Synthesis of Disaccharide Nucleosides Utilizing the Temporary Protection of the 2′,3′-cis-Diol of Ribonucleosides by a Boronic Ester



Abstract
:1. Introduction
2. Results and Discussion
2.1. O-Glycosylation of Nucleosides with Thioglycosyl Donors
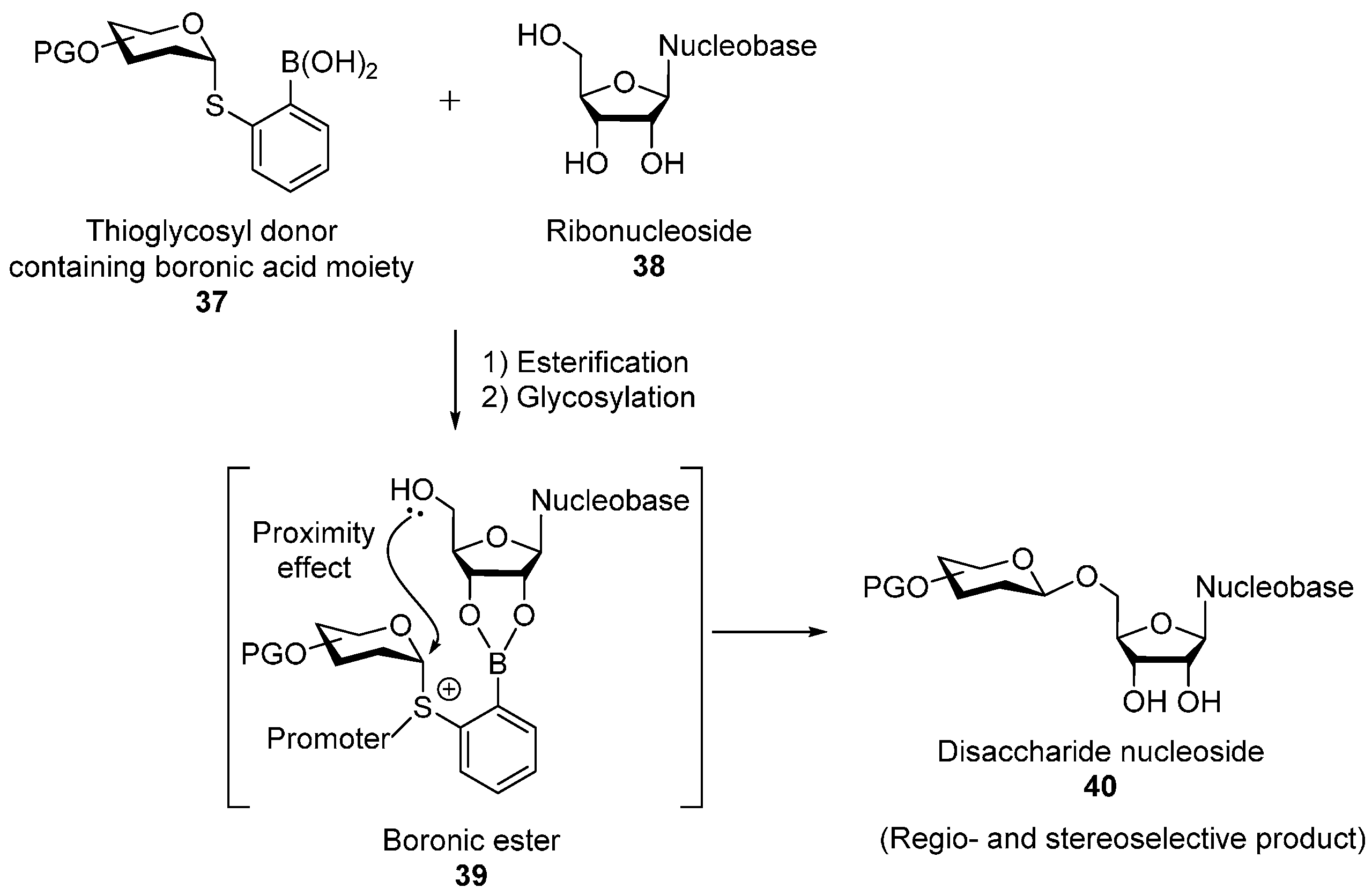
2.2. O-Glycosylation of Nucleosides with Thioglycosyl Donors Containing the Boronic Acid Moiety on the Leaving Group
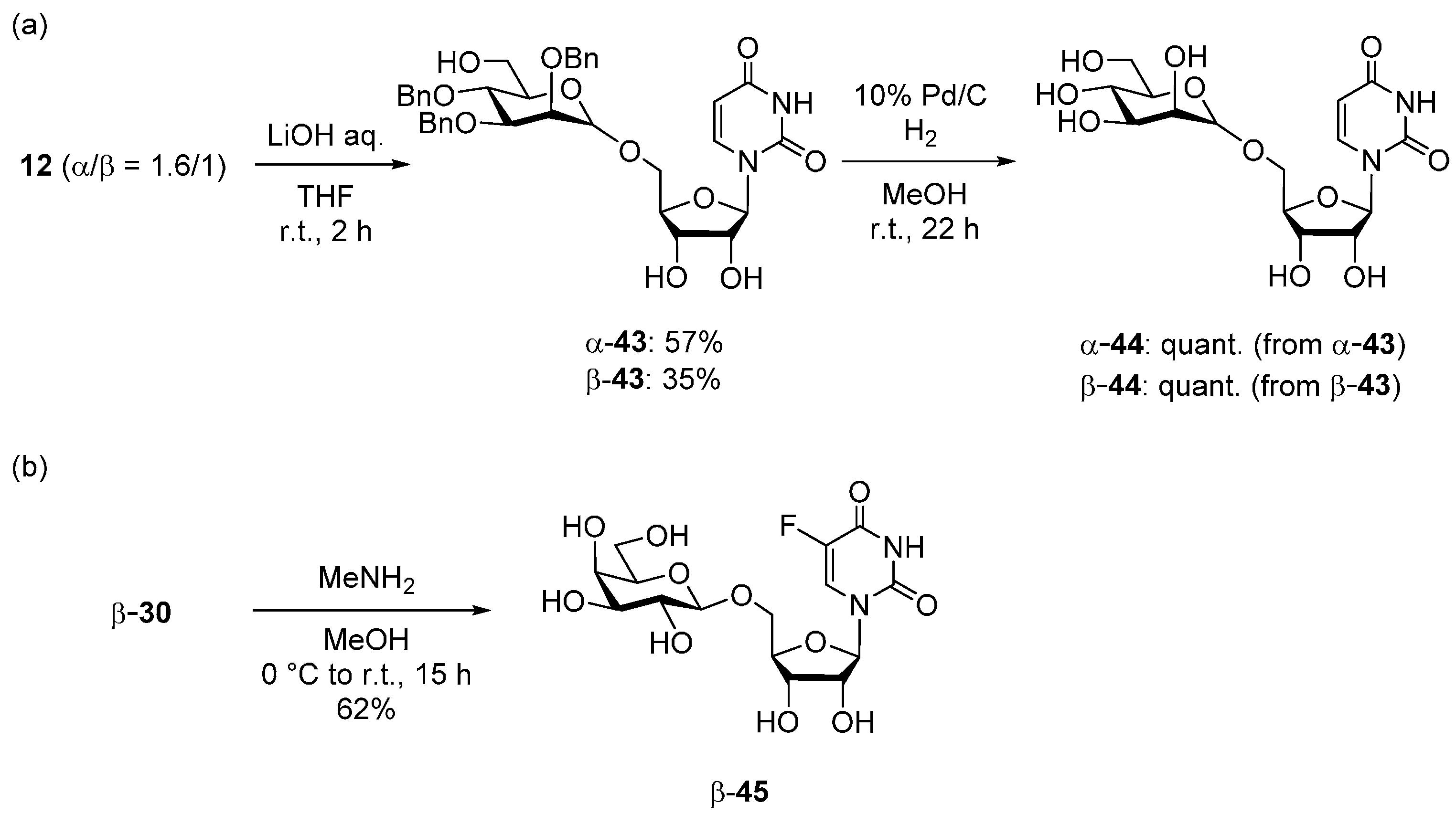
2.3. Deprotection of the Glycosylation Products
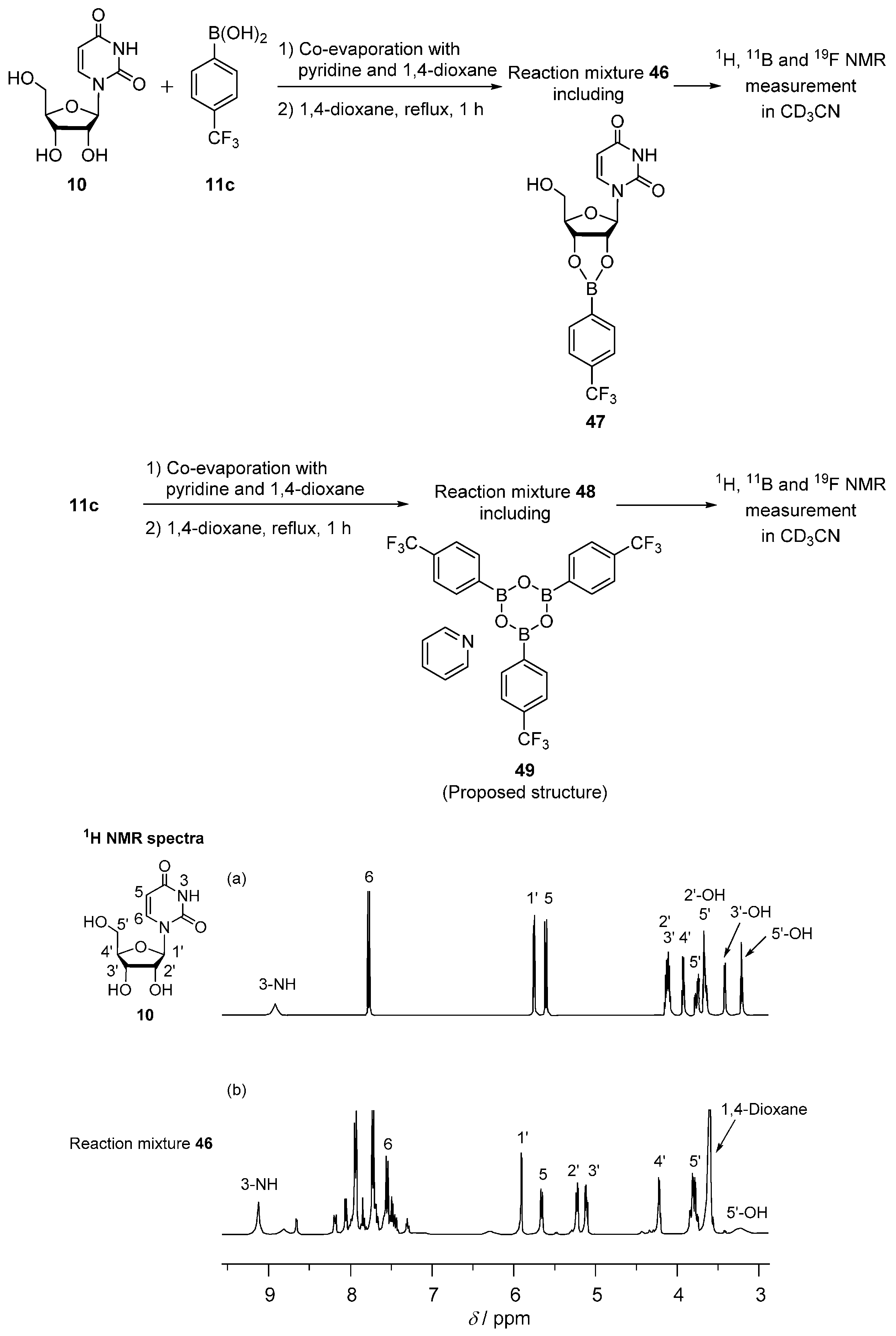
2.4. Interaction of Uridine and 4-(Trifluoromethyl)phenylboronic Acid Studied by 1H, 11B and 19F NMR Spectroscopy
3. Materials and Methods
3.1. General Information
3.2. Synthesis of Compounds
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kobayashi, J.; Doi, Y.; Ishibashi, M. Shimofuridin A, a nucleoside derivative embracing an acylfucopyranoside unit isolated from the okinawan marine tunicate Aplidium multiplicatum. J. Org. Chem. 1994, 59, 255–257. [Google Scholar] [CrossRef]
- Takahashi, M.; Tanzawa, K.; Takahashi, S. Adenophostins, newly discovered metabolites of penicillium brevicompactum, act as potent agonists of the inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 1994, 269, 369–372. [Google Scholar] [PubMed]
- Haneda, K.; Shinose, M.; Seino, A.; Tabata, N.; Tomoda, H.; Iwai, Y.; Omura, S. Cytosaminomycins, new anticoccidial agents produced by Strevtomvces sp. KO-8119 I. taxonomy, production, isolation and physico-chemical and biological properties. J. Antibiot. 1994, 47, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, K.; Haneda, K.; Tomoda, H.; Iwai, Y.; Omura, S. Cytosaminomycins, new anticoccidial agents produced by Streptomyces sp. KO-8119 II. structure elucidation of cytosaminomycins A, B, C and D. J. Antibiot. 1994, 47, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Knapp, S. Synthesis of complex nucleoside antibiotics. Chem. Rev. 1995, 95, 1859–1876. [Google Scholar] [CrossRef]
- Efimtseva, E.V.; Kulikova, I.V.; Mikhailov, S.N. Disaccharide nucleosides as an important group of natural compounds. Mol. Biol. 2009, 43, 301–312. [Google Scholar] [CrossRef]
- Huang, R.M.; Chen, Y.N.; Zeng, Z.; Gao, C.H.; Su, X.; Peng, Y. Marine nucleosides: Structure, bioactivity, synthesis and biosynthesis. Mar. Drugs 2014, 12, 5817–5838. [Google Scholar] [CrossRef] [PubMed]
- Efimtseva, E.V.; Mikhailov, S.N. Disaccharide nucleosides and oligonucleotides on their basis. New tools for the study of enzymes of nucleic acid metabolism. Biochemistry (Moscow) 2002, 67, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Mikhailov, S.N.; Efimtseva, E.V. Disaccharide nucleosides. Russ. Chem. Rev. 2004, 73, 401–414. [Google Scholar]
- Kimura, K.; Bugg, T.D.H. Recent advances in antimicrobial nucleoside antibiotics targeting cell wall biosynthesis. Nat. Prod. Rep. 2003, 20, 252–273. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.; Goss, R.J.M.; Kimura, K.; Bugg, T.D.H. Antimicrobial nucleoside antibiotics targeting cell wall assembly: Recent advances in structure-function studies and nucleoside biosynthesis. Nat. Prod. Rep. 2010, 27, 279–304. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Kagasaki, T.; Hosoya, T.; Takahashi, S. Adenophostins A and B: Potent agonists of inositol-1,4,5-trisphosphate receptor produced by Penicillium brevicompactum. Taxonomy, fermentation, isolation, physico-chemical and biological properties. J. Antibiot. 1993, 46, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Kinoshita, T.; Takahashi, M. Adenophostins A and B: Potent agonists of inositol-1,4,5-trisphosphate receptor produced by penicillium brevicompactum. Structure elucidation. J. Antibiot. 1994, 47, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Hotoda, H.; Takahashi, M.; Tanzawa, K.; Takahashi, S.; Kaneko, M. IP3 receptor-ligand. 1: Synthesis of adenophostin A. Tetrahedron Lett. 1995, 36, 5037–5040. [Google Scholar] [CrossRef]
- Hirota, J.; Michikawa, T.; Miyawaki, A.; Takahashi, M.; Tanzawa, K.; Okura, I.; Furuichi, T.; Mikoshiba, K. Adenophostin-medicated quantal Ca2+ release in the purified and reconstituted inositol 1,4,5-trisphosphate receptor type 1. FEBS Lett. 1995, 368, 248–252. [Google Scholar] [CrossRef]
- McCormick, J.; Li, Y.; McCormick, K.; Duynstee, H.I.; Van Engen, A.K.; Van der Marel, G.A.; Ganem, B.; Van Boom, J.H.; Meinwald, J. Structure and total synthesis of HF-7, a neuroactive glyconucleoside disulfate from the funnel-web spider Hololena curta. J. Am. Chem. Soc. 1999, 121, 5661–5665. [Google Scholar] [CrossRef]
- Bu, Y.Y.; Yamazaki, H.; Ukai, K.; Namikoshi, M. Anti-mycobacterial nucleoside antibiotics from a marine-derived Streptomyces sp. TPU1236A. Mar. Drugs 2014, 12, 6102–6112. [Google Scholar] [CrossRef] [PubMed]
- Knapp, S.; Gore, V.K. Synthesis of the ezomycin nucleoside disaccharide. Org. Lett. 2000, 2, 1391–1393. [Google Scholar] [CrossRef] [PubMed]
- Behr, J.B.; Gourlain, T.; Helimi, A.; Guillerm, G. Design, Synthesis and biological evaluation of hetaryl-nucleoside derivatives as inhibitors of chitin synthase. Bioorg. Med. Chem. Lett. 2003, 13, 1713–1716. [Google Scholar] [CrossRef]
- Binder, W.H.; Kӓhlig, H.; Schmid, W. Galactosylation by use of β-galactosidase: Enzymatic syntheses of disaccharide nucleosides. Tetrahedron: Asymmetry 1995, 6, 1703–1710. [Google Scholar] [CrossRef]
- Ye, M.; Yan, L.-Q.; Li, N.; Zong, M.-H. Facile and regioselective enzymatic 5-galactosylation of pyrimidine 2-deoxynucleosides catalyzed by β-glycosidase from bovine liver. J. Mol. Catal. B 2012, 79, 35–40. [Google Scholar] [CrossRef]
- Niedballa, U.; Vorbrüggen, H. A general synthesis of N-glycosides. III. Simple synthesis of pyrimidine disaccharide nucleosides. J. Org. Chem. 1974, 39, 3664–3667. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Shuto, S.; Matsuda, A. Synthesis of the C-glycosidic analog of adenophostin A, a potent IP3 receptor agonist, using a temporary silicon-tethered radical coupling reaction as the key step. Tetrahedron Lett. 2000, 41, 2391–2394. [Google Scholar] [CrossRef]
- Watanabe, K.A.; Matsuda, A.; Halat, M.J.; Hollenberg, D.H.; Nisselbaum, J.S.; Fox, J.J. Nucleosides. 114. 5′-O-Glucuronides of 5-fluorouridine and 5-fluorocytidine. Masked precursors of anticancer nucleosides. J. Med. Chem. 1981, 24, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.H.; O’Neill, R.A. Modern Methods in Carbohydrate Synthesis; Harwood Academic Publishers: Amsterdam, The Netherlands, 1996. [Google Scholar]
- Lindhorst, T.K. Essentials of Carbohydrate Chemistry and Biochemistry; Wiley-VCH Verlag Gmb-H & Co. KGaA: Weinheim, Germany, 2007. [Google Scholar]
- Demchenko, A.V. Handbook of Chemical Glycosylation; Wiley-VCH Verlag Gmb-H & Co. KGaA: Weinheim, Germany, 2008. [Google Scholar]
- Chen, X.; Halcomb, R.L.; Wang, P.G. Chemical Glycobiology (ACS Symposium Series 990); American Chemical Society: Washington, WA, USA, 2008. [Google Scholar]
- Toshima, K.; Tatsuta, K. Recent progress in O-glycosylation methods and its application to natural products synthesis. Chem. Rev. 1993, 93, 1503–1531. [Google Scholar] [CrossRef]
- Ito, Y. My stroll in the backyard of carbohydrate chemistry. Trend. Glycosci. Glycotechnol. 2010, 22, 119–140. [Google Scholar] [CrossRef]
- Yasomanee, J.P.; Demchenko, A.V. From stereocontrolled glycosylation to expeditious oligosaccharide synthesis. Trend. Glycosci. Glycotechnol. 2013, 25, 13–41. [Google Scholar] [CrossRef]
- Nakamura, M.; Fujita, S.; Ogura, H. Synthesis of disaccharide nucleoside derivatives of 3-deoxy-d-glycero-d-galacto-2-nonulosonic acid (KDN). Chem. Pharm. Bull. 1993, 41, 21–25. [Google Scholar] [CrossRef]
- Mikhailov, S.N.; Rodionov, A.A.; Efimtseva, E.V.; Ermolinsky, B.S.; Fomitcheva, M.V.; Padyukova, N.S.; Rothenbacher, K.; Lescrinier, E.; Herdewijn, P. Studies on disaccharide nucleoside synthesis. Mechanism of the formation of trisaccharide purine nucleosides. Nucleosides Nucleotides 1999, 18, 691–692. [Google Scholar] [CrossRef] [PubMed]
- Lichtenthaler, F.W.; Sanemitsu, Y.; Nohara, T. Synthesis of 5′-O-glycosyl-ribo-nucleosides. Angew. Chem. Int. Ed. 1978, 17, 772–774. [Google Scholar] [CrossRef]
- Knapp, S.; Gore, V.K. Synthesis of the shimofuridin nucleoside disaccharide. J. Org. Chem. 1996, 61, 6744–6747. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Knapp, S. Glycosylation of nucleosides. J. Org. Chem. 2016, 81, 2228–2242. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Niu, Q.; Li, C. Practical glucosylations and mannosylations using anomeric benzoyloxy as a leaving group activated by sulfonium ion. ACS Omega 2017, 2, 3698–3709. [Google Scholar] [CrossRef]
- Septak, M. Kinetic studies on depurination and detritylation of CPG-bound intermediates during oligonucleotide synthesis. Nucl. Acids Res. 1996, 24, 3053–3058. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, I.V.; Neuvonen, K.; Lönnberg, H.; Rodionov, A.A.; Shcheveleva, E.V.; Bobkov, G.V.; Efimtseva, E.V.; Mikhailov, S.N. Effective anomerisation of 2′-deoxyadenosine derivatives during disaccharide nucleoside synthesis. Nucleosides Nucleotides Nucleic Acids 2004, 23, 1849–1864. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Fukumoto, T.; Itoh, T.; Kurihara, M.; Saito, S.; Komabiki, S. Synthesis of disaccharide nucleosides by the O-glycosylation of natural nucleosides with thioglycoside donors. Chem. Asian J. 2015, 10, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Ferrier, R.J. 450. The interaction of phenylboronic acid with hexosides. J. Chem. Soc. (Resumed) 1961, 2325–2330. [Google Scholar] [CrossRef]
- Ferrier, R.J. Carbohydrate boronates. Adv. Carbohyd. Chem. Biochem. 1978, 35, 31–80. [Google Scholar]
- Duggan, P.J.; Tyndall, E.M. Boron acids as protective agents and catalysts in synthesis. J. Chem. Soc. Perkin Trans. 1 2002, 1325–1339. [Google Scholar] [CrossRef]
- Yamada, K.; Hayakawa, H.; Wada, T. Method for preparation of 2′-O-alkylribonucleosides by regioselective alkylation of 2′,3′-O-(arylboronylidene) ribonucleosides. JPN. Patent JP 2009/256335A, 5 November 2009. [Google Scholar]
- Lee, D.; Taylor, M.S. Borinic acid-catalyzed regioselective acylation of carbohydrate derivatives. J. Am. Chem. Soc. 2011, 133, 3724–3727. [Google Scholar] [CrossRef] [PubMed]
- Gouliaras, C.; Lee, D.; Chan, L.; Taylor, M.S. Regioselective activation of glycosyl acceptors by a diarylborinic acid-derived catalyst. J. Am. Chem. Soc. 2011, 133, 13926–13929. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H.; Manabe, S. Design of chemical glycosyl donors: Does changing ring conformation influence selectivity/reactivity? Chem. Soc. Rev. 2013, 42, 4297–4309. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, B.; Gu, X.; Chen, G.; Chen, L.; Wang, X.; Xiong, B.; You, Q.-D.; Chen, Y.-L.; Shen, J. 1,2-trans-1-Dihydroxyboryl benzyl S-glycoside as glycosyl donor. Carbohydr. Res. 2014, 398, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, A.; Tanaka, M.; Hanamura, S.; Takahashi, D.; Toshima, K. Regioselective and 1,2-cis-α-stereoselective glycosylation utilizing glycosyl-acceptor-derived boronic ester catalyst. Angew. Chem. Int. Ed. 2015, 127, 11085–11089. [Google Scholar] [CrossRef]
- Tanaka, M.; Nashida, J.; Takahashi, D.; Toshima, K. Glycosyl-acceptor-derived borinic ester-promoted direct and β-stereoselective mannosylation with a 1,2-anhydromannose donor. Org. Lett. 2016, 18, 2288–2291. [Google Scholar] [CrossRef] [PubMed]
- Nishi, N.; Nashida, J.; Kaji, E.; Takahashi, D.; Toshima, K. Regio- and stereoselective β-mannosylation using a boronic acid catalyst and its application in the synthesis of a tetrasaccharide repeating unit of lipopolysaccharide derived from E. Coli O75. Chem. Commun. 2017, 53, 3018–3021. [Google Scholar] [CrossRef] [PubMed]
- Mancini, R.S.; Leea, J.B.; Taylor, M.S. Boronic esters as protective groups in carbohydrate chemistry: Processes for acylation, silylation and alkylation of glycoside-derived boronates. Org. Biomol. Chem. 2017, 15, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Mancini, R.S.; Lee, J.B.; Taylor, M.S. Sequential functionalizations of carbohydrates enabled by boronic esters as switchable protective/activating groups. J. Org. Chem. 2017, 82, 8777–8791. [Google Scholar] [CrossRef] [PubMed]
- Lemanski, G.; Ziegler, T. Synthesis of 4-O-d-mannopyranosyl-α-d-glucopyranosides by intramolecular glycosylation of 6-O-tethered mannosyl donors. Tetrahedron 2000, 56, 563–579. [Google Scholar] [CrossRef]
- Huang, X.; Huang, L.; Wang, H.; Ye, X.-S. Iterative one-pot synthesis of oligosaccharides. Angew. Chem. Int. Ed. 2004, 43, 5221–5224. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.P.; Wang, C.-C. Highly stereoselective glycosyl-chloride-mediated synthesis of 2-deoxyglucosides. Chem. Eur. J. 2013, 19, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Lian, G.; Zhang, X.; Yu, B. Thioglycosides in carbohydrate research. Carbohydr. Res. 2015, 403, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Wulff, G.; Röhle, G. Results and problems of O-glycoside synthesis. Angew. Chem. Int. Ed. 1974, 13, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, A.; Stauch, T.; Boons, G.-J. Solvent and other effects on the stereoselectivity of thioglycoside glycosidations. Synlett 1997, 818–820. [Google Scholar] [CrossRef]
- Zhu, X.-F.; Williams, H.J., Jr.; Scott, A.I. An improved transient method for the synthesis of N-benzoylated nucleosides. Synth. Commun. 2003, 33, 1233–1243. [Google Scholar] [CrossRef]
- Eisenführ, A.; Arora, P.S.; Sengle, G.; Takaoka, L.R.; Nowickb, J.S.; Famuloka, M. A ribozyme with michaelase activity: Synthesis of the substrate precursors. Bioorg. Med. Chem. 2003, 11, 235–249. [Google Scholar] [CrossRef]
- Welch, C.J.; Bazin, H.; Heikkilä, J.; Chattopadhyaya, J. Synthesis of C-5 and N-3 arenesulfenyl uridines. Preparation and properties of a new class of uracil protecting group. Acta Chem. Scand. 1985, B 39, 203–212. [Google Scholar] [CrossRef]
- Samuels, E.R.; McNary, J.; Aguilar, M.; Awad, A.M. Effective synthesis of 3′-deoxy-3′-azido nucleosides for antiviral and antisense ribonucleic guanidine (RNG) applications. Nucleosides Nucleotides Nucleic Acids 2013, 32, 109–123. [Google Scholar] [CrossRef] [PubMed]
- France, R.R.; Rees, N.V.; Wadhawan, J.D.; Fairbanks, A.J.; Compton, R.G. Selective activation of glycosyl donors utilising electrochemical techniques: a study of the thermodynamic oxidation potentials of a range of chalcoglycosides. Org. Biomol. Chem. 2004, 2, 2188–2194. [Google Scholar] [CrossRef] [PubMed]
- Tam, P.-H.; Lowary, T.L. Synthesis of deoxy and methoxy analogs of octyl α-d-mannopyranosyl-(1→6)-α-d-mannopyranoside as probes for mycobacterial lipoarabinomannan biosynthesis. Carbohydr. Res. 2007, 342, 1741–1772. [Google Scholar] [CrossRef] [PubMed]
- Shuto, S.; Horne, G.; Marwood, R.D.; Potter, B.V.L. Total synthesis of nucleobase-modified adenophostin A mimics. Chem. Eur. J. 2001, 7, 4937–4946. [Google Scholar] [CrossRef]
- Wunderlich, C.H.; Spitzer, R.; Santner, T.; Fauster, K.; Tollinger, M.; Kreutz, C. Synthesis of (6-13C)pyrimidine nucleotides as spin-labels for RNA dynamics. J. Am. Chem. Soc. 2012, 134, 7558–7569. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.C.; Aman, N.; Borstel, R.V.; Darsley, M.; Kamireddy, B.; Kenten, J.; Morris, G.; Titmas, R. Conjugates of COL-1 monoclonal antibody and β-d-galactosidase can specifically kill tumor cells by generation of 5-fluorouridine from the prodrug β-d-galactosyl-5-fluorouridine. Cell Biophys. 1994, 24/25, 127–133. [Google Scholar] [CrossRef]
- Yalpani, M.; Boeseb, R. The structure of amine adducts of triorganylboroxines. Chem. Ber. 1983, 116, 3347–3358. [Google Scholar] [CrossRef]
- McKinley, N.F.; O’Shea, D.F. Efficient synthesis of aryl vinyl ethers exploiting 2,4,6-trivinylcyclotriboroxane as a vinylboronic acid equivalent. J. Org. Chem. 2004, 69, 5087–5092. [Google Scholar] [CrossRef] [PubMed]
- Iovine, P.M.; Fletcher, M.N.; Lin, S. Condensation of arylboroxine structures on Lewis basic copolymers as a noncovalent strategy toward polymer functionalization. Macromolecules 2006, 39, 6324–6326. [Google Scholar] [CrossRef]
- Chen, T.-B.; Huzak, M.; Macura, S.; Vuk-Pavlović, S. Somatostatin analogue octreotide modulates metabolism and effects of 5-fluorouracil and 5-fluorouridine in human colon cancer spheroids. Cancer Lett. 1994, 86, 41–51. [Google Scholar] [CrossRef]
- Agudo, R.; Arias, A.; Pariente, N.; Perales, C.; Escarmís, C.; Jorge, A.; Marina, A.; Domingo, E. Molecular characterization of a dual inhibitory and mutagenic activity of 5-fluorouridine triphosphate on viral RNA synthesis. Implications for lethal mutagenesis. J. Mol. Biol. 2008, 382, 652–666. [Google Scholar] [CrossRef] [PubMed]
- Kirienko, D.R.; Revtovich, A.V.; Kirienko, N.V. A high-content, phenotypic screen identifies fluorouridine as an inhibitor of pyoverdine biosynthesis and Pseudomonas aeruginosa virulence. mSphre 2016, 1, e00217-16. [Google Scholar]
- Wu, Q.; Xia, A.; Lin, X. Synthesis of monosaccharide derivatives and polymeric prodrugs of 5-fluorouridine via two-step enzymatic or chemo-enzymatic highly regioselective strategy. J. Mol. Catal. B: Enzymatic 2008, 54, 76–82. [Google Scholar] [CrossRef]
- Brusa, P.; Dosio, F.; Coppo, S.; Pacchioni, D.; Arpicco, S.; Crosasso, P.; Cattel, L. In vitro and in vivo antitumor activity of immunoconjugates prepared by linking 5-fluorouridine to antiadenocarcinoma monoclonal antibody. Farmaco 1997, 52, 71–81. [Google Scholar] [PubMed]
- Ozaki, S.; Akiyama, T.; Morita, T.; Kumegawa, M.; Nagase, T.; Uehara, N.; Hoshi, A. 5-Fluorouracil derivatives XX.: Synthesis and antitumor activity of 5′-O-unsaturated acyl-5-fluorouridines. Chem. Pharm. Bull. 1990, 38, 3164–3166. [Google Scholar] [CrossRef] [PubMed]
- Martino, M.M.; Jolimaitre, P.; Martino, R. The prodrugs of 5-fluorouracil. Curr. Med. Chem. Anti-Cancer Agents 2002, 2, 267–310. [Google Scholar] [CrossRef]
Sample Availability: Sample of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Boronic Acid b | Solvent | Condition | Yield (for 3 Steps) c |
|---|---|---|---|---|
| 1 a | - | MeCN | −20 °C, 1.5 h | <16% (complex mixture) |
| 2 a,d | PhB(OH)2 (11a) | MeCN | −20 °C, 1.5 h | 41% (α/β = 1.6/1) |
| 3 a,e | 11a | MeCN | −20 °C, 1.5 h | 45% (α/β = 1.6/1) |
| 4 a,e | 4-MeOPhB(OH)2 (11b) | MeCN | −20 °C, 1.5 h | 39% (α/β = 1.8/1) |
| 5 a,e | 4-CF3PhB(OH)2 (11c) | MeCN | −20 °C, 1.5 h | 51% (α/β = 1.8/1) |
| 6 a,e | 2,4-F2PhB(OH)2 (11d) | MeCN | −20 °C, 1.5 h | 46% (α/β = 1.8/1) |
| 7 a,e | 11c | 1,4-Dioxane | r.t., 1.5 h | 27% (α/β = 3.3/1) |
| 8 a,e | 11c | CH2Cl2 | −40 °C, 1.5 h | trace |
| 9 a,e | 11c | EtCN | −40 °C, 1.5 h | 61% (α/β = 1.6/1) |
| 10 e,f | 11c | EtCN | −40 °C, 1.5 h | 57% (α/β = 1.5/1) |
| 11 a,e | 4-CH3(CH2)5PhB(OH)2 (11e) | EtCN | −40 °C, 1.5 h | 30% (α/β = 1.6/1) |

| Entry a | Boronic Acid b | Solvent | Condition | Yield of 14 (for 3 Steps) c | Yield of 15 (for 3 Steps) |
|---|---|---|---|---|---|
| 1 | - | MeCN | −20 °C, 1.5 h | <10% (complex mixture) | not isolated |
| 2 d | PhB(OH)2 (11a) | MeCN | −20 °C, 1.5 h | 14% (α/β = 1/1.0) | 6% |
| 3 d | 4-CF3PhB(OH)2 (11c) | EtCN | −40 °C, 1.5 h | 11% (α/β = 1/1.2) | 27% |

| Entry a | Acceptor | Product | Yield (for 2 Steps) |
|---|---|---|---|
| 1 | 13 (Ade) | β-24 | 42% |
| 2 | 16 (AdeBz) | β-25 | 30% |
| 3 | 17 (Gua) | β-26 | 12% |
| 4 | 18 (GuaiBu) | β-27 | 44% |
| 5 | 10 (Uri) | β-28 | 42% (ca. 15%: nucleobase = 5-STol-Uri) |
| 6 | 19 (5-Me-Uri) | β-29 | 53% |
| 7 | 20 (5-F-Uri) | β-30 | 61% |
| 8 | 21 (Cyt) | β-31 | 55% |
| 9 | 22 (CytBz) | β-32 | 40% |

| Entry a | Donor | Product | Yield (for 2 Steps) |
|---|---|---|---|
| 1 | 33 (β-Glc) | β-35 | 54% |
| 2 b | 23 (β-Gal) | β-30 | 61% |
| 3 | 34 (α-Man) | α-36 | <39% (mixture) |

| Entry a | Donor | Acceptor | Product | Yield (for 3 Steps) b |
|---|---|---|---|---|
| 1 | 41 (Ar = 2-PhB(OH)2) (α form) | 10 (Nucleobase = Uri) | 12 | 44% (α/β = 1.9/1) |
| 2 | 41 (Ar = 2-PhB(OH)2) (α form) | 13 (Nucleobase = Ade) | 14 | 16% (α/β = 1.3/1) |
| 3 | 42 (Ar = 4-PhB(OH)2) (α/β = 1/1.0) | 10 (Nucleobase = Uri) | 12 | 36% (α/β = 2.1/1) |
| 4 | 42 (Ar = 4-PhB(OH)2) (α/β = 1/1.0) | 13 (Nucleobase = Ade) | 14 | 14% (α/β = 1.1/1) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Someya, H.; Itoh, T.; Aoki, S. Synthesis of Disaccharide Nucleosides Utilizing the Temporary Protection of the 2′,3′-cis-Diol of Ribonucleosides by a Boronic Ester. Molecules 2017, 22, 1650. https://doi.org/10.3390/molecules22101650
Someya H, Itoh T, Aoki S. Synthesis of Disaccharide Nucleosides Utilizing the Temporary Protection of the 2′,3′-cis-Diol of Ribonucleosides by a Boronic Ester. Molecules. 2017; 22(10):1650. https://doi.org/10.3390/molecules22101650
Chicago/Turabian StyleSomeya, Hidehisa, Taiki Itoh, and Shin Aoki. 2017. "Synthesis of Disaccharide Nucleosides Utilizing the Temporary Protection of the 2′,3′-cis-Diol of Ribonucleosides by a Boronic Ester" Molecules 22, no. 10: 1650. https://doi.org/10.3390/molecules22101650
APA StyleSomeya, H., Itoh, T., & Aoki, S. (2017). Synthesis of Disaccharide Nucleosides Utilizing the Temporary Protection of the 2′,3′-cis-Diol of Ribonucleosides by a Boronic Ester. Molecules, 22(10), 1650. https://doi.org/10.3390/molecules22101650