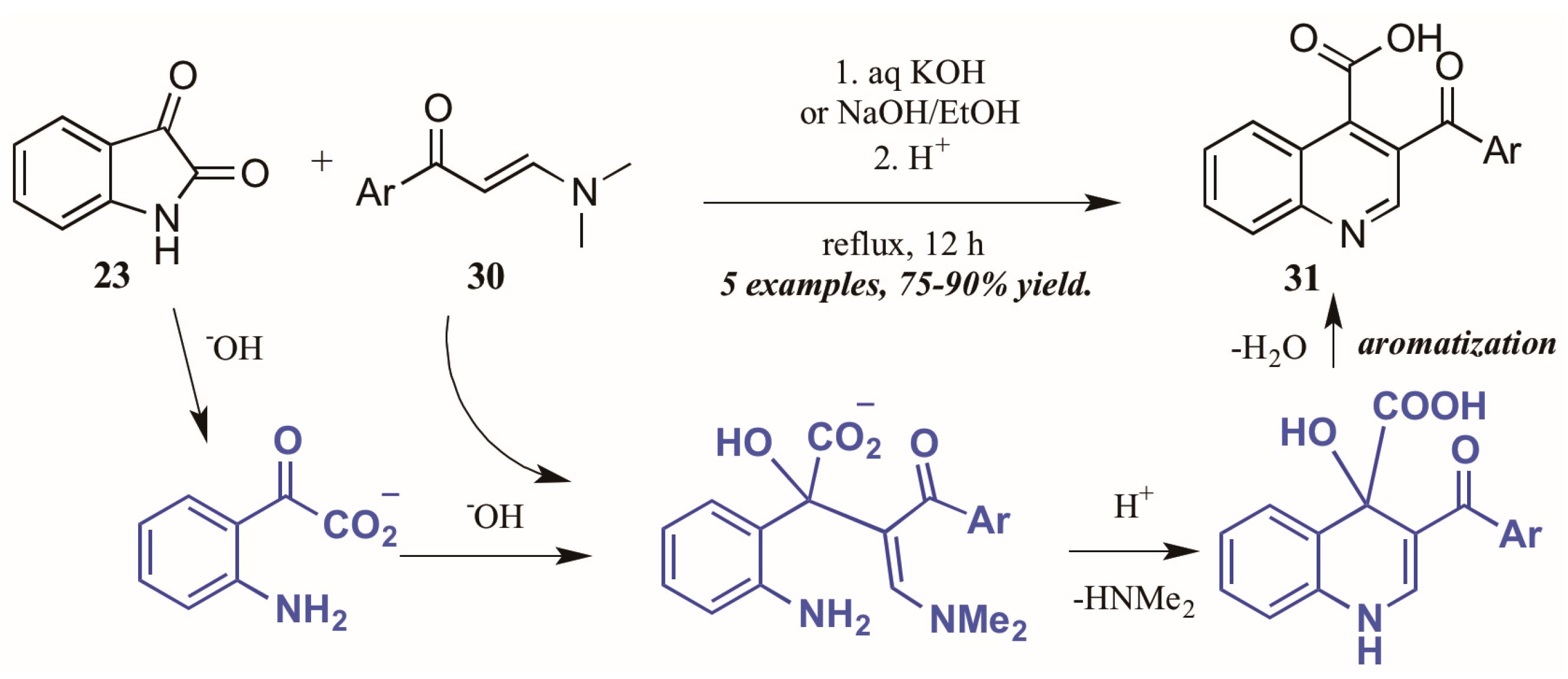
- Elghamry, I.; Al-Faiyz, Y. A simple one-pot synthesis of quinoline-4-carboxylic acids by the Pfitzinger reaction of isatin with enaminones in water. Tetrahedron Lett. 2016, 57, 110–112. [Google Scholar] [CrossRef]
- Marco-Contelles, J.; Perez-Mayoral, E.; Samadi, A.; do Carmo Carreiras, M.; Soriano, E. Recent advances in the Friedlander Reaction. Chem. Rev. 2009, 109, 2652–2671. [Google Scholar] [CrossRef] [PubMed]
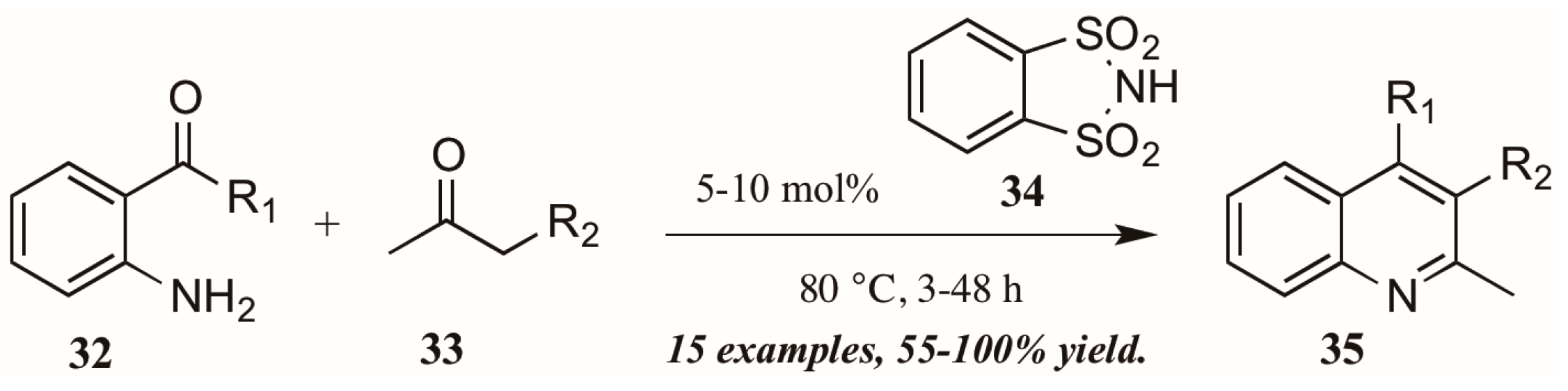
- Barbero, M.; Bazzi, S.; Cadamuro, S.; Dughera, S. o-Benzenedisulfonimide as a reusable Bronsted acid catalyst for an efficient and facile synthesis of quinolines via Friedlander annulation. Tetrahedron Lett. 2010, 51, 2342–2344. [Google Scholar] [CrossRef]
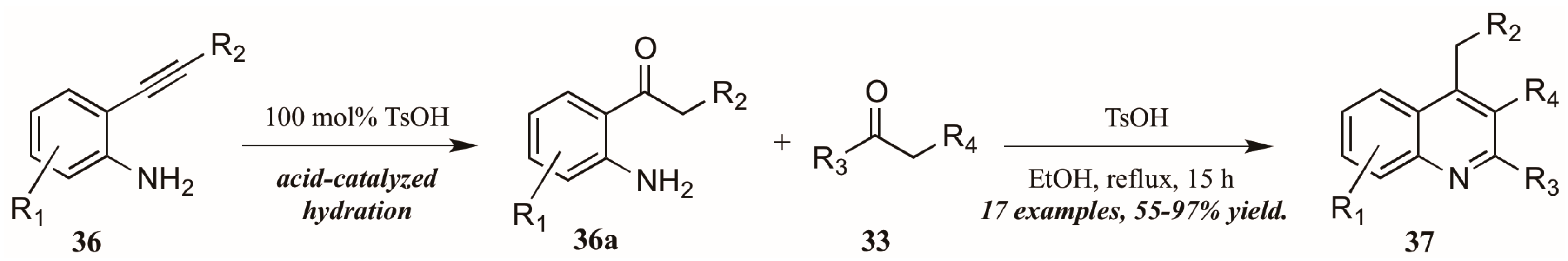
- Peng, C.; Wang, Y.; Liu, L.; Wang, H.; Zhao, J.; Zhu, Q. p-Toluenesulfonic acid promoted annulation of 2-alkynylanilines with activated ketones: Efficient synthesis of 4-alkyl-2,3-disubstituted quinolines. Eur. J. Org. Chem. 2010, 5, 818–822. [Google Scholar] [CrossRef]
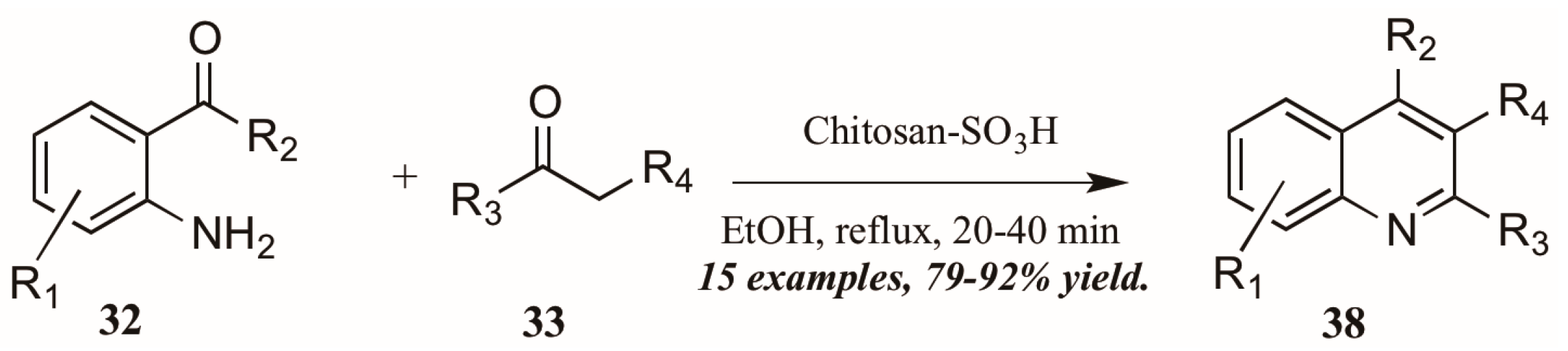
- Subba Reddy, B.V.; Venkateswarlu, A.; Niranjan Reddy, G.; Rami Reddy, Y.V. Chitosan-SO3H: An efficient, biodegradable, and recyclable solid acid for the synthesis of quinoline derivatives via Friedlander annulation. Tetrahedron Lett. 2013, 54, 5767–5770. [Google Scholar] [CrossRef]
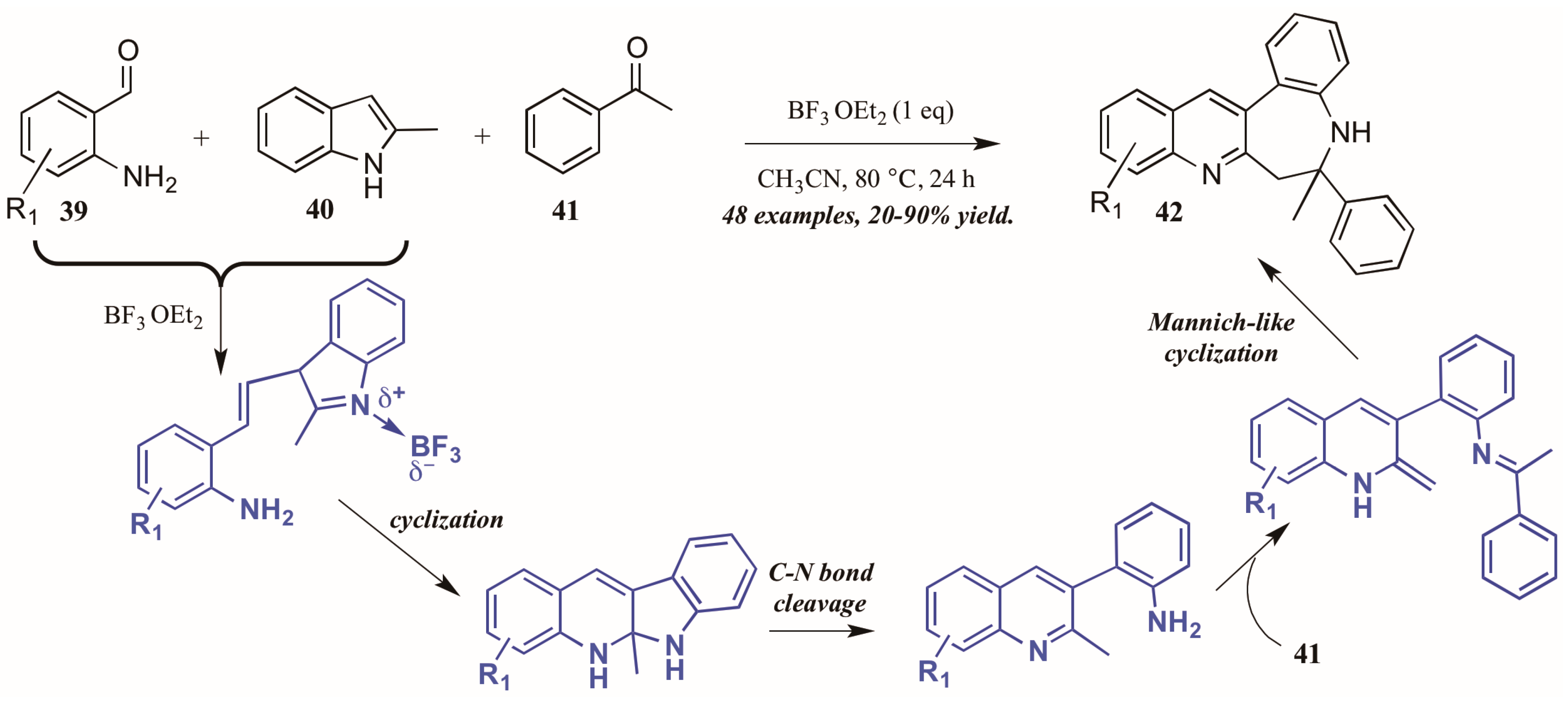
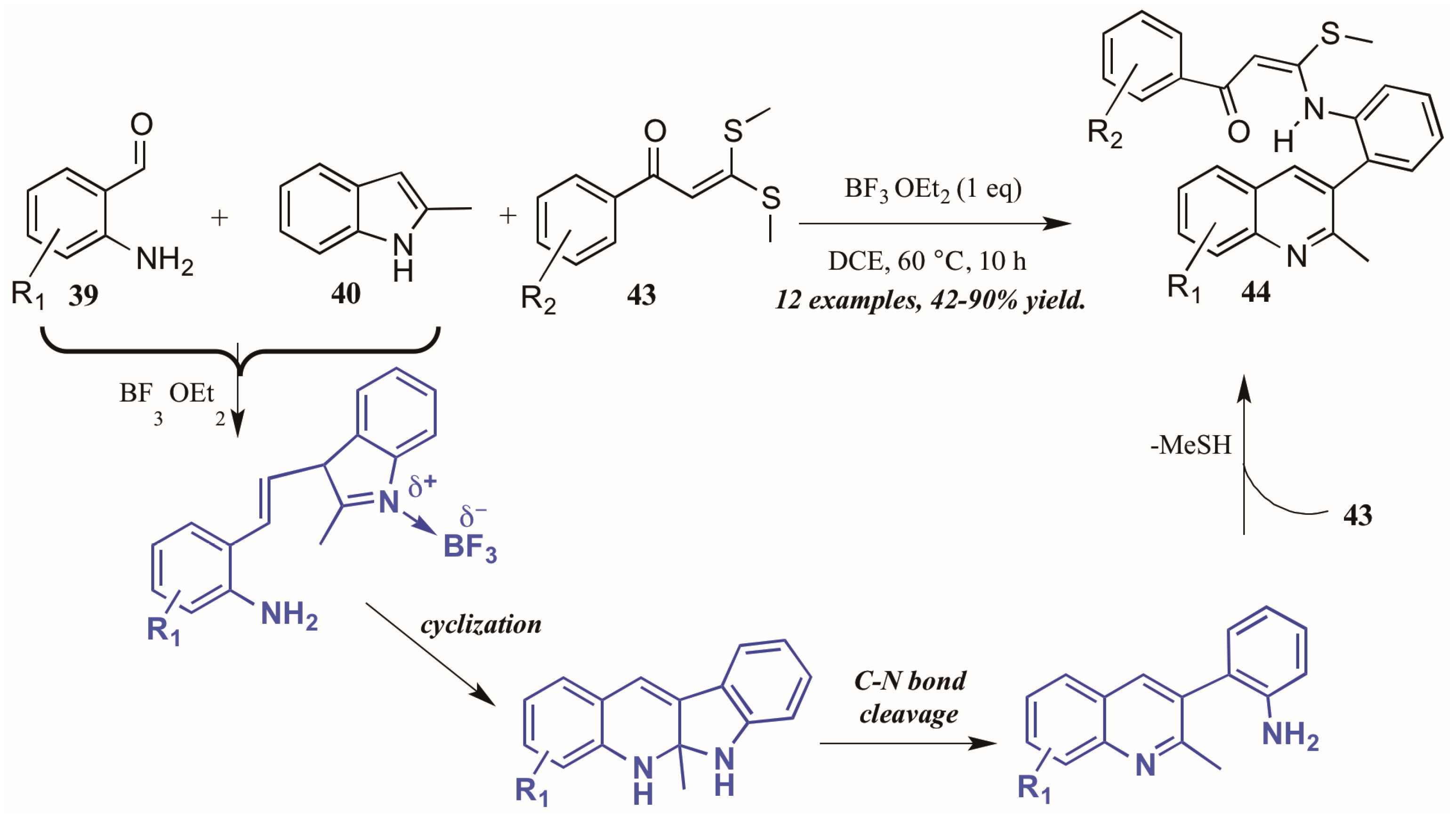
- Min, L.; Pan, B.; Gu, Y. Synthesis of quinoline-fused 1-benzazepines through a Mannich-type reaction of a C,N-bisnucleophile generated from 2-aminobenzaldehyde and 2-methylindole. Org. Lett. 2016, 18, 364–367. [Google Scholar] [CrossRef] [PubMed]
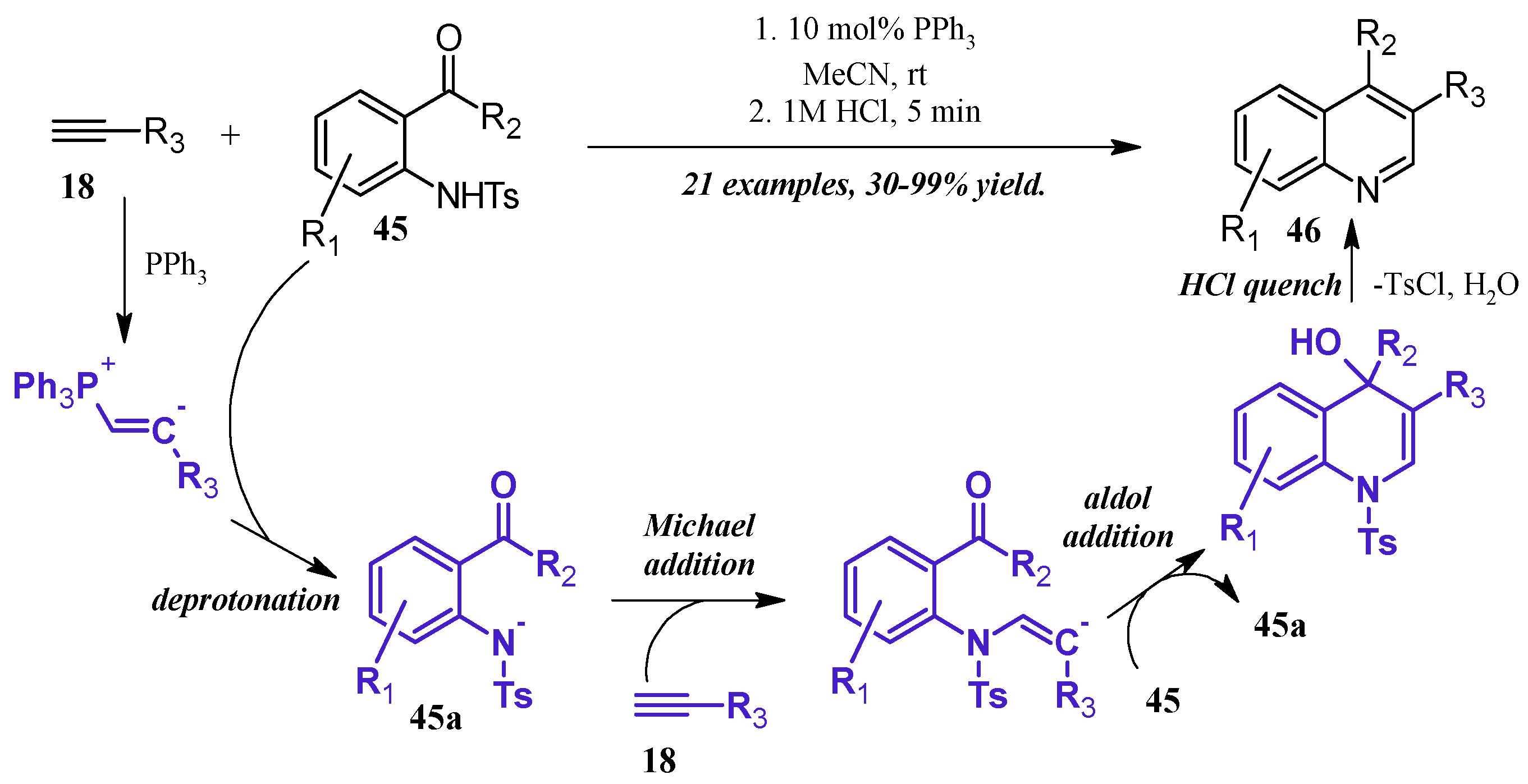
- Khong, S.; Kwon, O. One-pot phosphine-catalyzed syntheses of quinolines. J. Org. Chem. 2012, 77, 8257–8267. [Google Scholar] [CrossRef] [PubMed]
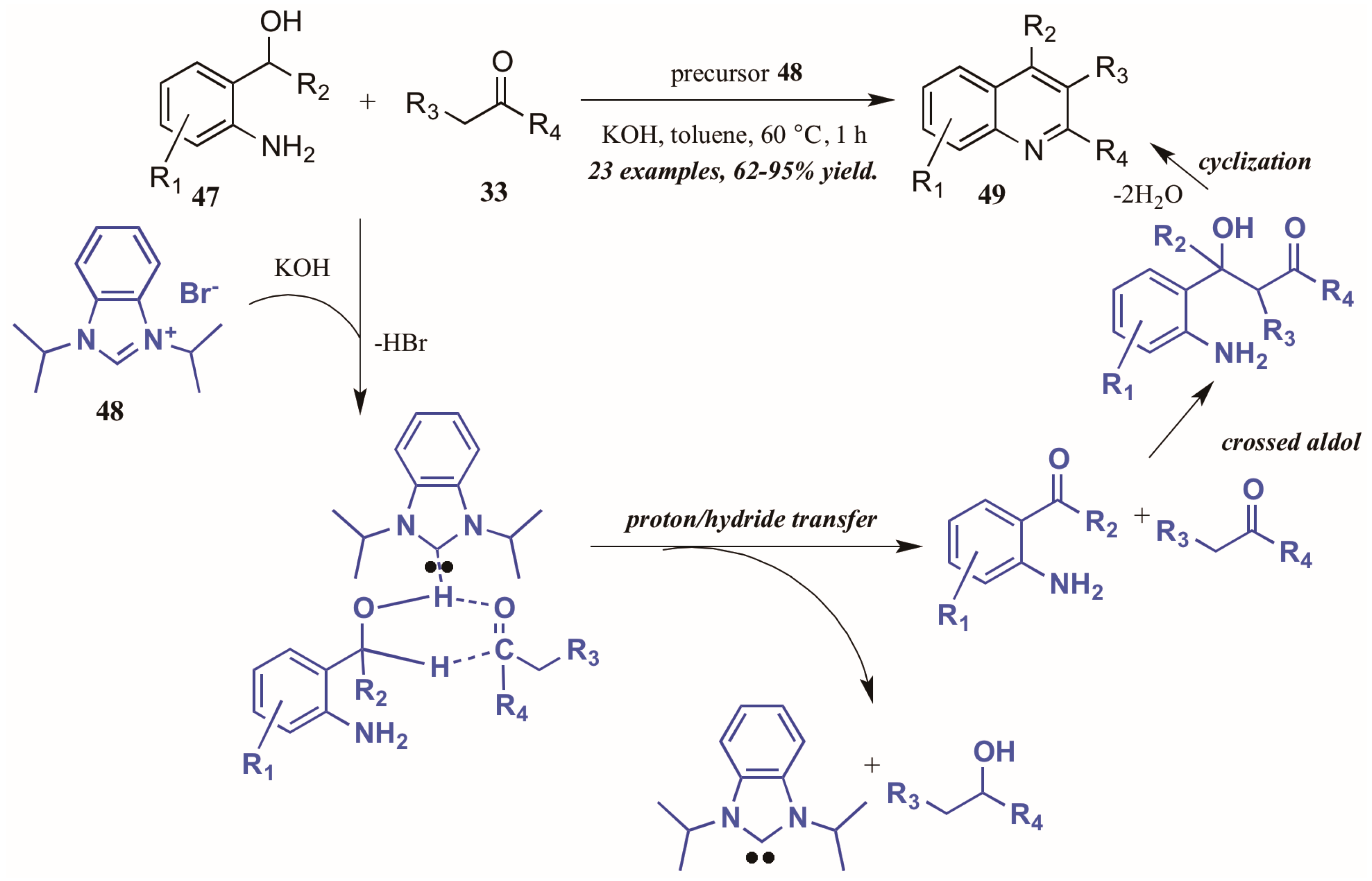
- Zhu, Y.; Cai, C. An N-heterocyclic carbene-catalyzed approach to the indirect Friedlander quinoline synthesis. RSC Adv. 2014, 4, 52911–52914. [Google Scholar] [CrossRef]
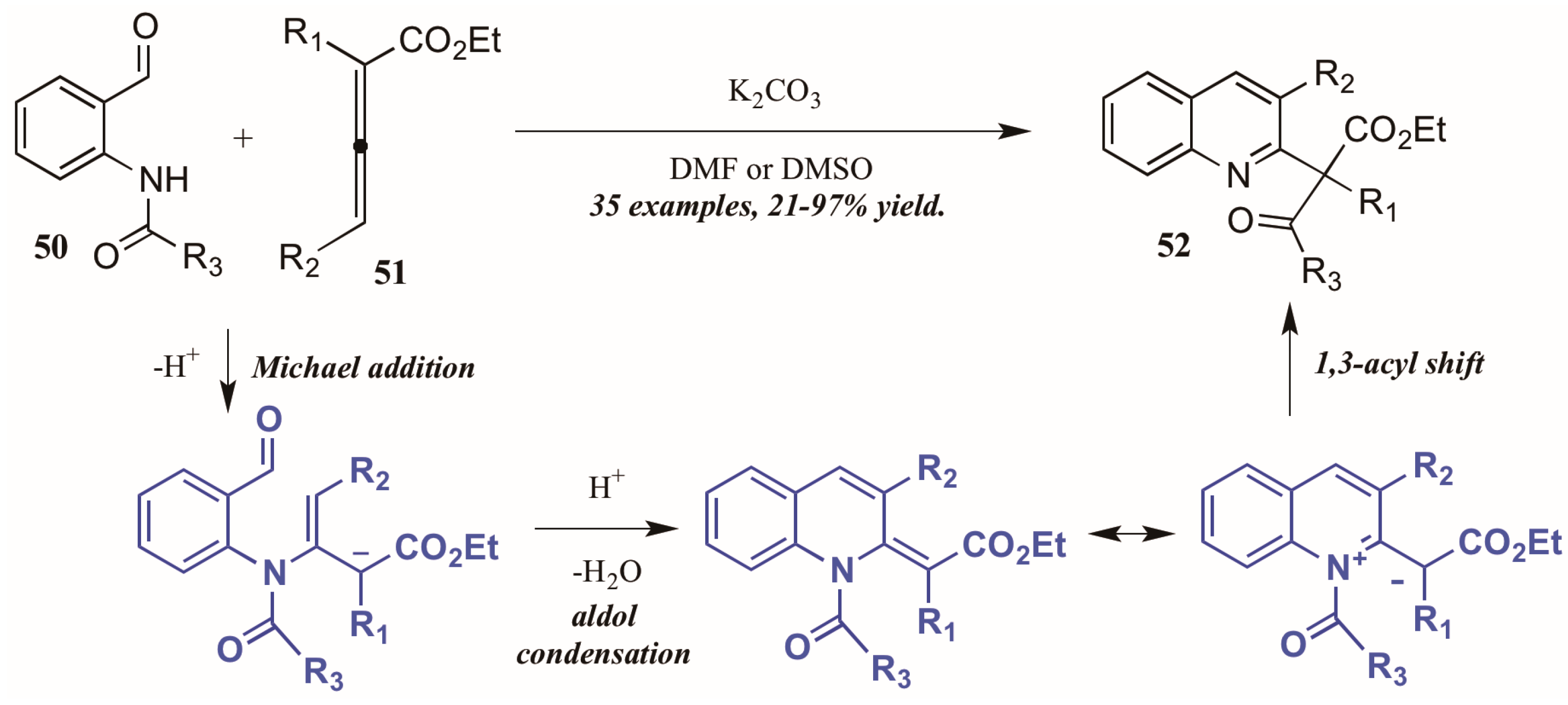
- Selig, P.; Raven, W. A convenient allenoate-based synthesis of 2-quinolin-2-yl malonates and β-ketoesters. Org. Lett. 2014, 16, 5192–5195. [Google Scholar] [CrossRef] [PubMed]
- Tajik, H.; Niknam, K.; Sarrafan, M. 1-Butyl-3-methylimidazolium hydrogen sulfate ([bmim]-HSO4)-mediated synthesis of polysubstituted quinolines. Synth. Commun. 2011, 41, 2103–2114. [Google Scholar] [CrossRef]
- Sarma, P.; Dutta, A.K.; Gogoi, P.; Sarma, B.; Borah, R. 3-Methyl-1-sulfoimidazolium ionic liquids as recyclable medium for efficient synthesis of quinoline derivatives by Friedlander annulation. Monatsh. Chem. 2015, 146, 173–180. [Google Scholar] [CrossRef]
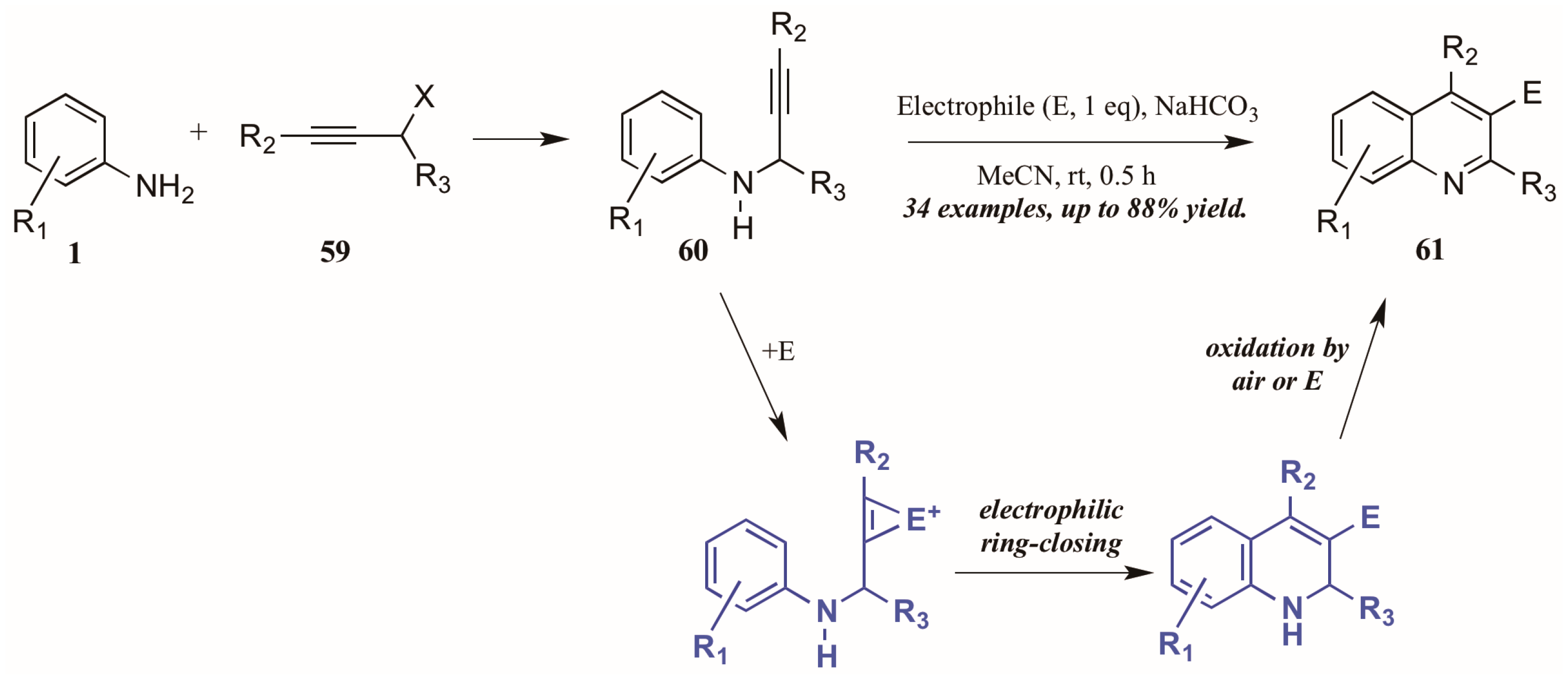
- Zhang, X.; Yao, T.; Campo, M.A.; Larock, R.C. Synthesis of substituted quinolines by the electrophilic cyclization of n-(2-alkynyl)anilines. Tetrahedron 2010, 66, 1177–1187. [Google Scholar] [CrossRef] [PubMed]
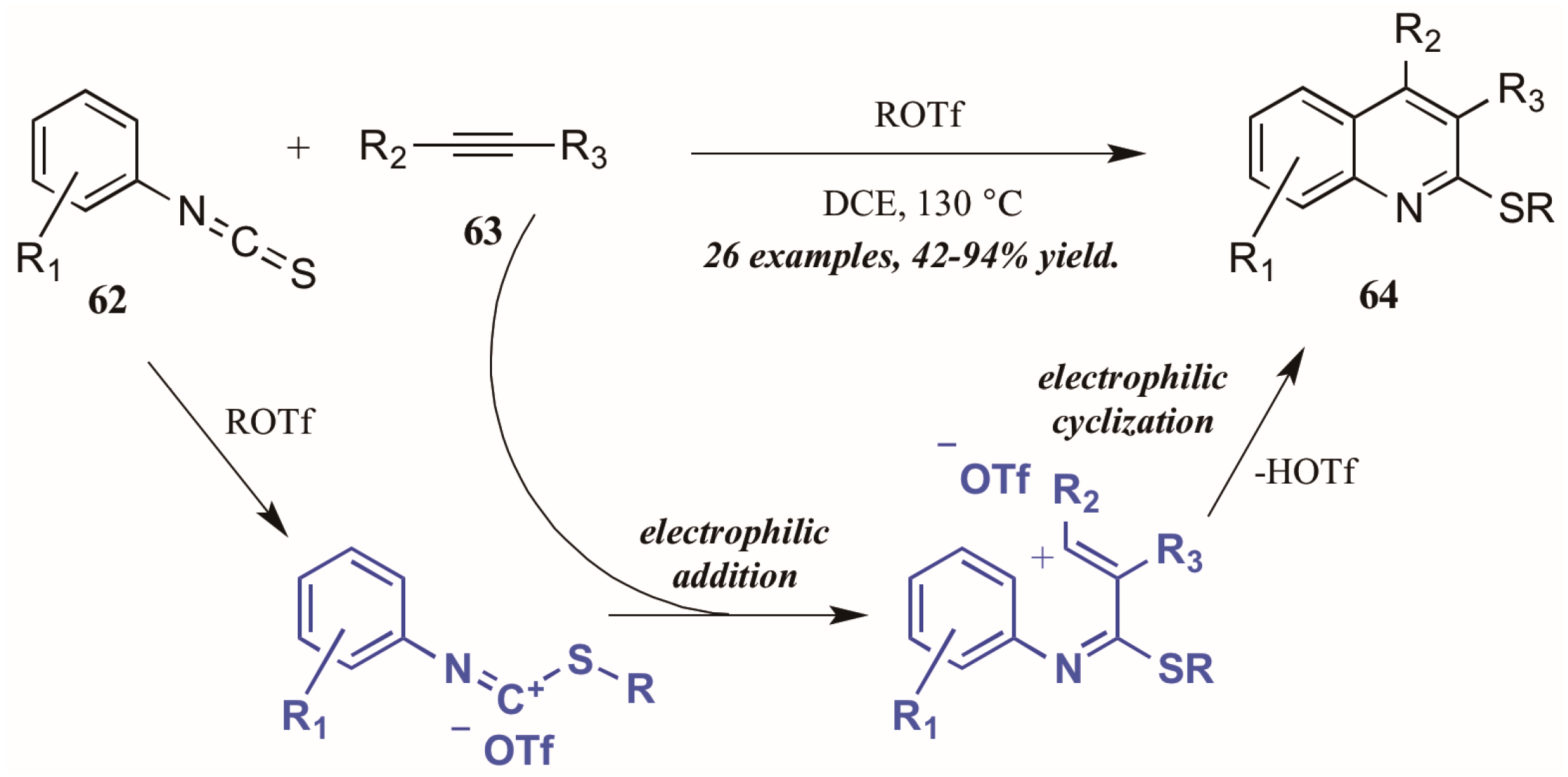
- Zhao, P.; Yan, X.; Yin, H.; Xi, C. Alkyltriflate-triggered annulation of arylisothiocyanates and alkynes leading to multiply substituted quinolines through domino electrophilic activation. Org. Lett. 2014, 16, 1120–1123. [Google Scholar] [CrossRef] [PubMed]
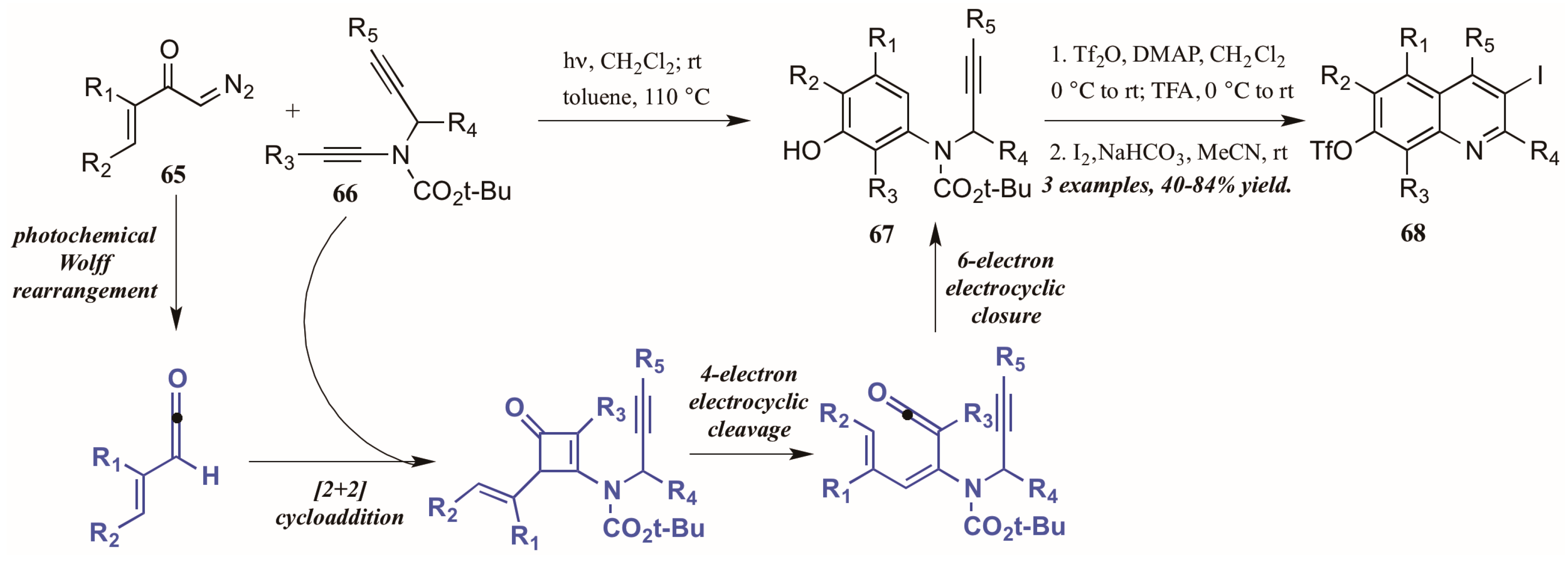
- Willumstad, T.P.; Boudreau, P.D.; Danheiser, R.L. Synthesis of highly substituted quinolines via a tandem ynamide benzannulation/iodocyclization strategy. J. Org. Chem. 2015, 80, 11794–11805. [Google Scholar] [CrossRef] [PubMed]
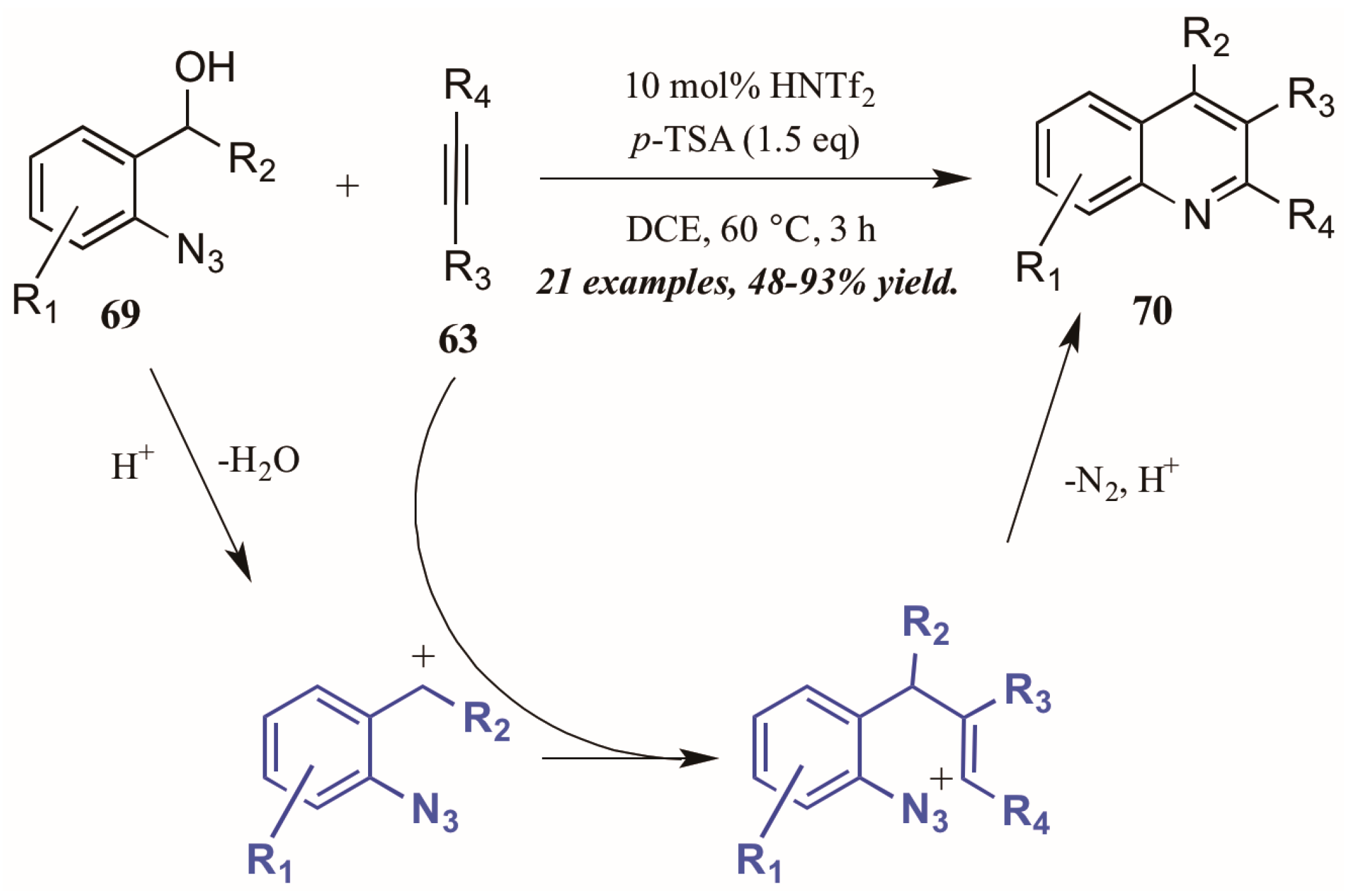
- Stopka, T.; Niggemann, M. Metal free carboamination of internal alkynes—An easy access to polysubstituted quinolines. Chem. Commun. 2016, 52, 5761–5764. [Google Scholar] [CrossRef] [PubMed]
- Wezeman, T.; Zhong, S.; Nieger, M.; Brase, S. Synthesis of highly functionalized 4-aminoquinolines. Angew. Chem. Int. Ed. 2016, 55, 3823–3827. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Huang, Z.; Liu, C.; Zhang, H.; Lei, A. Aerobic C-N bond activation: A simple strategy to construct pyridines and quinolines. Chem. Commun. 2015, 51, 2286–2289. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Yu, L.; Lv, S.; Yao, J.; Liu, J.; Jia, X. An unexpected construction of 2-arylquinolines from N-cinnamylanilines through sp3 C−H aerobic oxidation induced by a catalytic radical cation salt. Adv. Synth. Catal. 2016, 358, 459–465. [Google Scholar] [CrossRef]
- Rehan, M.; Hazra, G.; Ghorai, P. Synthesis of polysubstituted quinolines via transition-metal-free oxidative cycloisomerization of o-cinnamylanilines. Org. Lett. 2015, 17, 1668–1671. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.-H.; Zheng, H.-F.; Yuan, W.; Tang, Z.-L.; Zhang, A.-D.; Shi, D.-Q. An unexpected one-pot synthesis of multi-substituted quinolines via a cascade reaction of Michael/Staudinger/aza-Wittig/aromatization of ortho-azido-beta-styrenes with various carbonyl compounds. Tetrahedron 2013, 69, 8137–8141. [Google Scholar] [CrossRef]
- Qu, F.; He, P.; Hu, R.-F.; Cheng, X.-H.; Wang, S.; Wu, J. Efficient synthesis of quinolines via a Knoevenagel/Staudinger/aza-Wittig sequence. Synth. Commun. 2015, 45, 2802–2809. [Google Scholar] [CrossRef]
- Wang, W.-X.; Zhang, Q.-Z.; Zhang, T.-Q.; Li, Z.-S.; Zhang, W.; Yu, W. N-Bromosuccinimide-mediated radical cyclization of 3-arylallyl azides: Synthesis of 3-substituted quinolines. Adv. Synth. Catal. 2014, 357, 221–226. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, X.; Wu, Z.; Cao, J.; Ma, C.; He, Y.; Huang, G. Metal free synthesis of 2,4-diarylquinoline derivatives with enamides and imines. RSC Adv. 2015, 5, 88214–88217. [Google Scholar] [CrossRef]
- Saunthwal, R.K.; Patel, M.; Verma, A.K. Metal- and protection-free [4 + 2] cycloadditions of alkynes with azadienes: Assembly of functionalized quinolines. Org. Lett. 2016, 18, 2200–2203. [Google Scholar] [CrossRef] [PubMed]
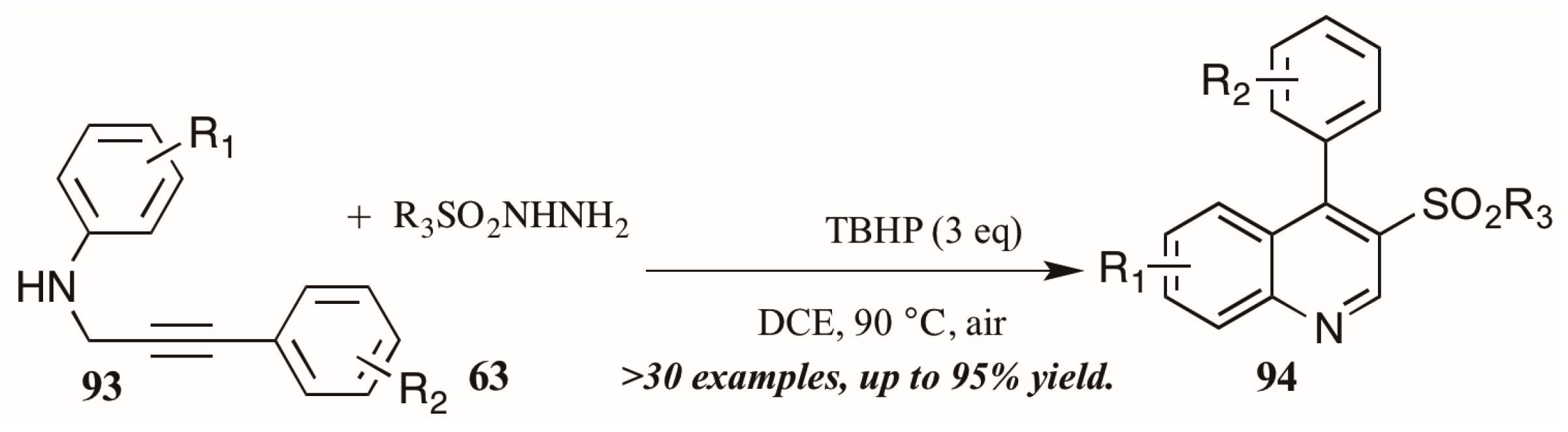
- Zhang, L.; Chen, S.; Gao, Y.; Zhang, P.; Wu, Y.; Tang, G.; Zhao, Y. tert-Butyl hydroperoxide mediated cascade synthesis of 3-arylsulfonylquinolines. Org. Lett. 2016, 18, 1286–1289. [Google Scholar] [CrossRef] [PubMed]









































© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramann, G.A.; Cowen, B.J. Recent Advances in Metal-Free Quinoline Synthesis. Molecules 2016, 21, 986. https://doi.org/10.3390/molecules21080986
Ramann GA, Cowen BJ. Recent Advances in Metal-Free Quinoline Synthesis. Molecules. 2016; 21(8):986. https://doi.org/10.3390/molecules21080986
Chicago/Turabian StyleRamann, Ginelle A., and Bryan J. Cowen. 2016. "Recent Advances in Metal-Free Quinoline Synthesis" Molecules 21, no. 8: 986. https://doi.org/10.3390/molecules21080986
APA StyleRamann, G. A., & Cowen, B. J. (2016). Recent Advances in Metal-Free Quinoline Synthesis. Molecules, 21(8), 986. https://doi.org/10.3390/molecules21080986