Design and Experimental Evolution of trans-Splicing Group I Intron Ribozymes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Group I Intron Ribozymes
3. trans-Splicing with Group I Intron Ribozymes
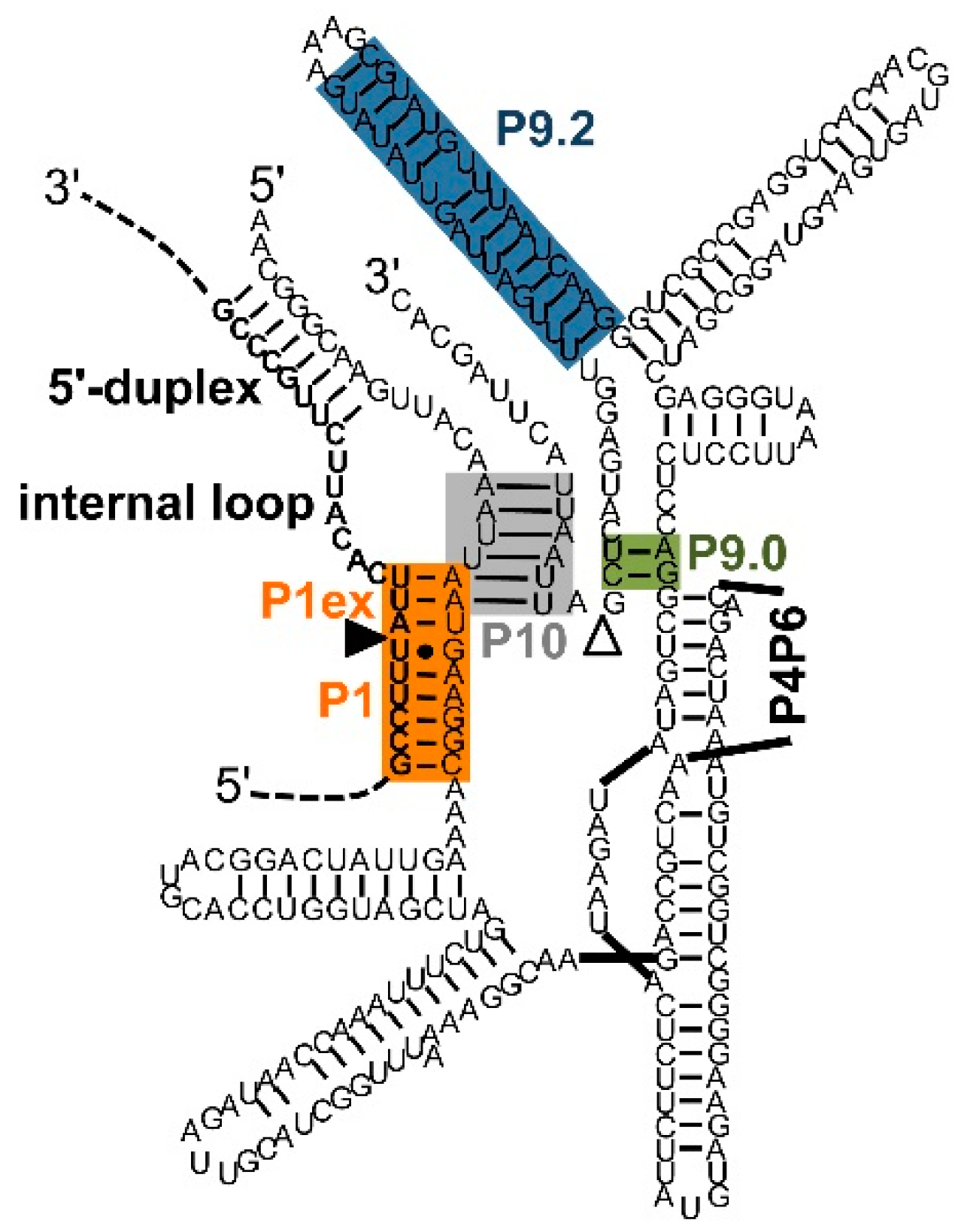
4. Identification of Efficient Splice Sites on Target RNAs
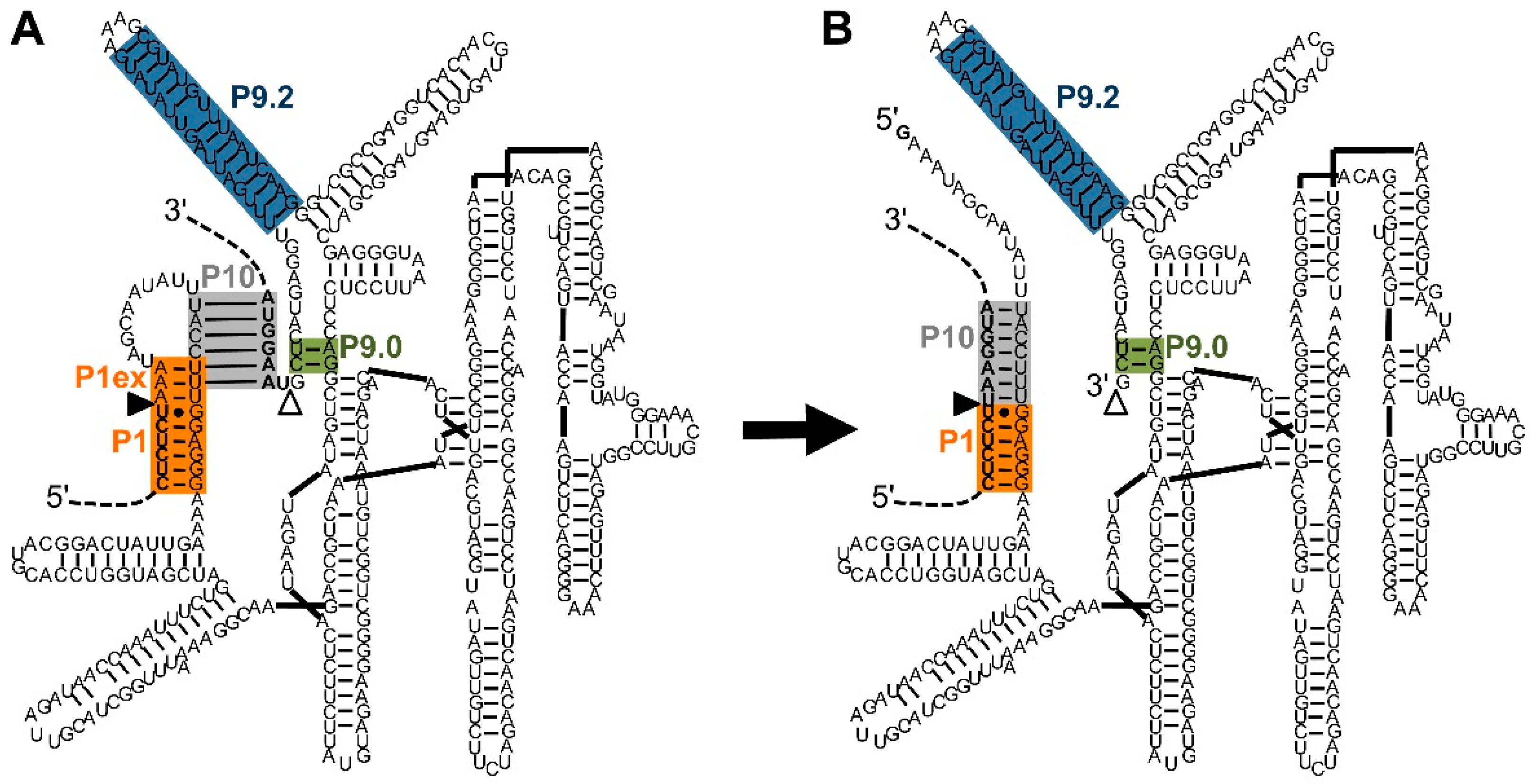
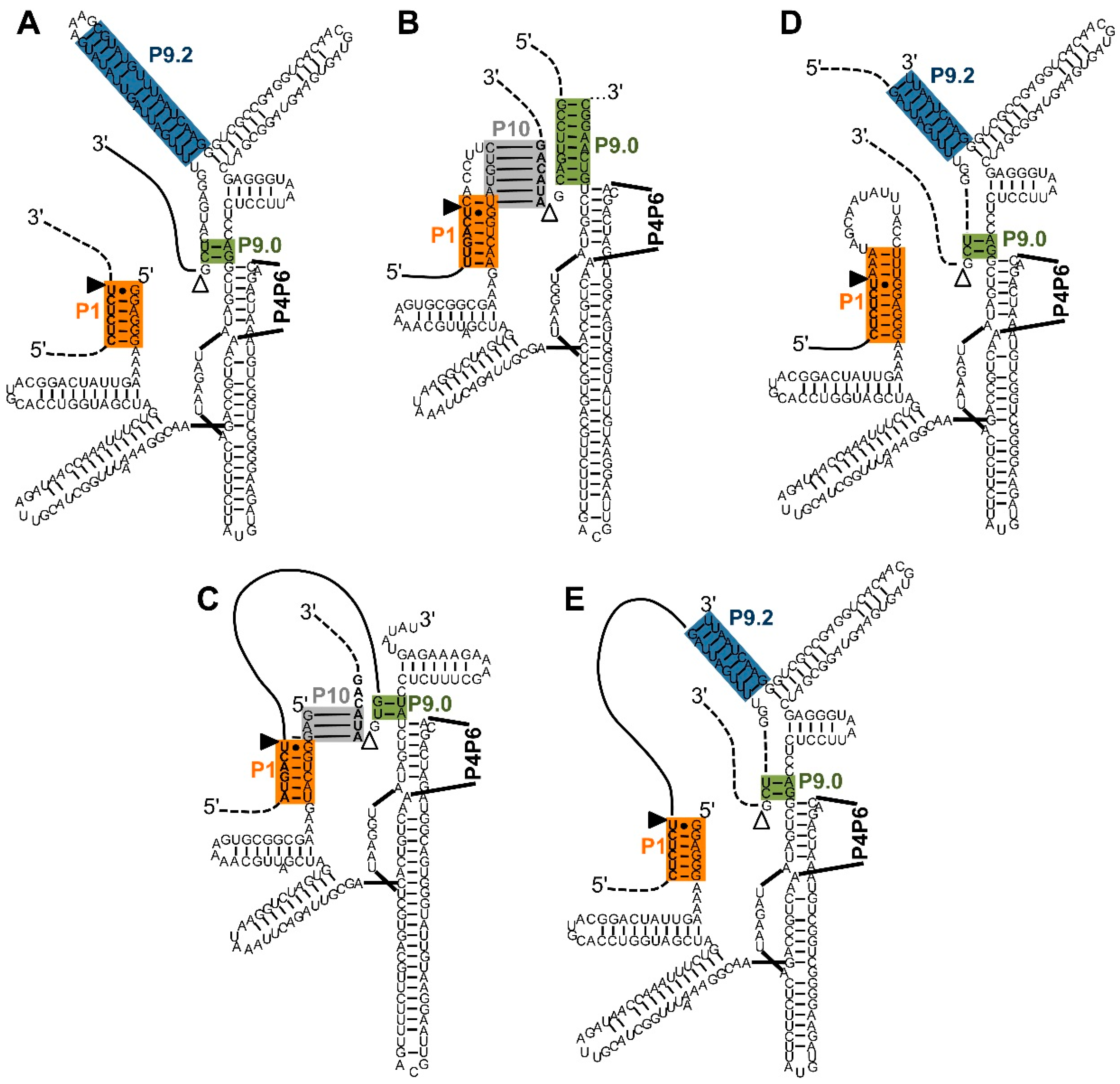
5. Identification of Efficient Extended Guide Sequences on the Ribozyme 5′-Termini
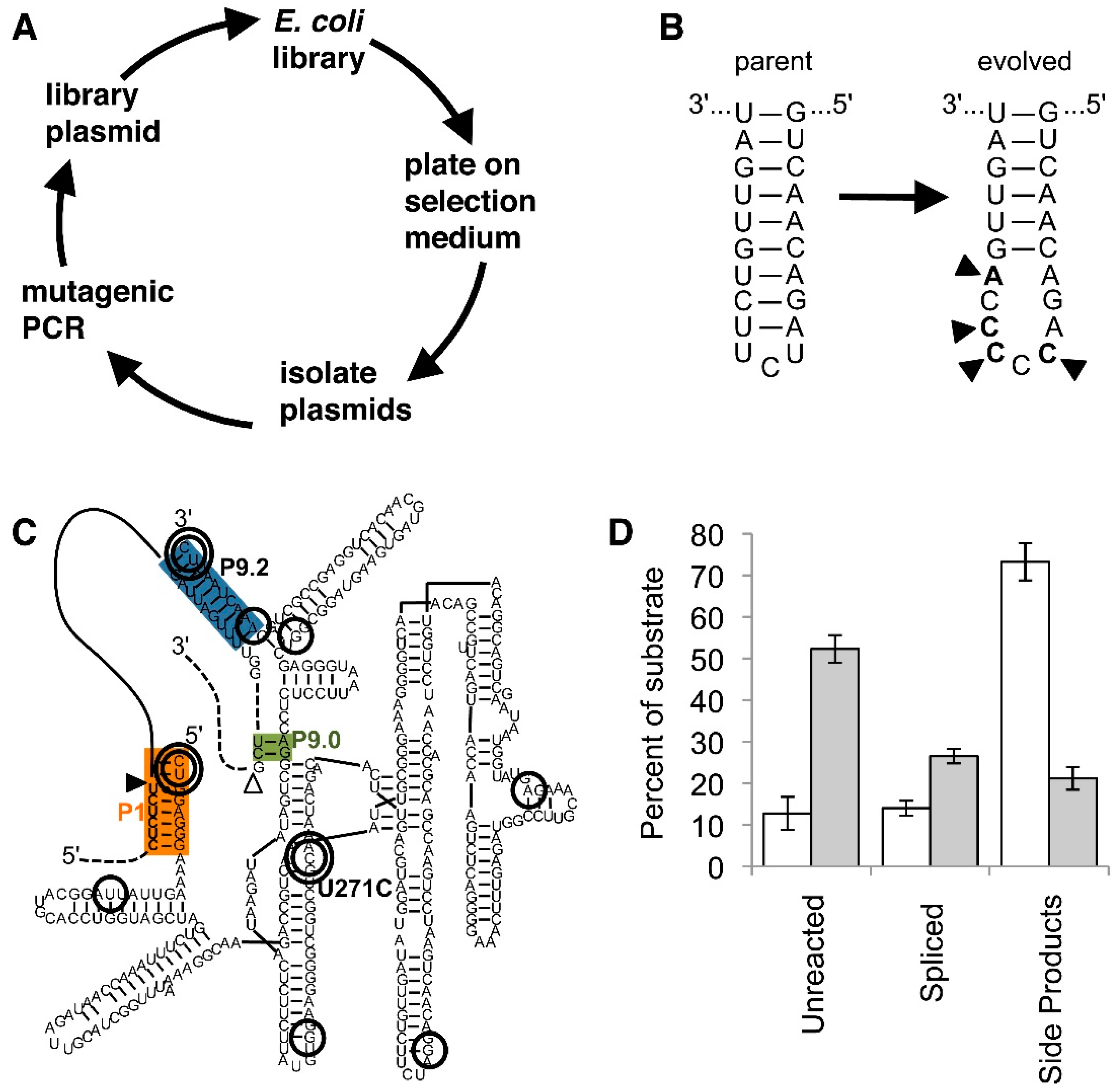
6. Selection Systems for Improved trans-Splicing Group I Intron Ribozymes
7. Evolution of Improved trans-Splicing Group I Intron Ribozymes
8. Possible Applications of Evolved trans-Splicing Ribozymes
Conflicts of Interest
References
- Kruger, K.; Grabowski, P.J.; Zaug, A.J.; Sands, J.; Gottschling, D.E.; Cech, T.R. Self-splicing RNA: Autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 1982, 31, 147–157. [Google Scholar] [CrossRef]
- Guerrier-Takada, C.; Gardiner, K.; Marsh, T.; Pace, N.; Altman, S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983, 35, 849–857. [Google Scholar] [CrossRef]
- Pombert, J.F.; Otis, C.; Turmel, M.; Lemieux, C. The mitochondrial genome of the prasinophyte Prasinoderma coloniale reveals two trans-spliced group I introns in the large subunit rRNA gene. PLoS ONE 2013, 8, e84325. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Sullivan, F.X.; Cech, T.R. Intermolecular exon ligation of the rRNA precursor of Tetrahymena: Oligonucleotides can function as 5′ exons. Cell 1985, 43, 431–437. [Google Scholar] [CrossRef]
- Sullenger, B.A.; Cech, T.R. Ribozyme-mediated repair of defective mRNA by targeted, trans-splicing. Nature 1994, 371, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Alexander, R.C.; Baum, D.A.; Testa, S.M. 5′ Transcript replacement in vitro catalyzed by a group I intron-derived ribozyme. Biochemistry 2005, 44, 7796–7804. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.A.; Johnson, A.K.; Testa, S.M. Ribozyme-catalyzed excision of targeted sequences from within RNAs. Biochemistry 2002, 41, 15327–15333. [Google Scholar] [CrossRef] [PubMed]
- Amini, Z.N.; Olson, K.E.; Muller, U.F. Spliceozymes: Ribozymes that remove introns from pre-mRNAs in trans. PLoS ONE 2014, 9, e101932. [Google Scholar] [CrossRef] [PubMed]
- Cate, J.H.; Gooding, A.R.; Podell, E.; Zhou, K.; Golden, B.L.; Szewczak, A.A.; Kundrot, C.E.; Cech, T.R.; Doudna, J.A. RNA tertiary structure mediation by adenosine platforms. Science 1996, 273, 1696–1699. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.L.; Stahley, M.R.; Kosek, A.B.; Wang, J.; Strobel, S.A. Crystal structure of a self-splicing group I intron with both exons. Nature 2004, 430, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Gooding, A.R.; Cech, T.R. Structure of the Tetrahymena ribozyme: Base triple sandwich and metal ion at the active site. Mol. Cell 2004, 16, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Golden, B.L.; Kim, H.; Chase, E. Crystal structure of a phage Twort group I ribozyme-product complex. Nat. Struct. Mol. Biol. 2005, 12, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Lipchock, S.V.; Strobel, S.A. A relaxed active site after exon ligation by the group I intron. Proc. Natl. Acad. Sci. USA 2008, 105, 5699–5704. [Google Scholar] [CrossRef] [PubMed]
- Narlikar, G.J.; Herschlag, D. Isolation of a local tertiary folding transition in the context of a globally folded RNA. Nat. Struct. Biol. 1996, 3, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Woodson, S.A. Folding mechanisms of group I ribozymes: Role of stability and contact order. Biochem. Soc. Trans. 2002, 30, 1166–1169. [Google Scholar] [CrossRef] [PubMed]
- Bhaskaran, H.; Russell, R. Kinetic redistribution of native and misfolded RNAs by a DEAD-box chaperone. Nature 2007, 449, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Solomatin, S.V.; Greenfeld, M.; Chu, S.; Herschlag, D. Multiple native states reveal persistent ruggedness of an RNA folding landscape. Nature 2010, 463, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Behrouzi, R.; Roh, J.H.; Kilburn, D.; Briber, R.M.; Woodson, S.A. Cooperative tertiary interaction network guides RNA folding. Cell 2012, 149, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Cech, T.R.; Herschlag, D.; Piccirilli, J.A.; Pyle, A.M. RNA catalysis by a group I ribozyme. Developing a model for transition state stabilization. J. Biol. Chem. 1992, 267, 17479–17482. [Google Scholar] [PubMed]
- Hougland, J.L.; Sengupta, R.N.; Dai, Q.; Deb, S.K.; Piccirilli, J.A. The 2′-hydroxyl group of the guanosine nucleophile donates a functionally important hydrogen bond in the tetrahymena ribozyme reaction. Biochemistry 2008, 47, 7684–7694. [Google Scholar] [CrossRef] [PubMed]
- Forconi, M.; Lee, J.; Lee, J.K.; Piccirilli, J.A.; Herschlag, D. Functional identification of ligands for a catalytic metal ion in group I introns. Biochemistry 2008, 47, 6883–6894. [Google Scholar] [CrossRef] [PubMed]
- Stahley, M.R.; Strobel, S.A. RNA splicing: Group I intron crystal structures reveal the basis of splice site selection and metal ion catalysis. Curr. Opin. Struct. Biol. 2006, 16, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Cech, T.R.; Zaug, A.J.; Grabowski, P.J. In vitro splicing of the ribosomal RNA precursor of Tetrahymena: Involvement of a guanosine nucleotide in the excision of the intervening sequence. Cell 1981, 27, 487–496. [Google Scholar] [CrossRef]
- Cech, T.R.; Damberger, S.H.; Gutell, R.R. Representation of the secondary and tertiary structure of group I introns. Nat. Struct. Biol. 1994, 1, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Fiskaa, T.; Lundblad, E.W.; Henriksen, J.R.; Johansen, S.D.; Einvik, C. RNA reprogramming of α-mannosidase mRNA sequences in vitro by myxomycete group IC1 and IE ribozymes. FEBS J. 2006, 273, 2789–2800. [Google Scholar] [CrossRef] [PubMed]
- Vicens, Q.; Paukstelis, P.J.; Westhof, E.; Lambowitz, A.M.; Cech, T.R. Toward predicting self-splicing and protein-facilitated splicing of group I introns. RNA 2008, 14, 2013–2029. [Google Scholar] [CrossRef] [PubMed]
- Dotson, P.P., 2nd; Sinha, J.; Testa, S.M. A Pneumocystis carinii group I intron-derived ribozyme utilizes an endogenous guanosine as the first reaction step nucleophile in the trans excision-splicing reaction. Biochemistry 2008, 47, 4780–4787. [Google Scholar] [CrossRef] [PubMed]
- Dotson, P.P., 2nd; Johnson, A.K.; Testa, S.M. Tetrahymena thermophila and Candida albicans group I intron-derived ribozymes can catalyze the trans-excision-splicing reaction. Nucleic Acids Res. 2008, 36, 5281–5289. [Google Scholar] [CrossRef] [PubMed]
- Dolan, G.F.; Muller, U.F. trans-Splicing with the group I intron ribozyme from Azoarcus. RNA 2014, 20, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Baum, D.A.; Testa, S.M. In vivo excision of a single targeted nucleotide from an mRNA by a trans excision-splicing ribozyme. RNA 2005, 11, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Been, M.D.; Cech, T.R. One binding site determines sequence specificity of Tetrahymena pre-rRNA self-splicing, trans-splicing, and RNA enzyme activity. Cell 1986, 47, 207–216. [Google Scholar] [CrossRef]
- Waring, R.B.; Towner, P.; Minter, S.J.; Davies, R.W. Splice-site selection by a self-splicing RNA of Tetrahymena. Nature 1986, 321, 133–139. [Google Scholar] [CrossRef]
- Shi, X.; Mollova, E.T.; Pljevaljcic, G.; Millar, D.P.; Herschlag, D. Probing the dynamics of the P1 helix within the Tetrahymena group I intron. J. Am. Chem. Soc. 2009, 131, 9571–9578. [Google Scholar] [CrossRef] [PubMed]
- Fiskaa, T.; Birgisdottir, A.B. RNA reprogramming and repair based on trans-splicing group I ribozymes. New Biotechnol. 2010, 27, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Testa, S.M.; Haidaris, C.G.; Gigliotti, F.; Turner, D.H. A Pneumocystis carinii group I intron ribozyme that does not require 2′ OH groups on its 5′ exon mimic for binding to the catalytic core. Biochemistry 1997, 36, 15303–15314. [Google Scholar] [CrossRef] [PubMed]
- Dotson, P.P.; Testa, S.M. Group I intron-derived ribozyme recombination reactions. Recent Dev. Nucleic Acids Res. 2006, 2, 307–324. [Google Scholar]
- Been, M.D.; Cech, T.R. Sites of circularization of the Tetrahymena rRNA IVS are determined by sequence and influenced by position and secondary structure. Nucleic Acids Res. 1985, 13, 8389–8408. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.; Herschlag, D. Specificity from steric restrictions in the guanosine binding pocket of a group I ribozyme. RNA 1999, 5, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.A.; Sinha, J.; Johnson, A.K.; Testa, S.M. Enhancing the second step of the trans excision-splicing reaction of a group I ribozyme by exploiting P9.0 and P10 for intermolecular recognition. Biochemistry 2004, 43, 4323–4331. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.K.; Sinha, J.; Testa, S.M. Trans insertion-splicing: Ribozyme-catalyzed insertion of targeted sequences into RNAs. Biochemistry 2005, 44, 10702–10710. [Google Scholar] [CrossRef] [PubMed]
- Dotson, P.P., 2nd; Frommeyer, K.N.; Testa, S.M. Ribozyme mediated trans insertion-splicing of modified oligonucleotides into RNA. Arch. Biochem. Biophys. 2008, 478, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.; Lan, N.; Long, M.; Sullenger, B.A. Efficient and specific repair of sickle beta-globin RNA by trans-splicing ribozymes. RNA 2003, 9, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.T.; Sullenger, B.A. Evaluating and enhancing ribozyme reaction efficiency in mammalian cells. Nat. Biotechnol. 1997, 15, 902–905. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Cormack, B.P.; Szostak, J.W. RNA structure, not sequence, determines the 5′ splice-site specificity of a group I intron. Proc. Natl. Acad. Sci. USA 1989, 86, 7402–7406. [Google Scholar] [CrossRef] [PubMed]
- Strobel, S.A.; Cech, T.R. Minor groove recognition of the conserved G.U pair at the Tetrahymena ribozyme reaction site. Science 1995, 267, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Campbell, T.B.; Cech, T.R. Identification of ribozymes within a ribozyme library that efficiently cleave a long substrate RNA. RNA 1995, 1, 598–609. [Google Scholar] [PubMed]
- Jones, J.T.; Lee, S.W.; Sullenger, B.A. Tagging ribozyme reaction sites to follow trans-splicing in mammalian cells. Nat. Med. 1996, 2, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Lan, N.; Howrey, R.P.; Lee, S.W.; Smith, C.A.; Sullenger, B.A. Ribozyme-mediated repair of sickle β-globin mRNAs in erythrocyte precursors. Science 1998, 280, 1593–1596. [Google Scholar] [CrossRef] [PubMed]
- Ryu, K.J.; Kim, J.H.; Lee, S.W. Ribozyme-mediated selective induction of new gene activity in hepatitis C virus internal ribosome entry site-expressing cells by targeted trans-splicing. Mol. Ther. 2003, 7, 386–395. [Google Scholar] [CrossRef]
- Olson, K.E.; Muller, U.F. An in vivo selection method to optimize trans-splicing ribozymes. RNA 2012, 18, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Meluzzi, D.; Olson, K.E.; Dolan, G.F.; Arya, G.; Muller, U.F. Computational prediction of efficient splice sites for trans-splicing ribozymes. RNA 2012, 18, 590–602. [Google Scholar] [CrossRef] [PubMed]
- Milligan, J.F.; Groebe, D.R.; Witherell, G.W.; Uhlenbeck, O.C. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987, 15, 8783–8798. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 2000, 25, 169–193. [Google Scholar] [CrossRef] [PubMed]
- Kohler, U.; Ayre, B.G.; Goodman, H.M.; Haseloff, J. trans-Splicing ribozymes for targeted gene delivery. J. Mol. Biol. 1999, 285, 1935–1950. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Cech, T.R. In vivo selection of better self-splicing introns in Escherichia coli: The role of the P1 extension helix of the Tetrahymena intron. RNA 2002, 8, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Porschke, D. Elementary steps of base recognition and helix-coil transitions in nucleic acids. Mol. Biol. Biochem. Biophys. 1977, 24, 191–218. [Google Scholar] [PubMed]
- Garcia, M.A.; Meurs, E.F.; Esteban, M. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Suh, E.R.; Waring, R.B. Base pairing between the 3′ exon and an internal guide sequence increases 3′ splice site specificity in the Tetrahymena self-splicing rRNA intron. Mol. Cell. Biol. 1990, 10, 2960–2965. [Google Scholar] [CrossRef] [PubMed]
- Amini, Z.N.; Muller, U.F. Low selection pressure aids the evolution of cooperative ribozyme mutations in cells. J. Biol. Chem. 2013, 288, 33096–33106. [Google Scholar] [CrossRef] [PubMed]
- Ayre, B.G.; Kohler, U.; Turgeon, R.; Haseloff, J. Optimization of trans-splicing ribozyme efficiency and specificity by in vivo genetic selection. Nucleic Acids Res. 2002, 30, e141. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, S.; Choi, J.W.; Rao, J. Single-cell detection of trans-splicing ribozyme in vivo activity. J. Am. Chem. Soc. 2004, 126, 7158–7159. [Google Scholar] [CrossRef] [PubMed]
- Beaudry, A.A.; Joyce, G.F. Directed evolution of an RNA enzyme. Science 1992, 257, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Treiber, D.K.; Rook, M.S.; Zarrinkar, P.P.; Williamson, J.R. Kinetic intermediates trapped by native interactions in RNA folding. Science 1998, 279, 1943–1946. [Google Scholar] [CrossRef] [PubMed]
- Ohuchi, S.J.; Ikawa, Y.; Shiraishi, H.; Inoue, T. Artificial modules for enhancing rate constants of a Group I intron ribozyme without a P4-P6 core element. J. Biol. Chem. 2004, 279, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Hayden, E.J.; Ferrada, E.; Wagner, A. Cryptic genetic variation promotes rapid evolutionary adaptation in an RNA enzyme. Nature 2011, 474, 92–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, K.E.; Dolan, G.F.; Muller, U.F. In vivo evolution of a catalytic RNA couples trans-splicing to translation. PLoS ONE 2014, 9, e86473. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Giver, L.; Shao, Z.; Affholter, J.A.; Arnold, F.H. Molecular evolution by staggered extension process (StEP) in vitro recombination. Nat. Biotechnol. 1998, 16, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Amini, Z.N.; Muller, U.F. Increased efficiency of evolved group I intron spliceozymes by decreased side product formation. RNA 2015, 21, 1480–1489. [Google Scholar] [CrossRef] [PubMed]
- Lehman, N.; Unrau, P.J. Recombination during in vitro evolution. J. Mol. Evol. 2005, 61, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Phylactou, L.A.; Darrah, C.; Wood, M.J. Ribozyme-mediated trans-splicing of a trinucleotide repeat. Nat. Genet. 1998, 18, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Ayre, B.G.; Kohler, U.; Goodman, H.M.; Haseloff, J. Design of highly specific cytotoxins by using trans-splicing ribozymes. Proc. Natl. Acad. Sci. USA 1999, 96, 3507–3512. [Google Scholar] [CrossRef] [PubMed]
- Kwon, B.S.; Jung, H.S.; Song, M.S.; Cho, K.S.; Kim, S.C.; Kimm, K.; Jeong, J.S.; Kim, I.H.; Lee, S.W. Specific regression of human cancer cells by ribozyme-mediated targeted replacement of tumor-specific transcript. Mol. Ther. 2005, 12, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Kim, K.T.; Lee, S.J.; Hong, S.H.; Moon, J.Y.; Yoon, E.K.; Kim, S.; Kim, E.O.; Kang, S.H.; Kim, S.K.; et al. Image-aided Suicide Gene Therapy Utilizing Multifunctional hTERT-targeting Adenovirus for Clinical Translation in Hepatocellular Carcinoma. Theranostics 2016, 6, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.R.; Keith, J.H.; Barde, P.V.; Fraser, T.S.; Fraser, M.J., Jr. Targeting of highly conserved Dengue virus sequences with anti-Dengue virus trans-splicing group I introns. BMC Mol. Biol. 2010, 11, 84. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Coban, O.; Snead, N.M.; Trebley, J.; Hoeprich, S.; Guo, S.; Sun, Y. Engineering RNA for targeted siRNA delivery and medical application. Adv. Drug Deliv. Rev. 2010, 62, 650–666. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Liu, X.L.; Li, X.R. Research progress on siRNA delivery with nonviral carriers. Int. J. Nanomed. 2011, 6, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Gong, H.; Li, H.; Vu, G.P.; Lu, S.; Liu, F. Oral delivery of RNase P ribozymes by Salmonella inhibits viral infection in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 3222–3227. [Google Scholar] [CrossRef] [PubMed]
- Fica, S.M.; Tuttle, N.; Novak, T.; Li, N.S.; Lu, J.; Koodathingal, P.; Dai, Q.; Staley, J.P.; Piccirilli, J.A. RNA catalyses nuclear pre-mRNA splicing. Nature 2013, 503, 229–234. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, U.F. Design and Experimental Evolution of trans-Splicing Group I Intron Ribozymes. Molecules 2017, 22, 75. https://doi.org/10.3390/molecules22010075
Müller UF. Design and Experimental Evolution of trans-Splicing Group I Intron Ribozymes. Molecules. 2017; 22(1):75. https://doi.org/10.3390/molecules22010075
Chicago/Turabian StyleMüller, Ulrich F. 2017. "Design and Experimental Evolution of trans-Splicing Group I Intron Ribozymes" Molecules 22, no. 1: 75. https://doi.org/10.3390/molecules22010075
APA StyleMüller, U. F. (2017). Design and Experimental Evolution of trans-Splicing Group I Intron Ribozymes. Molecules, 22(1), 75. https://doi.org/10.3390/molecules22010075