
Synthesis of a Potent Aminopyridine-Based nNOS-Inhibitor by Two Recent No-Carrier-Added 18F-Labelling Methods


Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of Reference Compounds and Precursors for a “Build-Up” Labelling Procedure
2.2. 18F-Radiosynthesis of the nNOS-Inhibitor 10 via the “Build-Up” Procedure
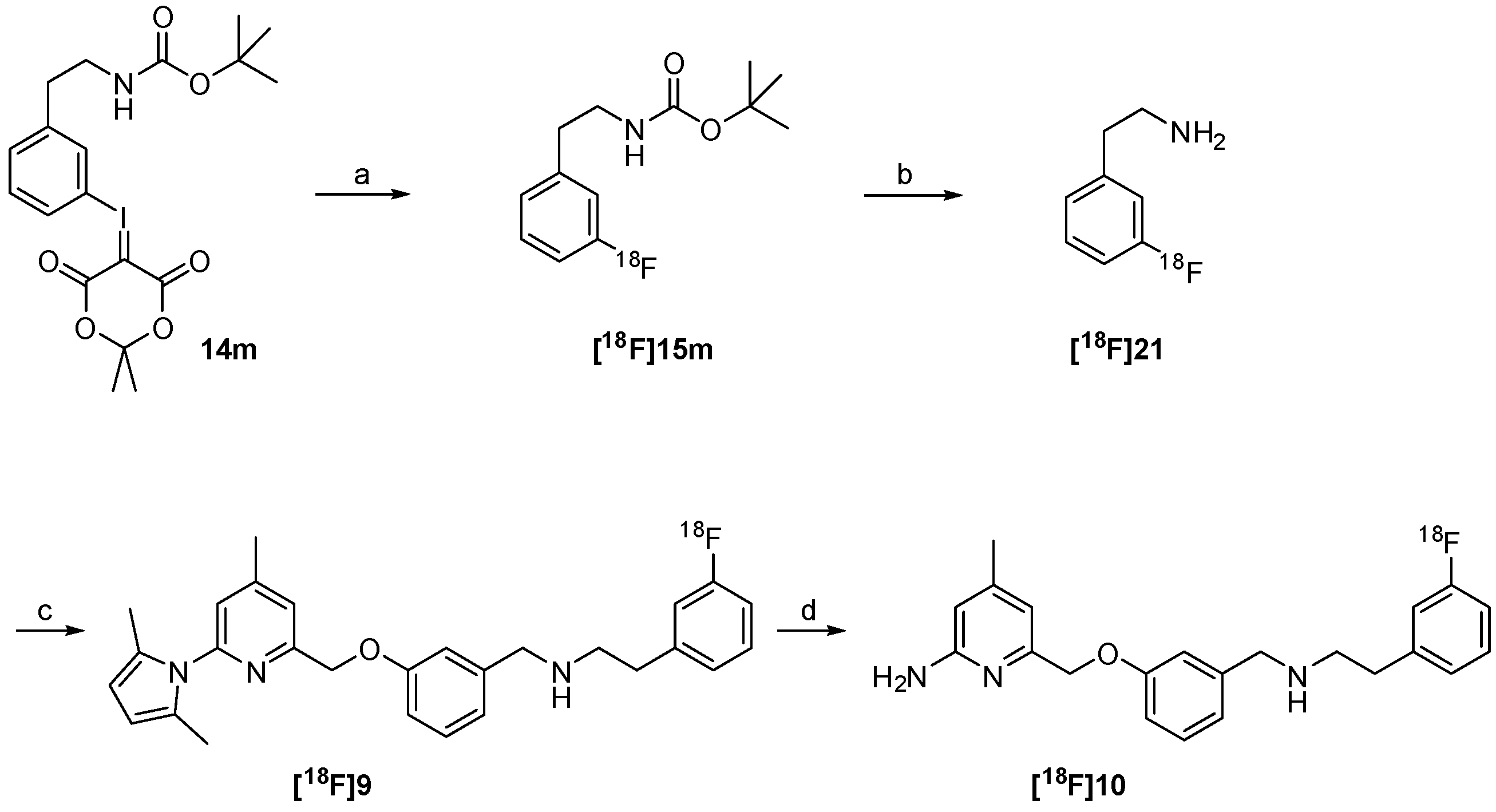
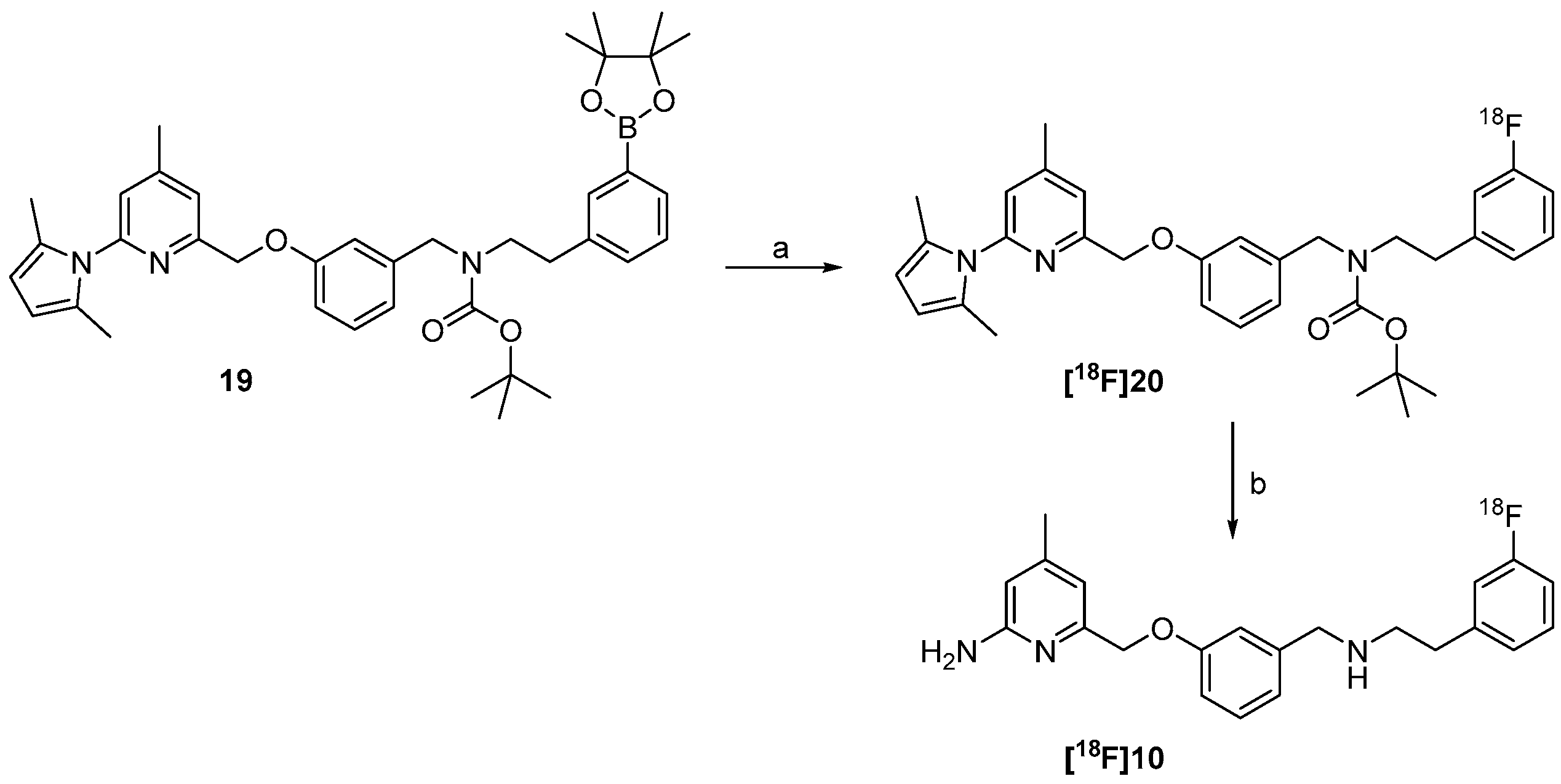
2.3. Synthesis of Reference Compound and Precursor for a “Late-Stage” 18F-Labelling Procedure
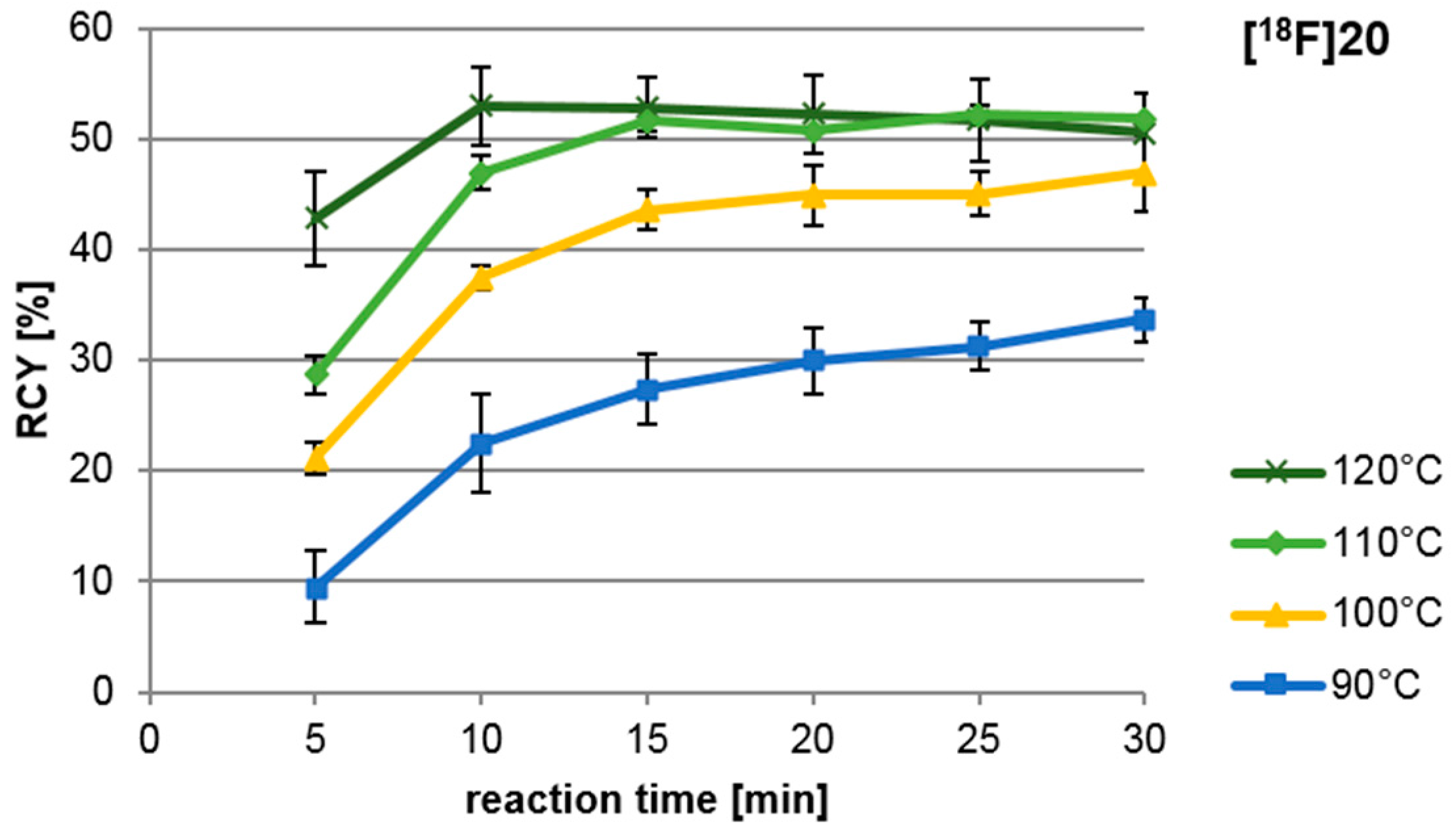
2.4. Radiosynthesis of the nNOS-Inhibitor [18F]10 by “Late-Stage” Nucleophilic 18F-Labelling
3. Materials and Methods
3.1. General Information
- a: Gemini 5 μm C18 110 Å with appropriate Gemini C18 Security Guard 250 mm × 4.6 mm (Phenomenex, Aschaffenburg, Germany)
- b: Luna 5 μm C18(2) 100 Å with appropriate Luna C18 Security Guard 250 mm × 4.6 mm (Phenomenex)
- c: Luna 5 μm PFP(2) 100 Å with appropriate Luna PFP(2) Security Guard 250 mm × 4.6 mm (Phenomenex)
3.2. Radiochemistry
3.2.1. Radiosynthesis of tert-butyl-([18F]fluorophenethyl)carbamate ([18F]15)
3.2.2. Radiosynthesis of 2-, 3- and 4-[18F]fluoroanisole
3.2.3. Radiosynthesis of [18F]10 via the “Build-up” Synthesis
3.2.4. Radiosynthesis of [18F]10 via “Late-Stage” Labelling
3.3. Precursor Syntheses
3.3.1. Synthesis of Iodonium Ylide Precursors
General procedure for the Reduction of Nitriles



General Procedure for Boc-Protection of 13






General Procedure for the Synthesis of Iodonium Ylides from Aryliodides






3.3.2. Synthesis of Reference Compounds and Iodinated Intermediates for the Preparation of the Boronic Ester Precursor





3.3.3. Synthesis of the Tetrakis(pyridine)copper(II)triflate-complex

3.3.4. Synthesis of the Boronic Ester Precursor tert-Butyl(3-((6-(2,5-dimethyl-1H-pyrrol-1-yl)-4-methylpyridin-2-yl)methoxy)benzyl)(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenylethyl)-carbamate (19)

4. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Thomas, D.D.; Ridnour, L.A.; Isenberg, J.S.; Flores-Santana, W.; Switzer, C.H.; Donzelli, S.; Hussain, P.; Vecoli, C.; Paolocci, N.; Ambs, S.; et al. The chemical biology of nitric oxide: Implications in cellular signaling. Free Radic. Biol. Med. 2008, 45, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Michel, T.; Feron, O. Nitric oxide synthases: Which, where, how, and why? J. Clin. Investig. 1997, 100, 2146–2152. [Google Scholar] [CrossRef] [PubMed]
- Titheradge, M.A. Nitric oxide in septic shock. Biochim. Biophys. Acta 1999, 1411, 437–455. [Google Scholar] [CrossRef]
- Ashutosh, K. Nitric oxide and asthma: A review. Curr. Opin. Pulm. Med. 2000, 6, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, A.J.; Higgs, A.; Moncada, S. Inhibition of nitric oxide synthase as a potential therapeutic target. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 191–220. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular pathophysiology of Parkinson’s disease. Annu. Rev. Neurosci. 2005, 28, 57–87. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Forstermann, U. Nitric oxide in the pathogenesis of vascular disease. J. Pathol. 2000, 190, 244–254. [Google Scholar] [CrossRef]
- Xu, W.; Liu, L.Z.; Loizidou, M.; Ahmed, M.; Charles, I.G. The role of nitric oxide in cancer. Cell Res. 2002, 12, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Lirk, P.; Rieder, J. Inducible nitric oxide synthase (iNOS) in tumor biology: The two sides of the same coin. Semin. Cancer Biol. 2005, 15, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Vannini, F.; Kashfi, K.; Nath, N. The dual role of iNOS in cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dawson, V.L.; Dawson, T.M. Role of nitric oxide in Parkinson’s disease. Pharmacol. Ther. 2006, 109, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.P.; Pozo-Rodrigalvarez, A.; Serrano, J.; Martinez-Murillo, R. Nitric oxide: Target for therapeutic strategies in Alzheimer’s disease. Curr. Pharm. Des. 2010, 16, 2837–2850. [Google Scholar] [CrossRef] [PubMed]
- Deckel, A.W. Nitric oxide and nitric oxide synthase in Huntington’s disease. J. Neurosci. Res. 2001, 64, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.J.; Lassmann, H. The role of nitric oxide in multiple sclerosis. Lancet Neurol. 2002, 1, 232–241. [Google Scholar] [CrossRef]
- Ashina, M. Nitric oxide synthase inhibitors for the treatment of chronic tension-type headache. Expert Opin. Pharmacother. 2002, 3, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.R.; Anderson, M.F. Mitochondrial contributions to tissue damage in stroke. Neurochem. Int. 2002, 40, 511–526. [Google Scholar] [CrossRef]
- Erdal, E.P.; Litzinger, E.A.; Seo, J.; Zhu, Y.; Ji, H.; Silverman, R.B. Selective neuronal nitric oxide synthase inhibitors. Curr. Top. Med. Chem. 2005, 5, 603–624. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yu, T.; Lian, Y.J.; Ma, R.; Yang, S.; Cho, J.Y. Nitric oxide synthase inhibitors: A review of patents from 2011 to the present. Expert Opin. Ther. Pat. 2015, 25, 49–68. [Google Scholar] [CrossRef] [PubMed]
- Maddaford, S.; Annedi, S.C.; Ramnauth, J.; Rakhit, S. Advancements in the development of nitric oxide synthase inhibitors. In Annual Reports in Medicinal Chemistry; John, E.M., Ed.; Academic Press: Foster City, CA, USA, 2009; Volume 44, pp. 27–50. [Google Scholar]
- Vallance, P.; Leiper, J. Blocking NO synthesis: How, where and why? Nat. Rev. Drug Discov. 2002, 1, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Lawton, G.R.; Ralay Ranaivo, H.; Chico, L.K.; Ji, H.; Xue, F.; Martasek, P.; Roman, L.J.; Watterson, D.M.; Silverman, R.B. Analogues of 2-aminopyridine-based selective inhibitors of neuronal nitric oxide synthase with increased bioavailability. Bioorg. Med. Chem. 2009, 17, 2371–2380. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Delker, S.L.; Li, H.; Martasek, P.; Roman, L.J.; Poulos, T.L.; Silverman, R.B. Exploration of the active site of neuronal nitric oxide synthase by the design and synthesis of pyrrolidinomethyl 2-aminopyridine derivatives. J. Med. Chem. 2010, 53, 7804–7824. [Google Scholar] [CrossRef] [PubMed]
- Xue, F.; Fang, J.; Lewis, W.W.; Martasek, P.; Roman, L.J.; Silverman, R.B. Potent and selective neuronal nitric oxide synthase inhibitors with improved cellular permeability. Bioorg. Med. Chem. Lett. 2010, 20, 554–557. [Google Scholar] [CrossRef] [PubMed]
- Jing, Q.; Li, H.; Fang, J.; Roman, L.J.; Martasek, P.; Poulos, T.L.; Silverman, R.B. In search of potent and selective inhibitors of neuronal nitric oxide synthase with more simple structures. Bioorg. Med. Chem. 2013, 21, 5323–5331. [Google Scholar] [CrossRef] [PubMed]
- Xue, F.; Gu, W.; Silverman, R.B. Concise route to the chiral pyrrolidine core of selective inhibitors of neuronal nitric oxide. Org. Lett. 2009, 11, 5194–5197. [Google Scholar] [CrossRef] [PubMed]
- Satyamurthy, N.; Barrio, J.R. No-Carrier-Added Nucleophilic [F-18]Fluorination of Aromatic Compounds. Patent PTC/US2010/001012, 17 May 2010. [Google Scholar]
- Cardinale, J.; Ermert, J.; Humpert, S.; Coenen, H.H. Iodonium ylides for one-step, no-carrier-added radiofluorination of electron rich arenes, exemplified with 4-(([18F]fluorophenoxy)phenylmethyl)-piperidine NET and SERT ligands. RSC Adv. 2014, 4, 17293. [Google Scholar] [CrossRef]
- Cardinale, J.; Ermert, J. Simplified synthesis of aryliodonium ylides by a one-pot procedure. Tetrahedron Lett. 2013, 54, 2067–2069. [Google Scholar] [CrossRef]
- Tredwell, M.; Preshlock, S.M.; Taylor, N.J.; Gruber, S.; Huiban, M.; Passchier, J.; Mercier, J.; Genicot, C.; Gouverneur, V. A general copper-mediated nucleophilic 18F-fluorination of arenes. Angew. Chem. Int. Ed. Engl. 2014, 53, 7751–7755. [Google Scholar] [CrossRef] [PubMed]
- Zlatopolskiy, B.D.; Zischler, J.; Krapf, P.; Zarrad, F.; Urusova, E.A.; Kordys, E.; Endepols, H.; Neumaier, B. Copper-mediated aromatic radiofluorination revisited: Efficient production of PET tracers on a preparative scale. Chemistry 2015, 21, 5972–5979. [Google Scholar] [CrossRef] [PubMed]
- Lam, P.Y.S.; Clark, C.G.; Saubern, S.; Adams, J.; Averill, K.M.; Chan, D.M.T.; Combs, A. Copper promoted aryl/saturated heterocyclic C-N bond cross-coupling with arylboronic acid and arylstannane. Synlett 2000, 5, 674–676. [Google Scholar] [CrossRef]
- Kügler, F.; Ermert, J.; Kaufholz, P.; Coenen, H.H. 4-[18F]Fluorophenylpiperazines by Improved Hartwig-Buchwald N-arylation of 4-[18F]fluoroiodobenzene, Formed via Hypervalent λ3-Iodane Precursors: Application to build-up of the dopamine D4 ligand [18F]FAUC 316. Molecules 2015, 20, 470–486. [Google Scholar] [CrossRef] [PubMed]
- Qaim, S.M.; Clark, J.C.; Crouzel, C.; Guillaume, M.; Helmeke, H.J.; Nebeling, B.; Pike, V.W.; Stöcklin, G. PET radionuclide production. In Radiopharmaceuticals for Positron Emission Tomography; Stöcklin, G., Pike, V.W., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1993; p. 9. [Google Scholar]
- Wyffels, L.; Muccioli, G.G.; De Bruyne, S.; Moerman, L.; Sambre, J.; Lambert, D.M.; De Vos, F. Synthesis, in vitro and in vivo evaluation, and radiolabeling of aryl anandamide analogues as candidate radioligands for in vivo imaging of fatty acid amide hydrolase in the brain. J. Med. Chem. 2009, 52, 4613–4622. [Google Scholar] [CrossRef] [PubMed]
- Landge, K.P.; Jang, K.S.; Lee, S.Y.; Chi, D.Y. Approach to the synthesis of indoline derivatives from diaryliodonium salts. J. Org. Chem. 2012, 77, 5705–5713. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.X.; Xu, Y.; Liu, D.; Costanzo, M.J. Polycyclic Compounds and Methods of Making and Using the Same. Patent PTC/US2013/073882, 19 June 2014. [Google Scholar]
- Haynes, J.S.; Rettig, S.J.; Sams, J.R.; Trotter, J.; Thompson, R.C. Pyrazine and pyridine complexes of copper(II) trifluoromethanesulfonate. Crystal-structure of Tetrakis(Pyridine)bis(Trifluoromethanesulfonato-O)Copper(II) and Magnetic Exchange in (Pyrazine)bis(Trifluoromethanesulfonato-O)Copper(II). Inorg. Chem. 1988, 27, 1237–1241. [Google Scholar] [CrossRef]
- Rotstein, B.H.; Stephenson, N.A.; Vasdev, N.; Liang, S.H. Spirocyclic hypervalent iodine(III)-mediated radiofluorination of non-activated and hindered aromatics. Nat. Commun. 2014, 5, 4365. [Google Scholar] [CrossRef] [PubMed]
- Coenen, H.H.; Ermert, J. Direct nucleophilic 18F-fluorination of electron rich arenes: Present limits of no-carrier-added reactions. Curr. Radiopharm. 2010, 3, 163–173. [Google Scholar] [CrossRef]
- Rotstein, B.H.; Wang, L.; Liu, R.Y.; Patteson, J.; Kwan, E.E.; Vasdev, N.; Liang, S.H. Mechanistic studies and radiofluorination of structurally diverse pharmaceuticals with spirocyclic iodonium(III) ylides. Chem. Sci. 2016, 7, 4407–4417. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HPLC Condition | Column | Eluent | Wavelength | Flow | Injection-Volume |
|---|---|---|---|---|---|
| 1 | c | 40 MeCN:60 H2O | 254 nm | 1 mL/min | 20 µL |
| 2 | b | 60 MeCN:40 H2O | 261 nm | 1 mL/min | 20 µL |
| 3 | c | a A: H2O + 0.1% TEA, B: MeCN + 0.1% TEA | 261 nm | 1 mL/min | 20 µL |
| 4 | a | 70 MeCN:30 H2O + 0.1% TEA | 261 nm | 1 mL/min | 20 µL |
| 5 | a | 60 ACN:40 H2O + 0.1% TEA | 261 nm | 1 mL/min | 20 µL |
| 6 | a | b A: H2O + 0.1% TEA B: MeCN + 0.1% TEA | 261 nm | 1 mL/min | 20 µL |
| 7 | a | c A: H2O + 0.1% TEA B: MeCN + 0.1% TEA | 261 nm | 1 mL/min | 20 µL |
| Compound | HPLC-System | Condition | k-Values |
|---|---|---|---|
| Fluoroanisole | Knauer | 1 | 6.7 (anisole), 7.3 (2-fluoroanisole), 9.3 (3-fluoroanisole), 8.2 (4-fluoroanisole) |
| 15 | Knauer | 2 | 3.2 |
| 21o | Dionex | 3 | 6.0 |
| 21m | Dionex | 3 | 7.0 |
| 21p | Dionex | 3 | 8.0 |
| 9 | Knauer | 4 | 6.7 |
| 9 | Knauer | 5 | 19.4 |
| 10 | Knauer | 5 | 2.9 |
| 9 | Dionex | 6 | 7.3 |
| 10 | Dionex | 6 | 2.7 |
| 9 | Dionex | 7 | 2.7 |
| 10 | Dionex | 7 | 7.2 |
| 20 | Dionex | 7 | 14.0 |
| Compound | Eluent | Rf |
|---|---|---|
| 9 | 9 EE:1 PE + 1% TEA | 0.8 |
| 10 | 9 EE:1 PE + 1% TEA | 0.5 |
| 15 | 1 EE:4 PE | 0.5 |
| 20 | 1 EE:4 PE | 0.7 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drerup, C.; Ermert, J.; Coenen, H.H. Synthesis of a Potent Aminopyridine-Based nNOS-Inhibitor by Two Recent No-Carrier-Added 18F-Labelling Methods. Molecules 2016, 21, 1160. https://doi.org/10.3390/molecules21091160
Drerup C, Ermert J, Coenen HH. Synthesis of a Potent Aminopyridine-Based nNOS-Inhibitor by Two Recent No-Carrier-Added 18F-Labelling Methods. Molecules. 2016; 21(9):1160. https://doi.org/10.3390/molecules21091160
Chicago/Turabian StyleDrerup, Christian, Johannes Ermert, and Heinz H. Coenen. 2016. "Synthesis of a Potent Aminopyridine-Based nNOS-Inhibitor by Two Recent No-Carrier-Added 18F-Labelling Methods" Molecules 21, no. 9: 1160. https://doi.org/10.3390/molecules21091160
APA StyleDrerup, C., Ermert, J., & Coenen, H. H. (2016). Synthesis of a Potent Aminopyridine-Based nNOS-Inhibitor by Two Recent No-Carrier-Added 18F-Labelling Methods. Molecules, 21(9), 1160. https://doi.org/10.3390/molecules21091160