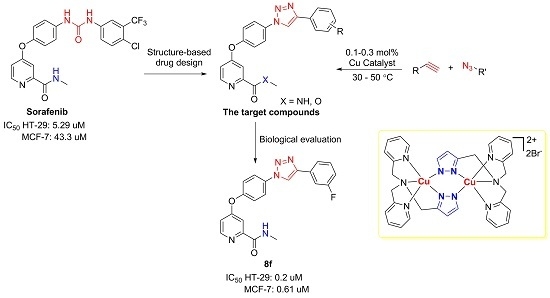
3.3. General Procedure for 2-Catalyzed 1,3-Dipolar Cycloaddition Reactions
Under N2 atmosphere, the mixture of alkyne (0.3 mmol), azide (0.3 mmol), Na ascorbate (0.006 mmol, 2 mol %), and 0.3 × 10−3 M solution of catalyst 2 in MeOH (0.1–0.3 mol %) was stirred in a 10 mL Schlenk tube at 25 °C for 16 h. Evaporation of the solvents followed by purification by short column chromatography on silica gel provided the desired 1,4-disubstituted triazole product. The unreacted alkyne and azide were first eluted out with 3/1 petroleum ether (PE)/ethyl acetate, and the pure 1,4-disubstituted triazole product was then obtained by elution with pure ethyl acetate.
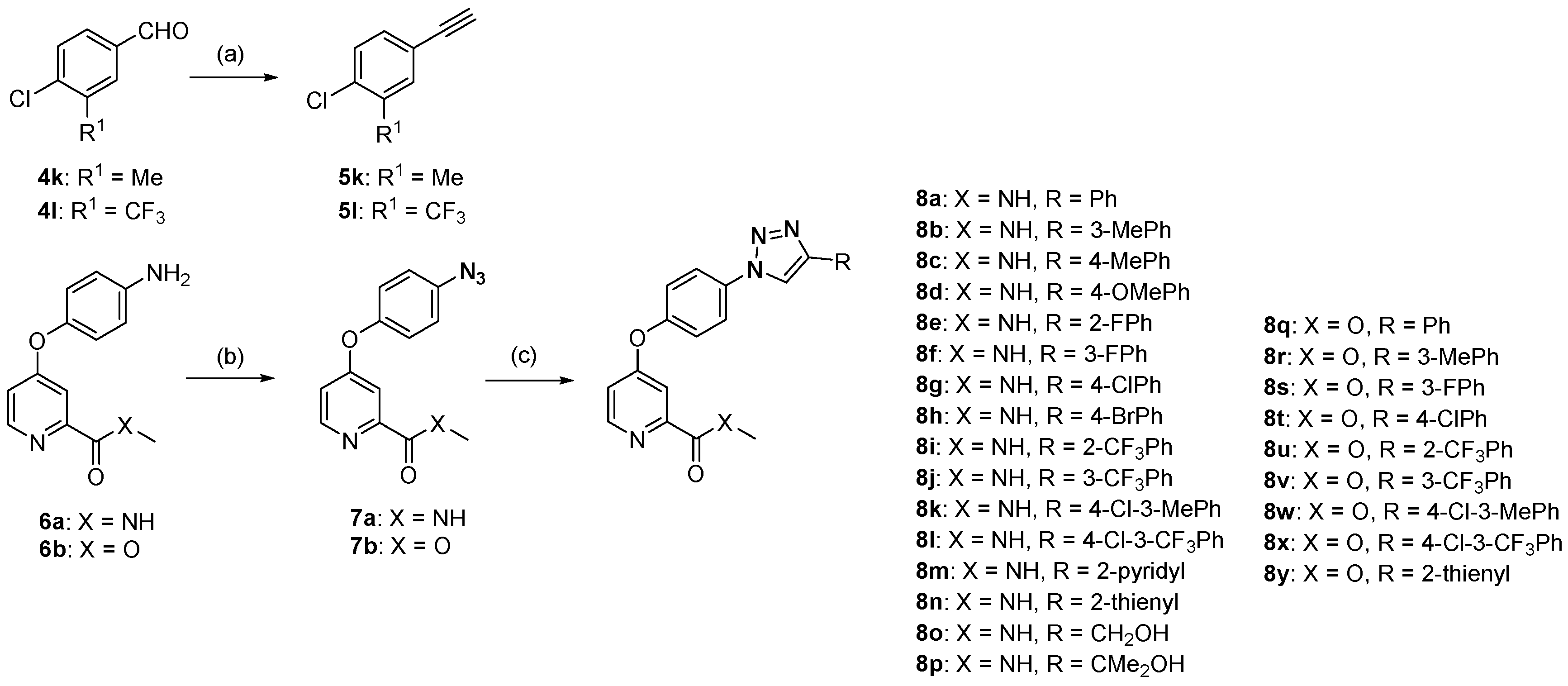
N-Methyl-4-(4-(4-(m-tolyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinamide (8b). Yield: 99%, white solid. m.p.: 185–187 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.33 (s, 1H, triazole-H), 8.82 (q, 1H, J = 4.8 Hz, NH), 8.57 (d, 1H, J = 5.6 Hz, ArH), 8.10 (dt, 2H, J = 8.9, 2.3 Hz, ArH), 7.79 (s, 1H, ArH), 7.75 (d, 1H, J = 7.7 Hz, ArH), 7.53 (dt, 2H, J = 8.9, 2.2 Hz, ArH), 7.49 (d, 1H, J = 2.6 Hz, ArH), 7.40 (t, 1H, J = 7.7 Hz, ArH), 7.27 (dd, 1H, J = 5.6, 2.6 Hz, ArH), 7.22 (d, 1H, J = 7.5 Hz, ArH), 2.80 (d, 3H, J = 4.9 Hz, NCH3), 2.39 (s, 3H, CCH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.18, 163.72, 153.28, 152.66, 150.67, 147.49, 138.21, 134.15, 130.13, 128.96, 125.91, 122.55, 122.37, 122.27, 119.80, 114.59, 109.34, 26.06, 21.12. HRMS Calcd for C22H19N5O2 [M]: 385.1539; found for C22H19N5O2 [M]: 385.1532.
N-Methyl-4-(4-(4-(p-tolyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinamide (8c). Yield: 99%, white solid. m.p.: 195–197 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.30 (s, 1H, triazole-H), 8.84 (q, 1H, J = 4.4 Hz, NH), 8.58 (d, 1H, J = 5.6 Hz, ArH), 8.10 (dt, 2H, J = 10.0, 3.2 Hz, ArH), 7.85 (d, 2H, J = 8.0 Hz, ArH), 7.54–7.49 (m, 3H, ArH), 7.33 (d, 2H, J = 7.9 Hz, ArH), 7.27 (dd, 1H, J = 5.6, 2.6 Hz, ArH), 2.80 (d, 3H, J = 4.9 Hz, NHCH3), 2.36 (s, 3H, CCH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.18, 163.70, 153.25, 152.65, 150.66, 147.45, 137.69, 134.17, 129.59, 127.44, 125.30, 122.36, 122.30, 119.46, 114.58, 109.32, 26.05, 20.90. HRMS Calcd for C22H19N5O2 [M]: 385.1539; found for C22H19N5O2 [M]: 385.1558.
4-(4-(4-(4-Methoxyphenyl)-1H-1,2,3-triazol-1-yl)phenoxy)-N-methylpicolinamide (8d). Yield: 97%, white solid. m.p.: 215–217 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.24 (s, 1H, triazole-H), 8.83 (d, 1H, J = 4.8 Hz, NH), 8.58 (d, 1H, J = 5.6 Hz, ArH), 8.10 (dt, 2H, J = 8.9, 3.2 Hz, ArH), 7.90 (dt, 2H, J = 8.8, 2.8 Hz, ArH), 7.54–7.49 (m, 3H, ArH), 7.27 (dd, 1H, J = 8.0, 2.6 Hz, ArH), 7.10 (dt, 2H, J = 8.8, 2.8 Hz, ArH), 3.82 (s, 3H, OCH3), 2.81(d, 3H, J = 4.9 Hz, NCH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.20, 163.72, 159.35, 153.21, 152.65, 150.68, 147.36, 134.21, 126.75, 122.75, 122.37, 122.26, 118.89, 114.58, 114.48, 109.33, 55.24, 26.07. HRMS Calcd for C22H19N5O3 [M]: 401.1488; found for C22H19N5O3 [M]: 401.1471.
4-(4-(4-(2-Fluorophenyl)-1H-1,2,3-triazol-1-yl)phenoxy)-N-methylpicolinamide (8e). Yield: 98%, white solid. m.p.: 182–183 °C. IR (KBr, cm−1): 3395.0, 1684.6, 1631.3, 1604.1, 1566.3, 1533.9, 1512.4, 901.8, 859.3, 837.4. 1H-NMR (600 MHz, DMSO-d6) δ 9.14 (d, 1H, J = 2.9 Hz, triazole-H), 8.83 (q, 1H, J = 4.4 Hz, NH), 8.57(d, 1H, J = 5.6 Hz, ArH), 8.21–8.14 (m, 3H, ArH), 7.52–7.46 (m, 4H, ArH), 7.42–7.37 (m, 2H, ArH), 7.27 (dd, 1H, J = 5.5, 2.6 Hz, ArH), 2.80 (d, 3H, J = 4.9 Hz, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.17, 163.72, 159.43, 157.79, 153.43, 152.66, 150.68, 141.01, 140.99, 134.01, 130.23, 130.18, 127.76, 127.74, 125.09, 125.07, 122.78, 122.29, 122.09, 122.02, 118.00, 117.91, 116.27, 116.13, 114.61, 109.34, 26.06. HRMS Calcd for C21H17N5O2 [M]: 389.1288; found for C21H17N5O2 [M]: 389.1304.
4-(4-(4-(3-Fluorophenyl)-1H-1,2,3-triazol-1-yl)phenoxy)-N-methylpicolinamide (8f). Yield: 99%, white solid. m.p.: 209–210 °C. IR (KBr, cm−1): 3403.3, 1676.7, 1630.7, 1610.4, 1589.8, 1568.9, 1538.8, 1514.7, 897.2, 862.4, 836.4. 1H-NMR (600 MHz, DMSO-d6) δ 9.42 (s, 1H, triazole-H), 8.83 (q, 1H, NH), 8.58 (d, 1H, J = 5.6 Hz, ArH), 8.09 (d, 2H, J = 8.9 Hz, ArH), 7.82 (d, 1H, J = 7.8 Hz, ArH), 7.76 (d, 1H, J = 10.1 Hz, ArH), 7.59–7.52 (m, 3H, ArH), 7.49 (d, 1H, J = 2.6 Hz, ArH), 7.27–7.22 (m, 2H, ArH), 2.80 (d, 3H, J = 4.9 Hz, CH3). 13C-NMR (150 MHz, DMSO-d6) δ 165.15, 163.72, 163.45, 161.83, 153.44, 152.67, 150.69, 146.29 (d, J = 2.7 Hz), 134.03, 132.61 (d, J = 8.6 Hz), 132.55 (d, J = 8.5 Hz), 131.30, 131.25, 122.44 (d, J = 2.8 Hz), 121.41 (d, J = 2.5 Hz), 120.71 (d, J = 2.6 Hz), 115.13, 114.99, 114.63, 112.03, 111.88, 109.36, 26.07. HRMS Calcd for C21H16FN5O2 [M]+: 389.1288; found for C21H16FN5O2 [M + H]+: 390.1355.
4-(4-(4-(4-Chlorophenyl)-1H-1,2,3-triazol-1-yl)phenoxy)-N-methylpicolinamide (8g). Yield: 94%, white solid. m.p.: 244–246 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.40 (s, 1H, triazole-H), 8.83 (d, 1H, J = 4.8 Hz, NH), 8.58 (d, 1H, J = 5.6 Hz, ArH), 8.09 (d, 2H, J = 8.9 Hz, ArH), 7.98 (d, 2H, J = 8.5 Hz, ArH), 7.60 (d, 2H, J = 8.5 Hz, ArH), 7.54 (d, 2H, J = 8.9 Hz, ArH), 7.49 (d, 1H, J = 2.5 Hz, ArH), 7.28 (dd, 1H, J = 5.6, 2.6 Hz, ArH), 2.81 (d, 3H, J = 4.9 Hz, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.16, 163.72, 153.39, 152.66, 150.69, 146.30, 134.06, 132.77, 129.17, 129.14, 127.05, 122.42, 122.40, 120.33, 120.31, 114.62, 109.35, 26.07. HRMS Calcd for C21H16ClN5O2 [M]: 405.0993; found for C21H16ClN5O2 [M]: 405.0991.
N-Methyl-4-(4-(4-(2-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinamide (8i). Yield: 99%, white solid. m.p.: 51–54 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.07 (s, 1H, triazole-H), 8.83 (q, 1H, J = 4.8 Hz, NH), 8.58 (d, 1H, J = 5.6 Hz, ArH), 8.14 (dt, 2H, J = 8.9, 3.2 Hz, ArH), 7.94 (d, 1H, J = 8.0 Hz, ArH), 7.84–7.83 (m, 2H, ArH), 7.73–7.71 (m, 1H, ArH), 7.53–7.50 (m, 3H, ArH), 7.27 (dd, 1H, J = 4.9, 2.5 Hz, ArH), 2.81 (d, 1H, J = 4.9 Hz, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.17, 163.71, 153.45, 152.67, 150.68, 144.84, 133.90, 132.76, 132.22, 129.36, 128.98, 127.08 (q, J = 30.2 Hz), 126.51 (q, J = 5.5 Hz), 124.89, 123.08, 122.59, 122.41, 114.59, 109.37, 26.06. HRMS Calcd for C21H16F3N5O2 [M]: 439.1256; found for C21H16F3N5O2 [M]: 439.1245.
N-Methyl-4-(4-(4-(3-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinamide (8j). Yield: 99%, white solid. m.p.: 216–218 °C. IR (KBr, cm−1): 3391.6, 1678.7, 1594.3, 1573.2, 1542.9, 835.8, 800.3. 1H-NMR (600 MHz, DMSO-d6) δ 9.56 (s, 1H, triazole-H), 8.83 (d, 1H, J = 4.9 Hz, NH), 8.59 (d, 1H, J = 5.6 Hz, ArH), 8.29 (d, 2H, J = 4.9 Hz, ArH), 8.11 (dt, 2H, J = 8.9, 2.0 Hz, ArH), 7.78–7.77 (m, 2H, ArH), 7.56 (dt, 2H, J = 8.9, 2.0 Hz, ArH), 7.50 (d, 1H, J = 2.5 Hz, ArH), 7.28 (dd, 1H, J = 5.6, 2.6 Hz, ArH), 2.81 (d, 3H, J = 4.9 Hz, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.14, 163.71, 153.47, 152.67, 150.69, 145.96, 134.00, 131.32, 130.36, 130.23 (q, JF = 31.8 Hz), 129.11, 126.87, 125.06, 124.84, 124.81, 123.26, 122.42, 122.38, 121.71, 121.68, 121.45, 120.96, 120.94, 114.64, 109.38, 26.06. HRMS Calcd for C21H16F3N5O2 [M]: 439.1256; found for C21H16F3N5O2 [M]: 439.1245.
4-(4-(4-(4-Chloro-3-methylphenyl)-1H-1,2,3-triazol-1-yl)phenoxy)-N-methylpicolinamide (8k). Yield: 99%, white solid. m.p.: 237–238 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.38 (s, 1H, triazole-H), 8.83 (d, 1H, J = 4.3 Hz, NH), 8.58 (d, 1H, J = 5.4 Hz, ArH), 8.09 (d, 1H, J = 8.5 Hz, ArH), 7.96 (s, 1H, ArH), 7.80 (d, 1H, J = 7.8 Hz, ArH), 7.57–7.49 (m, 4H, ArH), 7.28 (d, 1H, J = 2.9 Hz, ArH), 2.81 (d, 3H, J = 4.6 Hz, NCH3), 2.43 (s, 3H, CCH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.16, 163.71, 153.37, 152.66, 150.69, 146.42, 136.19, 134.07, 133.00, 129.59, 129.16, 127.93, 124.51, 122.39, 122.34, 120.20, 114.62, 109.35, 26.06, 19.75. HRMS Calcd for C22H18ClN5O2 [M]: 419.1149; found for C22H18ClN5O2 [M]: 419.1137.
4-(4-(4-(4-Chloro-3-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)phenoxy)-N-methylpicolinamide (8l). Yield: 98%, white solid. m.p.: 226–228 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.58 (s, 1H, triazole-H), 8.83 (d, 1H, J = 4.9 Hz, NH), 8.59 (d, 1H, J = 5.6 Hz, ArH), 8.38 (d, 1H, J = 1.9 Hz, ArH), 8.27 (dd, 1H, J = 8.3, 1.8 Hz, ArH), 8.10 (dt, 2H, J = 8.9, 3.2 Hz, ArH), 7.91 (d, 1H, J = 8.4 Hz, ArH), 7.56–7.50 (m, 3H, ArH), 7.28 (dd, 1H, J = 5.6, 2.6 Hz, ArH), 2.81 (d, 3H, J = 4.9 Hz, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.12, 163.71, 153.54, 152.67, 150.69, 145.06, 133.91, 132.62, 130.45, 130.09, 129.93, 127.52, 127.31, 124.27, 124.23, 123.70, 122.42, 122.41, 121.89, 121.28, 121.26, 114.65, 109.38, 26.06. HRMS Calcd for C22H15ClF3N5O2 [M]: 473.0866; found for C22H15ClF3N5O2 [M]: 473.0883.
N-Methyl-4-(4-(4-(pyridin-2-yl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinamide (8m). Yield: 94%, white solid. m.p.: 200–201 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.40 (s, 1H, triazole-H), 8.84 (q, 1H, J = 4.5 Hz, NH), 8.68 (d, 1H, J = 4.7 Hz, ArH), 8.58 (d, 1H, J = 5.6 Hz, ArH), 8.19–8.14 (m, 3H, ArH), 7.98 (dt, 1H, J = 7.7, 1.6 Hz, ArH), 7.52 (m, 3H, ArH), 7.43 (dd, 1H, J = 7.4, 4.9 Hz, ArH), 7.28 (dd, 1H, J = 5.5, 2.6 Hz, ArH), 2.81, 2.80 (d, 3H, J = 4.9 Hz, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.16, 163.72, 153.41, 152.65, 150.67, 149.73, 149.48, 148.29, 137.41, 134.05, 123.43, 122.54, 122.29, 121.52, 121.51, 119.86, 114.63, 109.36, 26.07. HRMS Calcd for C20H16N6O2 [M]: 372.1335; found for C20H16N6O2 [M]: 372.1340.
N-methyl-4-(4-(4-(thiophen-2-yl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinamide (8n). Yield: 92%, white solid. m.p.: 185–188 °C. IR (KBr, cm−1): 3381.7, 1677.0, 1631.4, 1587.3, 1571.9, 1530.8, 1511.8, 849.6, 838.6, 816.1. 1H-NMR (600 MHz, DMSO-d6) δ 9.26 (s, 1H, triazole-H), 8.83 (d, 1H, J = 4.8 Hz, NH), 8.58 (d, 1H, J = 5.6 Hz, ArH), 8.09 (d, 2H, J = 8.9 Hz, ArH), 7.63 (dd, 1H, J = 5.0, 0.8 Hz, ArH), 7.54–7.49 (m, 4H, ArH), 7.27 (dd, 1H, J = 5.5, 2.6 Hz, ArH), 7.21 (dd, 1H, J = 4.9, 3.6 Hz, ArH), 2.81 (d, 3H, J = 4.9 Hz, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.14, 163.69, 153.39, 152.65, 150.66, 142.79, 133.95, 132.30, 128.06, 126.01, 124.68, 122.43, 122.34, 119.14, 114.60, 109.35, 26.05. HRMS Calcd for C19H15N5O2S [M]: 377.0946; found for C19H15N5O2S [M]: 377.0938.
4-(4-(4-(Hydroxymethyl)-1H-1,2,3-triazol-1-yl)phenoxy)-N-methylpicolinamide (8o). Yield: 92%, white solid. m.p.: 191–192 °C. 1H-NMR (600 MHz, DMSO-d6) δ 8.82 (d, 1H, J = 4.9 Hz, NH), 8.74 (s, 1H, triazole-H), 8.57 (d, 2H, J = 5.6 Hz, ArH), 8.06 (dt, 2H, J = 8.9, 3.2 Hz, ArH), 7.49–7.46 (m, 3H, ArH), 7.26 (dd, 1H, J = 5.6, 2.6 Hz, ArH), 5.39 (t, 1H, J = 5.6 Hz, OH), 4.64 (d, 2H, J = 5.5 Hz, CH2), 2.80 (d, 3H, J = 4.9 Hz, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.18, 163.72, 153.11, 152.64, 150.66, 149.25, 134.27, 122.29, 122.26, 121.23, 114.58, 109.32, 54.98, 26.06. HRMS Calcd for C16H15N5O3 [M]: 325.1175; found for C16H15N5O3 [M]: 325.1191.
4-(4-(4-(2-Hydroxypropan-2-yl)-1H-1,2,3-triazol-1-yl)phenoxy)-N-methylpicolinamide (8p). Yield: 99%, white solid. m.p.: 150–152 °C. 1H-NMR (600 MHz, DMSO-d6) δ 8.83 (q, 1H, J = 4.5 Hz, NH), 8.65 (s, 1H, triazole-H), 8.57 (d, 1H, J = 5.6 Hz, ArH), 8.06 (dt, 2H, J = 8.9, 3.2 Hz, ArH), 7.48–7.45 (m, 3H, ArH), 7.25 (dd, 1H, J = 5.6, 2.6 Hz, ArH), 5.28 (s, 1H, OH), 2.81 (d, 3H, J = 4.9 Hz, NCH3), 1.55 (s, 6H, C(CH3)2). 13C-NMR (151 MHz, DMSO-d6) δ 165.19, 163.72, 157.03, 153.01, 152.64, 150.66, 134.34, 122.25, 122.17, 119.10, 114.57, 109.33, 67.02, 30.60, 26.06. HRMS Calcd for C18H19N5O3 [M]: 353.1488; found for C18H19N5O3 [M]: 353.1504.
Methyl 4-(4-(4-(m-tolyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinate (8r). Yield: 98%, white solid. m.p.: 147–149 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.34 (s, 1H, triazole-H), 8.65 (d, 1H, J = 5.5 Hz, ArH), 8.11 (d, 2H, J = 8.6 Hz, ArH), 7.79–7.74 (m, 2H, ArH), 7.56–7.52 (m, 3H, ArH), 7.41 (t, 1H, J = 7.6 Hz, ArH), 7.32 (dd, 1H, J = 5.2, 2.0 Hz, ArH), 7.22 (d, 1H, J = 7.44 Hz, ArH), 3.87 (s, 3H, OCH3), 2.40 (s, 3H, CCH3). 13C-NMR (151 MHz, DMSO-d6) δ164.82, 164.72, 153.22, 151.92, 149.74, 147.53, 138.24, 134.19, 130.13, 128.99, 125.93, 122.57, 122.28, 119.78, 115.51, 112.89, 52.67, 21.14. HRMS Calcd for C22H18N4O3 [M]: 386.1379; found for C22H18N4O3 [M]: 386.1370.
Methyl 4-(4-(4-(3-fluorophenyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinate (8s). Yield: 96%, white solid. m.p.: 154–157 °C. 1H-NMR (600 MHz, CDCl3) δ 9.42 (s, 1H, triazole-H), 8.65 (d, 1H, J = 5.5 Hz, ArH), 8.08 (d, 2H, J = 8.6 Hz, ArH), 7.81 (d, 1H, J = 7.6 Hz, ArH), 7.75 (d, 1H, J = 9.9 Hz, ArH), 7.58–7.53 (m, 4H, ArH), 7.32 (dd, 1H, J = 5.2, 2.0 Hz, ArH), 7.25 (t, 1H, J = 7.1 Hz, ArH), 3.86 (s, 3H, CH3). 13C-NMR (150 MHz, DMSO-d6) δ 164.84, 164.70, 163.47, 161.86, 153.39, 151.96, 149.76, 146.34, 134.07, 132.62, 132.56, 131.35, 131.29, 122.46, 122.34, 121.46, 120.70, 115.56, 115.17, 115.03, 112.93, 112.06, 111.91, 52.70. HRMS Calcd for C21H15FN4O3 [M]: 390.1128; found for C21H15FN4O3 [M]: 390.1142.
Methyl 4-(4-(4-(4-chlorophenyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinate (8t). Yield: 99%, white solid. m.p.: 173–175 °C. IR (KCl, cm−1): 3415.6, 1717.4, 1631.5, 1608.1, 1586.3, 884.0, 866.4, 855.6, 835.6. 1H-NMR (600 MHz, DMSO-d6) δ 9.40 (s, 1H, triazole-H), 8.65 (d, 1H, J = 5.6 Hz, ArH), 8.09 (d, 2H, J = 8.7 Hz, ArH), 7.98 (d, 2H, J = 8.3 Hz, ArH), 7.60 (d, 2H, J = 8.2 Hz, ArH), 7.56–7.53 (m, 3H, ArH), 7.32 (dd, 1H, J = 5.3, 1.7 Hz, ArH), 3.86 (s, 3H, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 164.82, 164.69, 153.34, 151.93, 149.75, 146.35, 134.09, 132.80, 129.18, 129.14, 127.07, 122.42, 122.30, 120.28, 115.53, 112.90, 52.67. HRMS Calcd for C21H15ClN4O3 [M]: 406.0833; found for C21H15ClN4O3 [M]: 406.0827.
Methyl 4-(4-(4-(2-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinate (8u). Yield: 99%, white solid. m.p.: 87–88 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.06 (s, 1H, triazole-H), 8.65 (d, 1H, J = 5.5 Hz, ArH), 8.14 (d, 2H, J = 8.8 Hz, ArH), 7.93 (d, 1H, J = 7.9 Hz, ArH), 7.83–7.82 (m, 2H, ArH), 7.72 (t, 1H, J = 6.8 Hz, ArH), 7.55–7.52 (m, 3H, ArH), 7.32 (dd, 1H, J = 5.5, 2.4 Hz, ArH), 3.86 (s, 3H, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 164.81, 164.71, 153.39, 151.92, 149.75, 144.88, 133.93, 132.78, 132.25, 129.39, 128.98, 127.11 (q, J = 30.0 Hz), 126.53 (q, J = 5.4 Hz), 124.91, 123.10, 122.58, 122.33, 115.51, 112.89, 52.67. HRMS Calcd for C22H15F3N4O3 [M]: 440.1096; found for C22H15F3N4O3 [M]: 440.1062.
Methyl 4-(4-(4-(3-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinate (8v). Yield: 93%, white solid. m.p.: 172–174 °C. 1H-NMR (600 MHz, CDCl3) δ 9.55 (s, 1H, triazole-H), 8.65 (d, 1H, J = 5.6 Hz, ArH), 8.28 (d, 2H, J = 5.0 Hz, ArH), 8.11 (dt, 2H, J = 8.9, 3.3 Hz, ArH), 7.77–7.76 (m, 2H, ArH), 7.55–7.53 (m, 3H, ArH), 7.32 (dd, 1H, J = 5.6, 2.6 Hz, ArH), 3.86 (s, 3H, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 164.78, 164.64, 153.39, 151.90, 149.73, 145.97, 134.00, 131.29, 130.33, 130.21 (q, J = 31.9 Hz), 129.08, 126.85, 125.05, 124.82, 124.80, 123.24, 122.34, 122.28, 121.69, 121.67, 121.44, 120.90, 120.88, 115.51, 112.90, 52.63. HRMS Calcd for C22H15F3N4O3 [M]: 440.1096; found for C22H15F3N4O3 [M]: 440.1102.
Methyl 4-(4-(4-(4-chloro-3-methylphenyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinate (8w). Yield: 99%, white solid. m.p.: 177–178 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.38 (s, 1H, triazole-H), 8.65 (d, 1H, J = 5.5 Hz, ArH), 8.10 (dt, 2H, J = 6.8, 3.3 Hz, ArH), 7.95 (d, 1H, J = 1.5 Hz, ArH), 7.80 (dd, 1H, J = 8.2, 1.8 Hz, ArH), 7.57–7.53 (m, 4H, ArH), 7.32 (dd, 1H, J = 5.6, 2.6 Hz, ArH), 3.86 (s, 3H, OCH3), 2.42 (s, 3H, CCH3). 13C-NMR (151 MHz, DMSO-d6) δ 164.84, 164.71, 153.33, 151.95, 149.76, 146.48, 136.23, 134.11, 133.06, 129.62, 129.16, 127.97, 124.55, 122.37, 122.32, 120.17, 115.55, 112.93, 52.70, 19.78. HRMS Calcd for C22H17ClN4O3 [M]: 420.0989; found for C22H17ClN4O3 [M]: 420.1007.
Methyl 4-(4-(4-(4-chloro-3-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinate (8x). Yield: 99%, white solid. m.p.: 175–177 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.59 (s, 1H, triazole-H), 8.66 (d, 1H, J = 5.6 Hz, ArH), 8.38 (d, 1H, J = 1.8 Hz, ArH), 8.27 (dd, 1H, J = 8.3, 1.9 Hz, ArH), 8.09 (dt, 2H, J = 8.9, 1.9 Hz, ArH), 7.92 (d, 1H, J = 8.4 Hz, ArH), 7.56–7.54 (m, 3H, ArH), 7.33 (dd, 1H, J = 5.5, 2.5 Hz, ArH), 3.87 (s, 3H, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 164.81, 164.64, 153.47, 151.93, 149.75, 145.09, 133.93, 132.62, 130.46, 130.12, 129.92, 127.52 (q, J = 30.8 Hz), 124.27, 123.71, 122.39, 122.31, 121.90, 121.20, 115.55, 112.94, 52.67. HRMS Calcd for C22H14ClF3N4O3 [M]: 474.0707; found for C22H14ClF3N4O3 [M]: 474.0730.
Methyl 4-(4-(4-(thiophen-2-yl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinate (8y). Yield: 99%, white solid. m.p.: 152–153 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.26 (s, 1H, triazole-H), 8.65 (d, 1H, J = 5.5 Hz, ArH), 8.09 (d, 2H, J = 8.9 Hz, ArH), 7.63 (d, 1H, J = 5.0 Hz, ArH), 7.56–7.52 (m, 4H, ArH), 7.32 (dd, 1H, J = 5.5, 2.5 Hz, ArH), 7.21 (dt, 1H, J = 3.7, 1.2 Hz, ArH), 3.87 (s, 3H, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 164.82, 164.69, 153.36, 151.93, 149.75, 142.85, 133.99, 132.29, 128.11, 126.06, 124.74, 122.46, 122.27, 119.12, 115.53, 112.91, 52.67. HRMS Calcd for C19H14N4O3S [M]: 378.0787; found for C19H14N4O3S [M]: 378.0797.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}