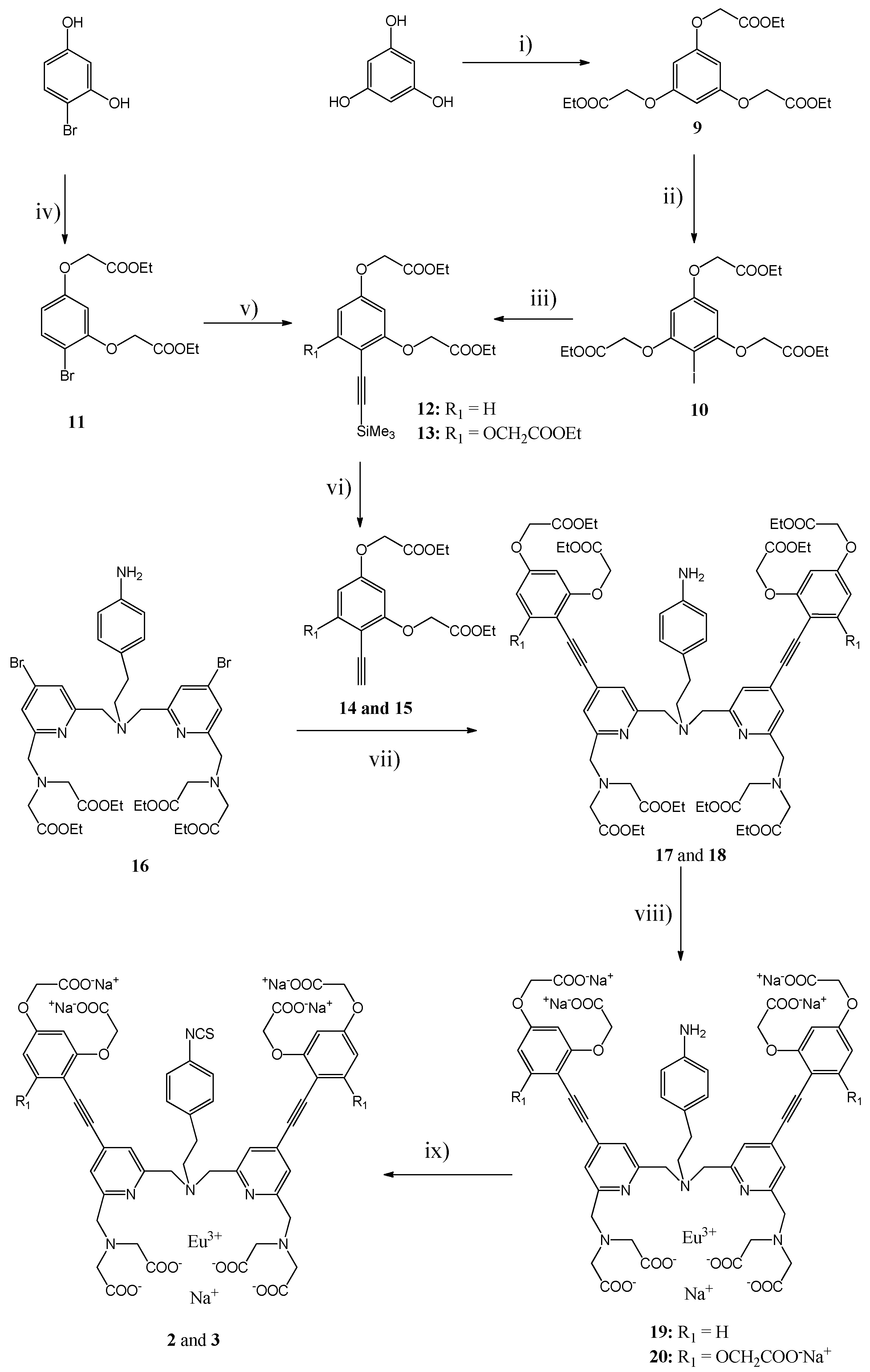
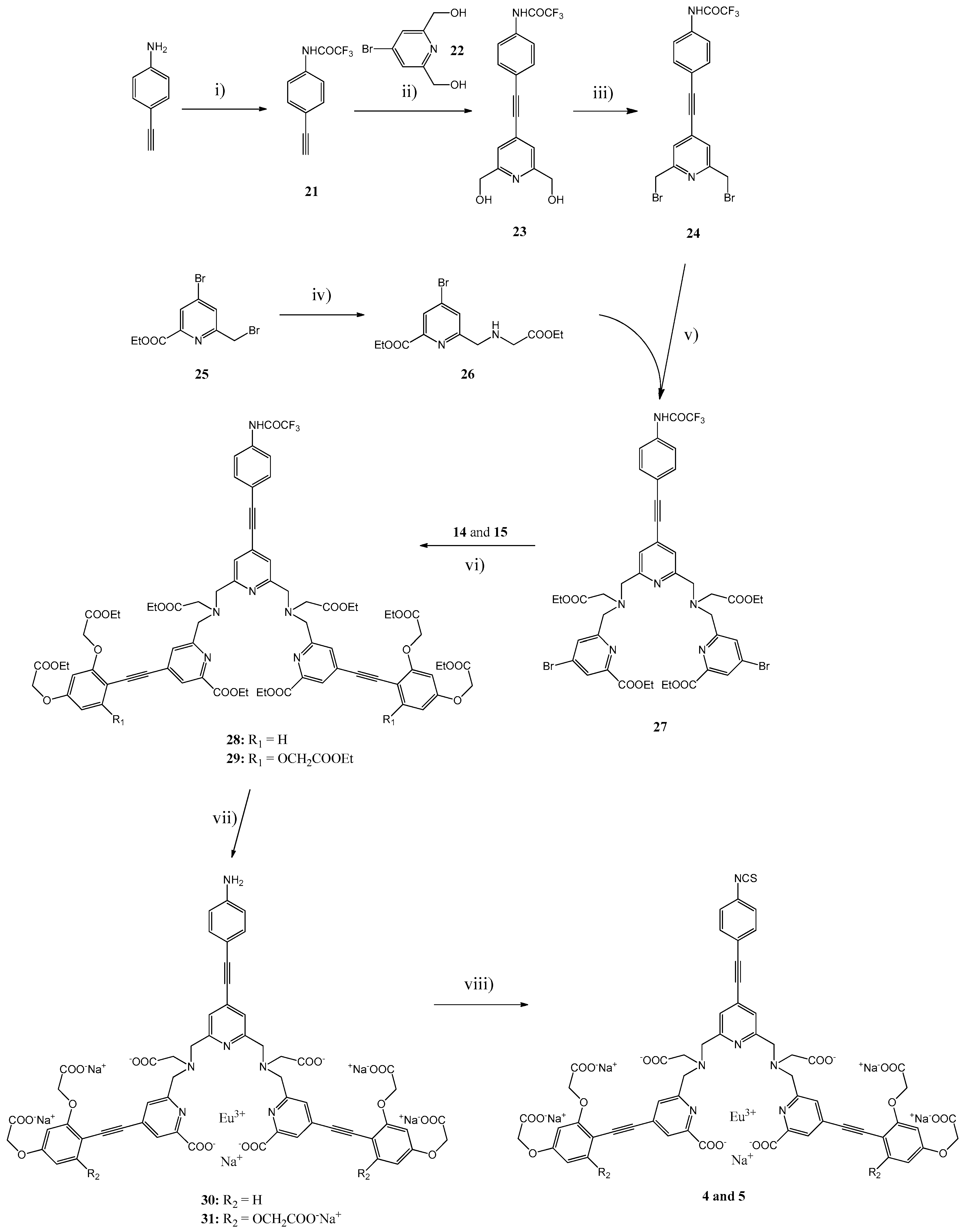
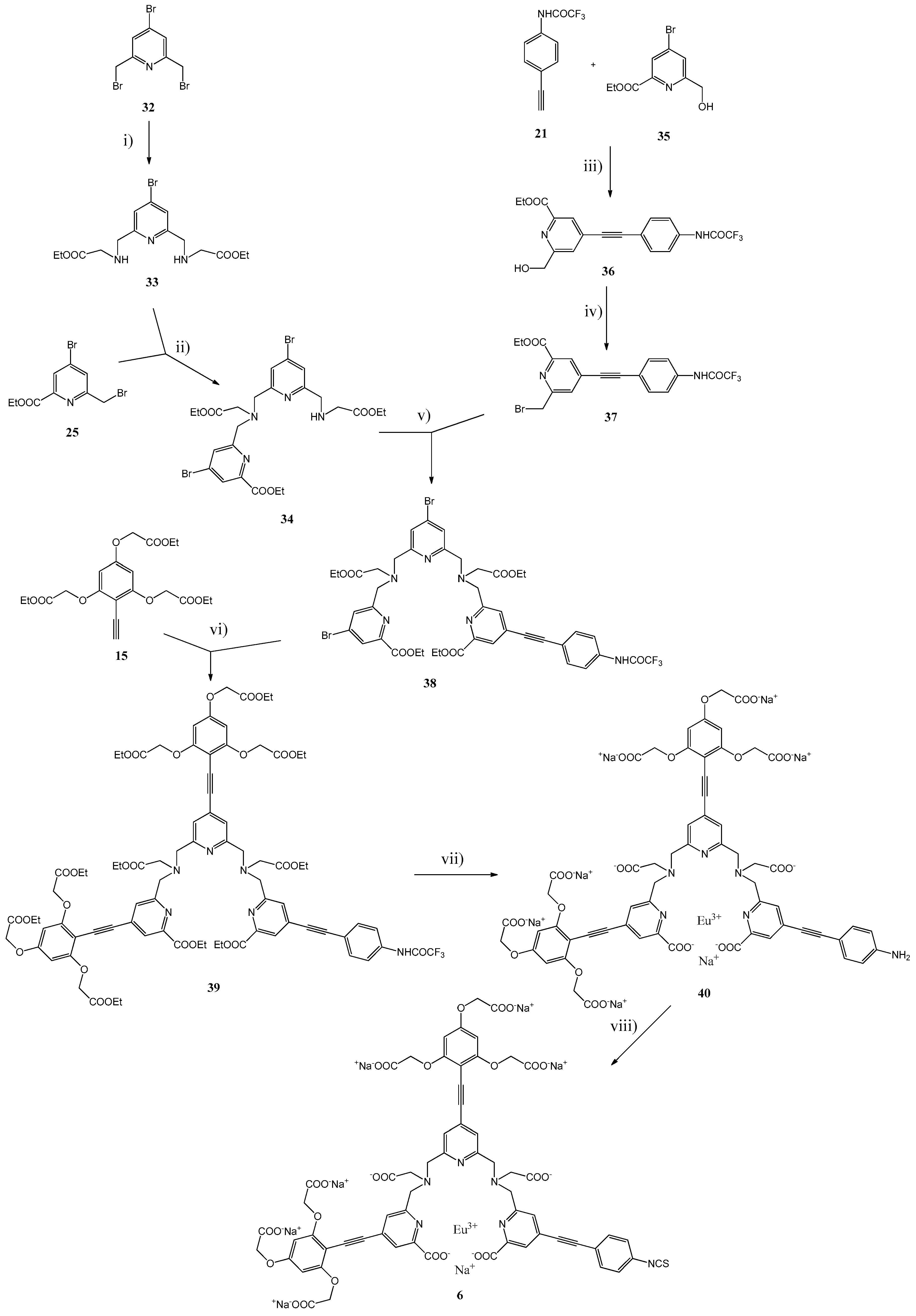
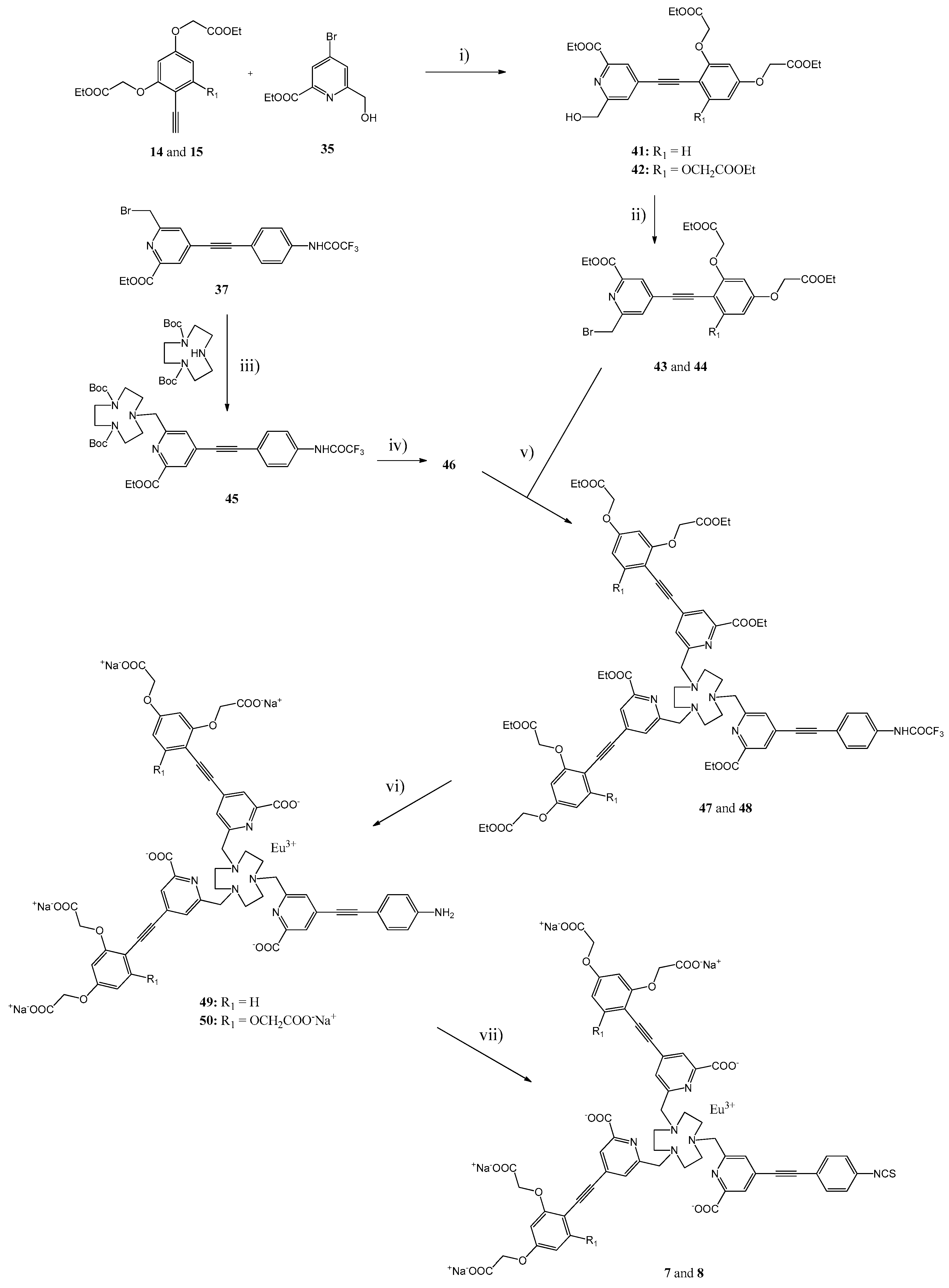
3.2. Syntheses
The general synthetic pathways used for the Eu(III) Chelates
2–
8 are shown in
Scheme 2,
Scheme 3,
Scheme 4 and
Scheme 5. The more detailed synthetic procedures and the results of the spectroscopic product characterization are presented in the Supporting information. Microwave assisted syntheses were performed with a Initiator+ microwave synthesizer from Biotage (Uppsala, Sweden).
The Eu(III) chelates were analysed and purified by using a reversed phase HPLC (2996 Photodiode Array Detector, 600 Controller, Delta 600 Fraction Collector III; Waters, Milford, MA, USA) with a RP-18 column. The solvents were A: triethylammonium acetate buffer (20 mM, pH 7) and B: 50% acetonitrile in triethylammonium acetate buffer (20 mM, pH 7). The gradient was started from 5% of solvent B and the amount of solvent B was linearly raised to 100% within 30 min.
The 1H and 13C-NMR spectra were recorded on an AVANCE 500 DRX (Bruker, Billerica, MA, USA). The chemical shifts are given in ppm from the internal tetramethylsilane. Mass spectra were recorded on Applied Biosystems QSTAR® XL ESI-TOF (electrospray ionization-time-of-flight mass analyzer) instrument (Thermo Fisher Scientific) using α-cyano-4-cinnamic acid matrix. UV-Vis spectra were recorded on an Ultrospec® 3300 pro (GE Healthcare Life Sciences, Chicago, IL, USA) or a UV-1800 spectrophotometer (Shimadzu, Kyoto, Japan).
Eu(III) Chelate (1a). A mixture of Eu(III) chelate 1 (2.5 μmol) and taurine (3.1 mg, 25 μmol) in aqueous 50 mM Na2CO3 buffer (0.125 mL, pH 9.8) was stirred overnight at RT (room temperature). The product was purified by HPLC. Rf(HPLC): 15.1 min. UV: 320 nm.
Triethyl 2,2′,2′′-[benzene-1,3,5-triyltris(oxy)]triacetate (9). Ethyl bromoacetate (14.6 mL, 132 mmol) was added within 2 h to a mixture of benzene-1,3,5-triol (5.04 g, 40 mmol) and dry K2CO3 (18.2 g, 132 mmol) in dry MeCN (300 mL) at 50 °C under argon. After stirring for ca. 20 h at 50 °C, the mixture was filtered, the solid material washed with MeCN and the filtrate evaporated to dryness. The product was purified by silica gel column chromatography using MeOH/CH2Cl2 (first 0:100, then 1:99) as an eluent. Yield: 10.8 g (70%). 1H-NMR [DMSO-d6 (dimethyl sulfoxide), δ ppm]: 6.13 (3H, s), 4.74 (6H, s), 4.17 (6 H, q, J = 7.1 Hz), 1.22 (9H, t, J = 7.1 Hz). 13C-NMR (DMSO-d6, δ ppm): 169.02, 159.84, 95.12, 65.27, 61.10, 14.51. MS(ESI-TOF) calculated for C18H24O9 [M + H]+: 385,15, found: 385.04.
Triethyl 2,2′,2′′-[(2-iodobenzene-1,3,5-triyl)tris(oxy)]triacetate (10). The compound 9 (8.36 g, 22 mmol) in CH3COOH (40 mL) was added to a mixture of N-chlorosuccinimide (2.95 g, 22 mmol) and NaI (3.30 g, 22 mol) in CH3COOH (65 mL). After stirring for 1.5 h at RT, the mixture was evaporated to dryness. The residue was dissolved in EtOAc (ethyl acetate, 320 mL) and neutralized with saturated NaHCO3 solution. The phases were separated, the aqueous phase was extracted with EtOAc (80 mL) and the combined organic phases were dried with Na2SO4. Yield: 11,2 g (100%). 1H-NMR (CDCl3, δ ppm): 6.10 (2H, s), 4.65 (4H, s), 4.55 (4H, s), 4.27 (2H, q, J = 7.15 Hz), 4.26 (4H, q, J = 7.15 Hz), 1.31 (3H, t, J = 7.15 Hz), 1.30 (6H, t, J = 7.15 Hz). 13C-NMR (CDCl3, δ ppm): 168.14, 168.01, 159.89, 158.65, 94.34, 66.61, 65.61, 61.52, 61.43, 14.05. MS(ESI-TOF) calculated for C18H23IO9 [M + H]+: 511.05, found: 510.93.
Diethyl 2,2′-[(4-bromo-1,3-phenylene)bis(oxy)]diacetate (11). A mixture of 4-bromobenxene-1,3-diol (3.18 g, 16.8 mmol), dry K2CO3 (5,12 g, 37,0 mmol) and ethyl bromoacetate (4.11 mL, 37.0 mmol) in dry MeCN (60 mL) was stirred for 24 h at 50 °C. The mixture was filtered, the solid material washed with MeCN and the filtrate evaporated to dryness. The product was purified by silica gel column chromatography using MeOH/CH2Cl2 (first 0:100, then 1:99) as an eluent. Yield: 5.97 g (98%). 1H-NMR (DMSO-d6, δ ppm): 7.46, (1H, d, J = 8.8 Hz), 6.65 (1H, d, J = 2.7 Hz), 6.51 (1H, dd, J = 2.7 and 8.8 Hz), 4.91 (2H, s), 4.79 (2H, s), 4.18 (2H, q, J = 7.1 Hz), 4.16 (2H, q, J = 7.1 Hz), 1.22 (3 H, t, J = 7.1 Hz), 1.21 (3 H, t, J = 7.1 Hz). 13C-NMR (DMSO-d6, δ ppm): 168.65, 158.62, 155.08, 133.55, 108.65, 102.64, 102.09, 65.81, 65.44, 61.21, 61.15, 14.49. MS(ESI-TOF) calculated for C14H17BrO6 [M + H]+: 361.03 and 363.03 found: 360.89 and 362.92.
General Procedure for the Synthesis of Compounds 12 and 13. A mixture of compound 10 or 11 (5.33 mmol), bis(triphenylphosphine)palladium(II) chloride (186 mg, 0.27 mmol), CuI (51 mg, 0.27 mmol), triphenylphosphine (142 mg, 0.54 mmol; only for compound 12) in diethylamine (10 mL) and dry DMF (5 mL) was de-aerated with argon. After addition of trimethylsilylacetylene (1.17 mL, 8.34 mmol), the mixture was stirred for 45 or 30 min for compound 10 and 11, respectively, at 100 °C using microwave heating. After evaporation to dryness, the residue was dissolved in CH2Cl2 (50 mL), washed with H2O (2 × 25 mL), dried with Na2SO4 and purified with column chromatography.
Diethyl 2,2′-{{4-[(trimethylsilyl)ethynyl]-1,3-phenylene}bis(oxy)}diacetate (12). The product was purified by silica gel column chromatography using EtOAc/petroleum ether (20:80) as an eluent. Yield: 94%. 1H-NMR (DMSO-d6, δ ppm): 7.31 (1H, d, J = 8.8 Hz), 6.52 (1 H, d, J = 2.4 Hz), 6.61 (1H, dd, J = 2.4 and 8.8 Hz), 4.85 (2H, s), 4.81 (2H, s), 4.18 (2H, q, J = 7.1 Hz), 4.17 (2H, q, J = 7.1 Hz), 1.23 (3H, t, J = 7.1 Hz), 1.21( 3H, t, J = 7.1 Hz), 0.22 (9H, s). 13C-NMR (DMSO-d6, δ ppm): 168.73, 160.08, 159.70, 134.94, 107.33, 105.18, 102.16, 100.66, 97.00, 65.66, 65.30, 61.17, 14.51, 14.49, 0.57. MS(ESI-TOF) calculated for C19H22O6Si [M + H]+: 379.16, found 379.39.
Triethyl 2,2′,2′′-{{2-[(trimethylsilyl)ethynyl]benzene-1,3,5-triyl}tris(oxy)}triacetate (13). The product was purified by silica gel column chromatography using EtOAc/petroleum ether (first 20:80, then 30:70) as an eluent. Yield: 70%. 1H-NMR (CDCl3, δ ppm): 6.10 (2H, s), 4.67 (4H, s), 4.55 (2H, s), 4.27 (2H, g, J = 7.15 Hz), 4.26 (4H, q, J = 7.15 Hz), 1.30 (6H, t, J = 7.15 Hz), 1.29 (3H, t, J = 7.15 Hz), 0.26 (9H, s). 13C-NMR (DMSO-d6, δ ppm): 168.60, 168.56, 160.98, 159.66, 101.34, 98.17, 95.29, 93.43, 65.77, 65.31, 61.06, 61.04, 14.41, 14.38, 0.56. MS(ESI-TOF) calculated for C23H32O9Si [M + H]+: 481.19, found: 481.99.
General Procedure for the Synthesis of Compounds 14 and 15. A mixture of compound 12 or 13 (10.4 mmol) and tetrabutylammonium fluoride (3.25 g, 12.4 mmol) in CH2Cl2 (200 mL) stirred for 1.5 h at RT under argon. Mixture was washed with aqueous 10% citric acid solution (200 mL), H2O (200 mL), died with Na2SO4 and used for the next step without further purification as no impurities were found with the TLC analysis.
Diethyl 2,2′-{[4-(ethynyl)-1,3-phenylene]bis(oxy)}diacetate (14). Yield: 100%. 1H-NMR (DMSO-d6, δ ppm): 7.34 (1H, d, J = 8.8 Hz), 6.53 (1H, d, J = 2.4 Hz), 6.52 (1H, dd, J = 2.4 and 8.8 Hz), 4.89 (2H, s), 4.81 (2H, s), 4.17 (2H, q, J = 7.1 Hz), 4.16 (2H, q, J = 7.1 Hz), 4.10 (1H, s), 1.22 (3H, t, J = 7.1 Hz), 1.21 (3H, t, J = 7.1 Hz). 13C-NMR (DMSO-d6, δ ppm): 168.84, 168.83, 160.29, 159.59, 134.90, 107.29, 104.62, 100.49, 83.70, 80.35, 65.40, 65.29, 61.17, 14.49. MS(ESI-TOF) calculated for C16H18O6 [M + H]+: 307.12, found: 307.01.
Triethyl 2,2′,2′′-{[2-(ethynyl]benzene-1,3,5-triyl]tris(oxy)}triacetate (15). Yield: 100%. 1H-NMR (CDCl3, δ ppm): 6.06 (2H, s), 4.69 (4H, s), 4.55 (2H, s), 4.27 (2H, q, J = 7.15 Hz), 4.26 (4H, q, J = 7.15 Hz), 3.50 (1H, s), 1.30 (3H, t, J = 7.15 Hz), 1.29 (6H, t, J = 7.15 Hz). 13C-NMR (CDCl3, δ ppm): 168.23, 168.10, 161.48, 159.39, 95.25, 94.18, 85.18, 66.42, 65.49, 61.64, 61.49, 14.14. MS(ESI-TOF) calculated for C20H24O9 [M + H]+: 409.15, found: 409.20.
General Procedure for the Synthesis of Ligand Esters 17 and
18. A mixture of compound
16 (0.22 g, 0.25 mmol; [
31]) and compound
14 or
15 (0.60 mmol) in dry TEA (2 mL) and THF (4 mL) was de-aerated with argon. After an addition of bis(triphenylphosphine)palladium(II) dichloride (11 mg, 16 μmol) and CuI (6 mg, 32 μmol), the mixture was stirred overnight at 55 °C and evaporated to dryness. The residue was dissolved in CH
2Cl
2 (30 mL), washed with H
2O (2 × 15 mL), dried with Na
2SO
4 and purified by column chromatography.
Ligand Ester 17. The product was purified by silica gel column chromatography using TEA/EtOAc/petroleum ether (20:40:40) as an eluent. Yield: 48%. 1H-NMR (DMSO-d6, δ ppm): 7.47 (2 H, d, J = 1.1 Hz), 7.45 (2H, d, J = 8.6 Hz), 7.43 (2H, d, J = 1.1 Hz), 6.79 (2H, d, J = 8.4 Hz), 6.61 (2H, d, J = 2.3 Hz), 6.55 (2H, dd, J = 8.4 and 2.3 Hz), 6.44 (2H, d, J = 8.4 Hz), 4.91 (4H, s), 4.83 (4H, s), 4.75 (2H, s), 4.17 (4H, q, J = 7.1 Hz), 4.17 (4H, q, J = 7.1 Hz), 4.05 (4H, q, J = 7.1 Hz), 3.94 (4H, s), 3.82 (4H, s), 3.56 (8H, s), 2.65 (4H, s), 1.23 (6H, t, J = 7.1 Hz), 1.20 (6H, t, J = 7.1 Hz), 1.16 (12H, t, J = 7.1 Hz). 13C-NMR (DMSO-d6, δ ppm): 171.14, 168.77, 168.702, 160.28, 160.14, 159.58, 159.21, 146.96, 134.86, 129.34, 129.28, 129.18, 127.50, 122.84, 122.50, 114.42, 107.61, 104.26, 100.64, 90.62, 90.38, 65.65, 65.35, 61.21, 61.19, 59.73, 59.60, 56.67, 54.98, 32.61, 14.50, 14.49, 14.46. MS(ESI-TOF) calculated for C70H84N6O20 [M + 2H]+: 1329.59, found: 1330.66.
Ligand Ester 18. The product purified by silica gel column chromatography using MeOH/CH2Cl2 (5:95) as an eluent. Yield: 73%. 1H-NMR (CDCl3, δ ppm): 7.56 (2H, s), 7.51 (2H, s), 6.88 (2H, d, J = 8.3 Hz), 6.56 (2H, d, J = 8.3 Hz), 5.98 (4H, s), 4.70 (8H, s), 4.52 (4H, s), 4.28 (4H, q, J = 7.1 Hz), 4.26 (8H, q, J = 7.1 Hz), 4.15 (8H, q, J = 7.1 Hz), 4.04 (4H, s), 3.87 (4H, s), 3.62 (8H, s), 2.80–2.70 (4H, m), 1.65 (2H, bs), 1.31 (6H, t, J = 7.1 Hz), 1.25 (12H, t, J = 7.1 Hz), 1.24 (12H, t. J = 7.1 Hz). 13C-NMR (CDCl3, δ ppm): 171.14, 168.45, 168.14, 160.91, 159.58, 159.52, 157.81, 144.59, 133.17, 130.33, 129.52, 123.52, 122.58, 115.26, 96.52, 95.52, 93.46, 85.52, 66.04, 65.29, 61.54, 61.32, 60.49, 60.35, 59.92, 56.02, 54.88, 32.58, 14.24, 14.16, 14.15. MS(ESI-TOF) calculated for C78H96N6O26 [M + 2H]+: 1533.65, found: 1532.96.
General Procedure for the Synthesis of Eu(III) Chelates 19 and 20. Compound 17 or 18 (52 μmol) in 0.5 M KOH in EtOH (5.4 mL) was stirred for 30 min at RT, H2O (2.7 mL) was added and the mixture was further stirred at RT for 2 h. After evaporation of EtOH and an additional stirring for 2 h at RT, the pH was adjusted to 6.5 by addition of 6 M HCl. EuCl3 (21 mg, 57 μmol) in water (0.5 mL) was added within 5 min and the pH was maintained at 6.0–6.5 with suitable additions of solid NaHCO3. After stirring overnight at RT, the pH was adjusted to 8.5 with 1 M NaOH. The precipitate was removed by centrifugation and the supernatant evaporated to dryness. The product was purified by HPLC.
Eu(III) Chelate 19. Yield: 88%. Rf(HPLC): 14.8 min. UV: 342 nm.
Eu(III) Chelate 20. Yield: 92%. Rf(HPLC): 12.3 min. UV: 346 nm.
General Procedure for the Synthesis of Eu(III) Chelates 2 and 3. An aqueous solution (1.3 mL) of the Eu(III) chelate 19 or 20 (55 μmol) was added within 5 min to a mixture of CSCl2 (30 µL, 0.39 mmol) and NaHCO3 (37 mg, 0.44 mmol) in CHCl3 (1.3 mL). After stirring for 45 min at RT, the two phases were separated and the aqueous phase was washed with CHCl3 (3 × 1.3 mL). The product was precipitated with acetone (ca. 45 mL), isolated by centrifugation, washed with acetone (2 × 10 mL) and dried overnight in vacuum desiccator. The products were used as such for next phase or for labeling the antibodies.
Eu(III) Chelate 2. Rf(HPLC): 18.4 min. UV: 346 nm.
Eu(III) Chelate 3. Rf(HPLC): 14.6 min. UV: 350 nm.
General Procedure for the Synthesis of Eu(III) Chelates 2a and 3a. A mixture of Eu(III) chelate 2 or 3 (2.5 μmol) and taurine (3.1 mg, 25 μmol) was stirred in aqueous 50 mM Na2CO3 buffer (0.125 mL, pH 9.8) for overnight at RT. The product was purified by HPLC.
Eu(III) Chelate 2a. Rf(HPLC): 15.1 min. UV: 340 nm.
Eu(III) Chelate 3a. Rf(HPLC): 12.5min. UV: 351 nm.
N-(4-Ethynylphenyl)-2,2,2-trifluoroacetamide (21). 4-Ethynylaniline (1.17 g, 10 mmol) was added in small portions to a ice-cold trifluoroacetic anhydride (5.6 mL, 40 mmol). After stirring for 2.5 h at RT, cold H2O was added, the mixture filtrated and the product washed with cold H2O. Yield: 2.12 g (98%). 1H-NMR (CDCl3, δ ppm): 8.08 (1H, bs), 7.55 (2H, d, J = 8.8 Hz), 7.51 (2 H, d, J = 8.8 Hz), 3.1 (1H, s). 13C-NMR (CDCl3, δ ppm): 154.72, 135.40, 133.23, 129.92, 120.25, 120.01, 82.67, 77.99. MS(ESI-TOF) calculated for C10H6F3NO [M + H]+: 213.04, found 213.05.
N-{4-{[2,6-Bis(hydroxymethyl)pyridin-4-yl]ethynyl}phenyl}-2,2,2-trifluoroacetamide (
23) A mixture of the compound
21 (0.55 g, 2.58 mmol) and 4-bromo-2,6-dihydroxymethylpyridine (
22; [
28]) (0.47 g, 2.15 mmol) in dry TEA (5 mL) and THF (10 mL) was de-aerated with argon. After addition of bis(triphenylphosphine)palladium(II) chloride (30 mg, 43 μmol) and CuI (16 mg, 86 μmol), the mixture was stirred for 19 h at 55 °C. After evaporation to dryness, the residue was treated with a cold mixture of CH
2Cl
2 (40 mL) and H
2O (20 mL), filtered and the product washed with cold H
2O (10 mL) and CH
2Cl
2 (10 mL). Yield: 0.56 g (75%).
1H-NMR (DMSO-
d6, δ ppm): 11.5 (1H, bs), 7.79 (2H, d,
J = 8.8 Hz), 7.68 (2H, d,
J = 8.8 Hz), 7.41 (2H, s), 5.49 (2H, bs), 4.56 (4H, s).
13C-NMR (DMSO-
d6, δ ppm): 162.25, 155.53, 155.23, 154.94, 154.64. 138.01, 133.12, 131.39, 121.52, 120.00, 119.58, 118.63, 117.29, 114.99, 92.93, 87.98, 64.39. MS(ESI-TOF) calculated for C
17H
13F
3N
2O
3 [M + H]
+: calculated 351.10, found 351.96.
N-{4-{[2,6-bis(bromomethyl)pyridin-4-yl]ethynyl}phenyl}-2,2,2-trifluoroacetamide (24). PBr3 (0.35 mL, 3.67 mmol) was added in a suspension of the compound 23 (0.86 g, 2.45 mmol) in CHCl3 (100 mL). After stirring for 20 h at 60 °C, the mixture was neutralized with 5% NaHCO3 (50 mL). The aqueous phase was extracted with CHCl3 (50 mL) and the combined organic phases were dried with Na2SO4, filtered and evaporated to dryness. The product 24 (1.09 g, 93%) was used for the next step without further purifications. 1H-NMR (CDCl3, δ ppm): 7.93 (1H, s), 7.63 (2H, d, J = 8.7 Hz), 7.59 (2H, d, J = 8.7 Hz), 7.47 (2H, s), 4.53 (4H, s). 13C-NMR (CDCl3, δ ppm): 157.02, 155.16, 154.85, 154.55, 154.23, 134.92, 133.36, 133.11, 124.55, 120.26, 119.73, 118.98, 116.68, 114.38, 112.10, 93.70, 86.77, 32.98. MS(ESI-TOF) calculated for C17H11Br2F3N2O [M + H]+: 474.93, 476.93, 478.92, found 475.38, 477.44, 479.45.
Ethyl-2-{[4-bromo-6-(carboxyethyl)pyridine-2-yl]methylenenitrilo}acetate (
26). A mixture of 4-bromo-6-bromomethyl-2-carboxyethylpyridine (
25; [
14]) (1.66 g, 5.15 mmol), glycine ethyl ester hydrochloride (3.60 g, 25.8 mmol) and di-isopropylethylamine (9.1 mL) in dry MeCN (70 mL) was stirred overnight at RT. The mixture was evaporated to dryness and the product was purified by silica gel column chromatography using MeOH/CH
2Cl
2 (3:97) as an eluent. Yield 1.55 g (87%).
1HNMR: (CDCl
3, δ ppm): 8.14 (1H, d,
J = 1.65 Hz), 7.85 (1H, d,
J = 1.60 Hz), 4.47 (2H, q,
J = 7.12 Hz), 4.20 (2H, q,
J = 7.13 Hz), 4.05 (2H, s), 3.47 (2H, s), 2.26 (1H, s), 1.43 (3H, t,
J = 7.13 Hz), 1.28 (3H, t,
J = 7.13 Hz).
13C-NMR (DMSO-
d6, δ ppm): 172.41, 164.08, 162.23, 148.76, 133.81, 128.53, 126.29, 62.07, 60.47, 53.65, 50.24, 14.59, 14.54. MS(ESI-TOF) calculated for C
13H
17BrN
2O
4 [M + H]
+: 345.05, 347.05, found 344.97, 346.97.
Synthesis of Compound 27. A mixture of compounds 24 (0.44 g, 0.94 mmol), 26 (0.65 g, 1.89 mmol), dry K2CO3 (0.52 g, 3.76 mmol) was stirred in dry MeCN (30 mL) for 27 h at 70 °C. The mixture was filtrated and the filtrate evaporated to dryness, and the product was purified by silica gel column chromatography using TEA/EtOAc/petroleum ether (1:69:30) as an eluent. Yield: 66%. 1H-NMR (CDCl3, δ ppm): 8.53 (1H, s), 8.14 (2H, d, J = 1.80 Hz), 8.11 (2H, d, J = 1.80 Hz), 7.68 (2 H, d, J = 8.73 Hz), 7.60 (2H, d, J = 8.73 Hz), 7.34 (2H, s), 4.44 (4H, q, J = 7.08 Hz), 4.18 (4 H, q, J = 7.15 Hz); 4.10 (4H, s), 3.98 (4 H, s), 3.50 (4 H, s), 1.40 (6H, t, J = 7.08 Hz), 1.28 (6H, t, J = 7.15 Hz). 13C-NMR (CDCl3, δ ppm): 171.02, 164.11, 161.85, 158.48, 155.23, 154.92, 154.62, 154.32, 148.52, 136.02, 134.37, 132.98, 132.16, 129.28, 128.35, 127.01, 123.43, 120.28, 119.04, 116.74, 114.45, 112.15, 92.79, 87.68, 62.23, 60.68, 59.82, 59.64, 55.41, 14.24. MS(ESI-TOF) calculated for C43H43Br2F3N6O9 [M + H]+: 1003.15, 1005.15, 1007.15, found 1003.71, 1005.48, 1007.58.
General Procedure for the Synthesis of Ligand Esters 28 and 29. A mixture of compound 27 (0.120 g, 0.148 mmol), compound 14 or 15 (0.355 mmol) in dry TEA (1 mL) and THF (2 mL) was de-aerated with argon. After an addition of bis(triphenylphosphine)palladium(II) dichloride (10 mg, 14 μmol) and CuI (6 mg, 28 μmol), the mixture was stirred overnight at 55 °C and evaporated to dryness. The residue was dissolved in CH2Cl2 (30 mL), washed with H2O (2 × 15 mL), dried with Na2SO4 and purified by silica gel column chromatography.
Ligand Ester 28. The product purified by silica gel column chromatography using 10% EtOH/CH2Cl2 as an eluent. Yield: 69%. 1H-NMR (DMSO-d6, δ ppm): 11.44 (1H, s), 7.85 (2H, s), 7.77 (2H, s), 7.69 (2H, d, J = 8.1 Hz), 7.57 (2H, d, J = 6.7 Hz), 7.45 (2H, d, J = 8.1 Hz), 7.43 (2H, s), 6.60 (2H, s), 6.53 (2H, d, J = 6.7 Hz), 4.91 (4H, s), 4.82 (4 H, s), 4.30 (4H, q, J = 6.7 Hz), 4.17 (8H, q, J = 7.1 Hz), 4.06 (4H, q, J = 6.9 Hz), 4.00 (4 H, s), 3.95 (4 H, s), 3.53 (4 H, s), 1.30 (6H, t, J = 6.7 Hz), 1.22 (6H, t. J = 7.1 Hz), 1.20 (6H, t, J = 7.1 Hz), 1.18 (6H, t, J = 6.9 Hz). 13C-NMR (DMSO-d6, δ ppm): 171.19, 168.67, 168.62, 164.58, 160.62, 160.32, 159.04, 155.47, 155.18, 154.89, 154.59, 147.74, 137.93, 134.95, 133.48, 132.98, 130.67, 129.50, 127.50, 124.78, 123.44, 121.34, 119.46, 118.50, 117.26, 114.96, 112.67, 107.69, 103.73, 100.64, 92.13, 89.65, 87.58, 65.70, 65.34, 61.84, 61.20, 60.48, 59.64, 56.50, 55.71, 55.38, 14.51, 14.46. MS(ESI-TOF) calculated for C75H77F3N6O21 [M + H]+: 1455.52, found 1455.36.
Ligand Ester 29. The product was purified by silica gel column chromatography using TEA/EtOAc/petroleum ether (1:69:30) as an eluent. Yield: 57%. 1H-NMR (CDCl3, δ ppm): 9.12 (1H, s), 8.09 (2H, d, J = 1.25 Hz), 8.06 (2H, d, J = 1.25 Hz), 7.57 (2 H, s), 7.34 (2H, d, J = 8.80 Hz), 7.30 (2H, d, J = 8.80 Hz), 5.97 (4H, s), 4.63 (8H, s), 4.60 (4H, s), 4.45 (4H, q, J = 7.12 Hz), 4.30 (4H, q, J = 7.12 Hz), 4,21 (8H, q, J = 7.12 Hz), 4.17 (4H, q, J = 7.12), 4.10 (4H, s), 4.02 (4H, s), 3.52 (4H, s), 1.42 (6H, t, J = 7.12 Hz), 1.33 (6H, t, J = 7.12 Hz), 1.29 (6H, J = 7.12 Hz), 1.25 (12H, t, J = 7.12 Hz). 13C-NMR (CDCl3, δ ppm): 171.13, 168.69, 168.69, 165.07, 161.08, 160.53, 159.98, 159.03, 154.95, 154.66, 147.65, 136.35, 133.99, 132.56, 132.17, 128.47, 127.64, 125.43, 123.12, 119.95, 116.90, 114.59, 96.39, 94.76, 93.71, 92.65, 87.75, 87.21, 66.12, 65.22, 61.91, 61.89, 61.51, 60.57, 60.10, 59.94, 55.11, 14.32, 14.27, 14.13. MS(ESI-TOF) calculated for C83H89F3N6O27 [M + H]+: 1659.58, found 1659.81.
General Procedure for the Synthesis of Eu(III) Chelates 30 and 31. Compound 28 or 29 (90 μmol) was stirred in 0.5 M KOH in EtOH (13 mL) for 30 min at RT. H2O (10 mL) was added and the mixture was further stirred at RT for 3 h. After evaporation of EtOH and an additional overnight stirring at RT, the pH was adjusted to 6.5 by addition of 6 M HCl. EuCl3 (33 mg, 90 μmol) in water (0.5 mL) was added within 5 min and the pH was maintained at 6.0–6.5 with suitable additions of solid NaHCO3. After stirring overnight at RT, the pH was adjusted to 8.5 with 1 M NaOH. The precipitate was removed by centrifugation and the supernatant evaporated to dryness. The product was purified by HPLC.
Eu(III) Chelate 30. Yield: 100%. Rf(HPLC): 14.2 min. UV: 340 nm.
Eu(III) Chelate 31. Yield: 100%. Rf(HPLC): 14.5 min. UV: 359 nm.
General Procedure for the Synthesis of Eu(III) Chelates 4 and 5. An aqueous solution (2.7 mL) of the Eu(III) chelate 30 or 31 (0.10 mmol) was added within 5 min to a mixture of CSCl2 (53 µL, 0.70 mmol) and NaHCO3 (67 mg, 0.80 mmol) in CHCl3 (2.7 mL). After stirring for 30 min at RT, the two phases were separated and the aqueous phase was washed with CHCl3 (3 × 3 mL). The product was precipitated with acetone (ca. 45 mL), isolated by centrifugation, washed with acetone (2 × 10 mL) and dried overnight in vacuum desiccator.
Eu(III) Chelate 4. Rf(HPLC): 18.4 min. UV: 346 nm.
Eu(III) Chelate 5. Rf(HPLC): 20.9 min. UV: 325 (sh), 340 and 362 (sh) nm.
General Procedure for the Synthesis of Eu(III) Chelates 4a and 5a. A mixture of Eu(III) chelate 4 or 5 (6.3 μmol) and taurine (8 mg, 63 μmol) was stirred in aqueous 50 mM Na2CO3 buffer (0.64 mL, pH 9.8) and DMF (0.64 mL) for overnight at RT. The product was purified by HPLC.
Eu(III) Chelate 4a. Rf(HPLC): 16.7 min. UV: 340 nm.
Eu(III) Chelate 5a. Rf(HPLC): 14.3 min. UV: 349 nm.
Diethyl 2,2′-{[(4-bromopyridine-2,6-diyl)bis(methylene)]bis(azanediyl)}diacetate (
33). A mixture of 4-bromo-2,6-dibromomethylpyridine (
32, [
28]) (3.50 g, 10 mmol), glycine ethyl ester hydrochloride (14.0 g, 0.10 mol) and di-isopropylethylamine (35 mL) in dry MeCN (130 mL) was stirred overnight at RT. After evaporation to dryness, the residue was dissolved in CH
2Cl
2 (100 mL), washed with H
2O (3 × 50 mL), dried with Na
2SO
4, and the product was purified by silica gel column chromatography using TEA/EtOAc/petroleum ether (first 10:20:70, then 15:30:55) as an eluent. Yield 2.48 g (64%).
1H-NMR (CDCl
3, δ ppm): 7.43 (2H, s), 4.20 (4H, q,
J = 7.2 Hz), 3.91 (4H, s), 3.46 (4H, s), 2.25–2.15 (2H, bs), 1.28 (6H, t,
J = 7.2 Hz).
13C-NMR (CDCl3, δ ppm): 172.00, 160.40, 133.83, 60.75, 54.00, 50.34, 14.18. MS(ESI-TOF) calculated for C
15H
22BrN
3O
4 [M + H]
+: 388.09 and 390.09, found 388.75 and 390.75.
Ethyl 4-bromo-6-{{{{4-bromo-6-{[(2-ethoxy-2-oxoethyl)amino]methyl}pyridin-2-yl}methyl}(2-ethoxy-2-oxo-ethyl)amino}methyl}picolinate (
34). 4-Bromo-6-bromomethyl-2-carboxyethylpyridine (
25, [
14]) (2.06 g, 6.39 mmol) was added in small portions to a mixture of the compound
33 (2.49 h, 6.39 mmol), dry K
2CO
3 (3.53 g, 25.6 mmol) in dry MeCN (215 mL) within 2 h at RT. After stirring for 22 h at RT, the mixture was filtrated and the filtrate evaporated to dryness. The product was purified by silica gel column chromatography using TEA/EtOAc/petroleum ether (first 10:25:65) as an eluent. Yield 2.03 g (50%).
1H-NMR (CDCl
3, δ ppm): 8.12 (1H, d,
J = 1.7 Hz), 8.07 (1H, d,
J = 1.7 Hz), 7.55 (1H, d,
J = 1.5 Hz), 7.43 (1H, d,
J = 1.5 Hz), 4.46 (2H. q,
J = 7.2 Hz), 4.19 (4H, q,
J = 7.2 Hz), 4.09 (2H, s), 3.95 (2H, s), 3.90 (2H, s), 2.3–2.1 (1H, bs), 1.43 (3H, t,
J = 7.2 Hz), 1.29 (3H, t,
J = 7.2 Hz), 1.29 (3H, t,
J = 7.2 Hz).
13C-NMR (CDCl3, δ ppm): 171.98, 170.81, 164.03, 161.57, 160.41, 159.70, 148.54, 134.26, 133.90, 129.13, 126.95, 124.60, 123.77, 62.16, 60.75, 60.63, 59.61, 59.48, 55.38, 54.00, 50.36, 14.19, 14.17, 14.14. MS(ESI-TOF) calculated for C
24H
30Br
2N
4O
6 [M + H]
+: 629.06, 631.05 and 633.06, found 629.54, 631.56 and 633.54.
Ethyl 6-(hydroxymethyl)-4-{[4-(2,2,2-trifluoroacetamido)phenyl]ethynyl}picolinate (
36). A mixture of the compound
21 (0.34 g, 1.60 mmol) and
35 (0.47 g, 2.15 mmol; [
14]) in dry TEA (5 mL) and THF (10 mL) was de-aerated with argon. After addition of bis(triphenylphosphine)palladium(II) chloride (19 mg, 27 μmol) and CuI (10 mg, 53 μmol), the mixture was stirred for 24 h at 55 °C. After evaporation to dryness, the product was purified by silica gel column chromatography using EtOH/CH
2Cl
2/TEA (10:89:1) as an eluent. Yield: 0.49 g (94%).
1H-NMR (CDCl
3, δ ppm): 8.49 (1H, s), 8.08 (1H, s), 7.66 (2H, d,
J = 8.7 Hz), 7.60 (1H, s), 7.59 (2H, d,
J = 8.7 Hz), 4.85 (2H, s), 4.49 (2 H, q,
J = 7.1 Hz), 1.43 (3H, t,
J = 7.1 Hz).
13C-NMR (CDCl
3, δ ppm): 164.55, 160.42, 155.24, 154.94, 154.64, 154.34, 147.42, 136.24, 133.10, 132.99, 125.58, 125.27, 120.30, 119.36, 118.94, 116.65, 114.35, 112.06, 94.24, 87.57, 64.30, 62.10, 14.18. MS(ESI-TOF) calculated for C
19H
15F
3N
2O
4 [M + H]
+ 393.18; found 394.16.
Ethyl 6-(bromomethyl)-4-{[4-(2,2,2-trifluoroacetamido)phenyl]ethynyl}picolinate (37). A mixture of compound 36 (2.10 g, 5.35 mmol) and PBr3 (0.75 mL, 8.03 mmol) in dry CHCl3 (180 mL) was stirred for 18 h at 55 °C, neutralized with aqueous 5% NaHCO3 (150 mL), the aqueous phase was extracted with CHCl3 (2 × 50 mL) and the combined organic phases were dried with Na2SO4. The product was purified by silica gel column chromatography using EtOH/CH2Cl2 (10:90) as an eluent. Yield: 2.01 g (84%). 1H-NMR (CDCl3, δ ppm): 8.25 (1H, s), 8.01 (1H, d, J = 1.1 Hz), 7.75 (1H, d, J = 1.1 Hz); 7.68 (2H, d, J = 8.7 Hz), 7.65 (2H, d, J = 8.7 Hz); 4.62 (2H, s), 4.50 (2H, q, J = 7.1 Hz), 1.45 (3H, t, J = 7.1 Hz). 13C-NMR (CDCl3, δ ppm): 164.32, 157.68, 155.16, 154.85, 154.55, 154.25, 148.06, 136.21, 133.59, 133.06, 128.36, 126.09, 120.26, 119.32, 118.92, 116.62, 114.32, 112.03, 94.61, 86.29, 62.25, 32.62, 14.20. MS(ESI-TOF) calculated for C19H14BrF3N2O3 [M + H]+ 455.02 and 457.02; found 455.78 and 457.73.
Compound 38. A mixture of compound 34 (2.02 g, 3.20 mmol), compound 37 (1.46 g, 3.20 mmol), dry K2CO3 (1.77 g, 12.8 mmol) in dry MeCN (70 mL) was stirred for 18 h at RT under argon. The mixture was filtrated and the filtrate evaporated to dryness. The product was purified by silica gel column chromatography using EtOH/CH2Cl2 (first 5:95, then 10:90) as an eluent. Yield: 3.03 g (94%). 1H-NMR (DMSO-d6, δ ppm): 11.46 (1H, s), 7.97, (1H, s), 7.90 (1H, s), 7.82–7.77 (4H, m), 7.65 (2H, d, J = 8.4 Hz), 7.58 (1H, s), 7.54 (1H, s), 4.35 (2H, q, J = 7.1 Hz), 4.32 (2H, q, J = 7.2 Hz), 4.07 (2H, q, J = 7.1 Hz), 4.06 (2H, q, J = 7.1 Hz), 4.01 (2H, s), 3.98 (2H, s), 3.90 (2 H, s), 3.89 (2H, s), 3.57 (2H, s), 3.53 (2H, s), 1.34 (3H, t, H = 7.1Hz),1.31 (3H, t, J = 7.2 Hz), 1.20 (3H, t, J = 7.1 Hz), 1.18 (3H, t, J = 7.1 Hz). 13C-NMR (DMSO-d6, δ ppm): 170.71, 170.69, 170.68, 170.56, 164.00, 163.38, 161.62, 160.41, 159.98, 159.81, 154.94, 154.64, 154.36, 154.03, 148.04, 147.33, 137.57, 135.94, 133.09, 132.78, 132.66, 132.62, 131.45, 128.42, 125.84, 120.81, 120.61, 118.07, 116.68, 114.39, 112.06, 93.81, 86.24, 61.43, 61.28, 61.14, 58.89, 58.19, 58,13, 58.94, 58.88, 55.32, 55.21, 14.07, 14.01, 13.96, 13.94. MS(ESI-TOF) calculated for C43H43Br2F3N6O9 [M + H]+ 1003.22, 1005.22 and 1007.22; found 1003.59, 1005.48 and 1007.40.
Ligand Ester 39. A mixture of compound 38 (0.23 g, 0.23 mmol), compound 15 (0.22 g, 0.55 mmol) in dry TEA (2 mL) and THF (4 mL) was de-aerated with argon. After an addition of bis(triphenyl-phosphine)palladium(II) dichloride (10 mg, 14 μmol) and CuI (6 mg, 28 μmol), the mixture was stirred overnight at 55 °C and evaporated to dryness. The product was purified by silica gel column chromatography using EtOH/CH2Cl2/TEA (10:89/1) as an eluent. Yield: 0.31 g (57%). 1H-NMR (DMSO-d6, δ ppm): 11.43 (1H, s), 7.90 (1H, s), 7.87 (1H, s), 7.83 (1H, s), 7.77 (1H, s), 7.71 (2H, d, J = 8.6 Hz), 7.60 (2H, d, J = 8.6 Hz), 7.38 (1H, s), 7.37 (1H, s), 4.87 (4H, s), 4.83 (4H, s), 4.82 (2H, s), 4.79 (2H, s), 4.32 (2H, q, J = 7.1 Hz), 4.31 (2H, q, J = 7.1 Hz), 4.18 (4H, q, J = 7.1 Hz), 4.17 (2H, s), 4.16 (4H, q, J = 7.1 Hz), 4.13 (4H, q, J = 7.1 Hz), 4.06 (2H, q, J = 7.2 Hz), 4.05 (2H, q, J = 7.2 Hz), 4.04 (2H, s), 3.96 (2H, s), 3.95 (2H, s), 3.54 (2H, s), 3.52 (2H, s), 1.31 (3H, t, J = 7.1 Hz), 1.30 (3H, t, J = 7.1 Hz), 1.22 (6H, t, J = 7.1 Hz), 1.20 (6H, t, J = 7.1 Hz), 1.18 (6H, t, J = 7.1 Hz), 1.17 (3H, t, J = 7.2 Hz), 1.16 (3H, t, J = 7.2 Hz). 13C-NMR (DMSO-d6, δ ppm): 171.27, 171.21, 168.60, 164.73, 164.62, 162.78, 161.07, 160.92, 160.66, 160.55, 158.80, 158.68, 155.53, 155.25, 154.96, 154.65, 147.81, 138.43, 133.45, 133.10, 132.74, 132.13, 127.72, 127.21, 124.93, 124.63, 123.14, 123.08, 121.35, 119.62, 118.07, 117.33, 115.04, 112.74, 94.65, 94.43, 94.29, 93.93, 93.88, 93.44, 93.42, 89.09, 87.53, 86,80, 65.83, 65.76, 65.46, 65.44, 61.82, 61.75, 61.19, 61.17, 61.14, 60.40, 60.35, 59.59, 59.53, 59.29, 59.25, 55.37, 55.02, 14.50, 14.48, 14.47, 14.46, 14.45, 14.44, 14.42. MS(ESI-TOF) calculated for C83H89F3N6O27 [M + H]+ 1659.58; found 1659.34.
Eu(III) Chelates 40. The compound 39 (0.28 g, 0.169 mmol) in 0.5 M KOH in EtOH (18.2 mL) was stirred for 30 min at RT, H2O (10 mL) was added and the mixture was further stirred at RT for 7 h. After evaporation of EtOH and an additional overnight stirring at RT, the pH was adjusted to 6.5 by addition of 6 M HCl. EuCl3 (68 mg, 0.186 μmol) in water (1.0 mL) was added within 5 min and the pH was maintained at 6.0–6.5 with suitable additions of solid NaHCO3. After stirring overnight at RT, the pH was adjusted to 8.5 with 1 M NaOH. The precipitate was removed by centrifugation and the supernatant evaporated to dryness. The product was purified by HPLC. Yield: 0.20 g (56%). Rf(HPLC): 15.2 min. UV: 358 nm.
Eu(III) Chelate 6. An aqueous solution (2.25 mL) of the Eu(III) chelate 40 (0.13 g, 61 μmol) was added within 5 min to a mixture of CSCl2 (64 µL, 0.85 mmol) and NaHCO3 (82 mg, 0.97 mmol) in CHCl3 (2.25 mL). After stirring for 15 min at RT, the two phases were separated and the aqueous phase was washed with CHCl3 (3 × 2.25 mL). The product was precipitated with acetone (ca. 45 mL), isolated by centrifugation, washed with acetone (2 × 10 mL) and dried overnight in vacuum desiccator over silica gel. Yield: 0.12 g. Rf(HPLC): 21.9 min. UV: 342 nm.
Eu(III) Chelate 6a. A mixture of Eu(III) chelate 6 (25 mg 11.7 μmol) and taurine (15 mg, 0.117 mmol) in aqueous 50 mM Na2CO3 buffer (0.88 mL, pH 9.8) was stirred for overnight at RT. The product was purified by HPLC. Rf(HPLC): 14.9 min. UV: 352 nm.
General Procedure for the Synthesis of Compounds 41 and 42. A mixture of compound
14 or
15 (1.00 mmol), and compound
35 (0.22 g, 0.84 mmol; [
28]) in dry TEA (5 mL) and THF (10 mL) was de-aerated with argon. After an addition of bis(triphenylphosphine)palladium(II) dichloride (10 mg, 14 μmol) and CuI (6 mg, 28 μmol), the mixture was stirred overnight at 55 °C and evaporated to dryness. The residue was dissolved in CH
2Cl
2 (30 mL) washed with H
2O (3 × 10 mL), dried with Na
2SO
4 and evaporated to dryness. The product was purified by silica gel column chromatography.
Diethyl 2,2′-{{4-[(2-(ethoxycarbonyl)-6-(hydroxymethyl)pyridin-4-yl]ethynyl)-1,3-phenylene}bis-(oxy)}-di-acetate (41). The product was purified by silica gel column chromatography using MeOH/CH2Cl2 (5:95) as an eluent. Yield: 80%. 1H-NMR (CDCl3, δ ppm): 8.08 (2H, s), 7.60 (2H, s), 7.45 (1H, d, J = 8.5 Hz), 6.50 (1H, dd, J = 2.0 and 8.5 Hz), 6.45 (1H, d, J = 2.0 Hz), 4.85 (2H, s), 4.71 (2H, s), 4.63 (2H, s), 4.47 (2H, q, J = 7.1 Hz), 4.30 (2H, q, J = 7.1 Hz), 4.29 (2H, q, J = 7.1 Hz), 1.44 (3H, t, J = 7.1 Hz), 1.32 (3H, t, J = 7.1 Hz), 1.31 (3H, t, J = 7.1 Hz). 13C-NMR (CDCl3, δ ppm): 168.11, 168.00, 164.63, 161.71, 160.08, 159.95, 147.27, 134.81, 127.02, 125.50, 106.42, 105.38, 100.99, 91.30, 89.67, 65.91, 65.34, 64.32, 62.24, 61.93, 61.54, 14.20, 14.19, 14.07. MS(ESI-TOF) calculated for C25H27NO9[M + H]+ 486.18; found 486.46.
Triethyl 2,2′,2′′-{{2-{[2-(ethoxycarbonyl)-6-(hydroxymethyl)pyridin-4-yl]ethynyl)}benzene-1,3,5-triyl}tris-(oxy)}triacetate (42). The product was purified by silica gel column chromatography using EtOAc/petroleum ether/TEA (from 69:30:1 to 89:10:1) as an eluent. Yield: 76%. 1H-NMR (CDCl3, δ ppm): 8.11 (1H, d, J = 0.5 Hz), 7.64 (1H, d, J = 0.5 Hz), 6.09 (2H, s), 4.84 (2 H, s), 4.70 (4H, s), 4.58 (2H, s), 4.47 (2H, q, J = 7.1 Hz), 4.29 (4H, q, J = 7.1 Hz), 4.28 (2 H, q, J = 7.1 Hz), 3.45 (1 H, bs), 1.44 (3H, t, J = 7.1 Hz), 1.32 (3H, t, J = 7.1 Hz), 1.31 (6H, t, J = 7.1 Hz). 13C-NMR (CDCl3, δ ppm): 167.97, 167.89, 164.70, 161.04, 160.22, 158.87, 147.21, 134.13, 125.42, 125.17, 96.17, 94.27, 93.80, 87.78, 66.21, 65.42, 64.29, 61.86, 61.61, 61.49, 14.20, 14.08, 14.06. MS(ESI-TOF) calculated for C29H33NO12[M + H]+ 588.56; found 589.03.
General Procedure for the Synthesis of Compounds 43 and 44. A mixture of compound 41 or 42 (0.613 mmol) and PBr3 (86 μL, 0.919 mmol) in dry CHCl3 (20 mL) was stirred for 2.5 h at RT, neutralized with aqueous 5% NaHCO3 (20 mL), the aqueous phase was extracted with CHCl3 (2 × 20 mL) and the combined organic phases were dried with Na2SO4. The product was purified by silica gel column chromatography.
Diethyl 2,2′-{{4-{[2-(bromomethyl)-6-(ethoxycarbonyl)pyridin-4-yl]ethynyl}-1,3-phenylene}bis-(oxy)}diacetate (43). The product was purified by silica gel column chromatography using EtOH/CH2Cl2 (10:90) as an eluent. Yield: 89%. 1H-NMR (CDCl3, δ ppm): 8.10 (1H, d, J = 1.2 Hz), 7.75 (1H, d, J = 1.2 Hz), 7.46 (1H, d, J = 8.5 Hz), 6.50 (1H, dd, J = 2.3 and 8.5 Hz), 6.46 (1H, d, J = 2.3 Hz), 4.72 (2H, s), 4.63 (2H, s), 4.60 (2H, s), 4.49 (2H, q, J = 7.1 Hz), 4.30 (2H, q, J = 7.1 Hz), 4.29 (2H, q, J = 7.1 Hz), 1.45 (3H, t, J = 7.1 Hz), 1.32 (3H, t, J = 7.1 Hz), 1.31 (3H, t, J = 7.1 Hz). 13C-NMR (CDCl3, δ ppm): 168.10, 167.97, 164.43, 160.05, 160.03, 157.41, 147.95, 134.88, 127.65, 126.00, 106.42, 105.28, 100.97, 91.97, 89.34, 65.92, 65.35, 62.40, 62.09, 61.54, 61.49, 32.88, 14.22, 14.09, 14.07. MS(ESI-TOF) calculated for C25H26BrNO8[M + H]+ 548.09 and 550.09; found 548.83 and 550.80.
Triethyl 2,2′,2′′-{{2-{[2-(bromomethyl)-6-(ethoxycarbonyl)pyridin-4-yl]ethynyl}benzene-1,3,5-triyl}-tris(oxy)}-triacetate (44). The product was purified by silica gel column chromatography using EtOH/CH2Cl2 (10:90) as an eluent using EtOH/CH2Cl2 (10:90) as an eluent. Yield: 82%. 1H-NMR (CDCl3, δ ppm): 8.12 (1H, d, J = 1.3 Hz), 7.79 (1H, d, J = 1.3 Hz), 6.08 (2H, s), 4.71 (4H, s), 4.59 (2H, s), 4.51 (2H, s), 4.48 (2H, q, J = 7.1 Hz), 4.30 (4H, q, J = 7.1 Hz), 4.29 (2H, q, J = 7.1 Hz), 1.43 (3H, t, J = 7.1 Hz), 1.31 (9 H, t, J = 7.1 Hz). 13C-NMR (CDCl3, δ ppm): 167.92, 167.79, 164.52, 156.29, 133.27, 125.98, 125.08, 96.09, 93.92, 93.76, 88.21, 66.22, 65.59, 65.43, 62.04, 61.62, 61.51, 32.96, 14.22, 14.07, 14.03. MS(ESI-TOF) calculated for C29H32BrNO11[M + H]+ 650.13 and 652.13; found 651.08 and, 653.02.
Compound 45. A mixture of compound 37 (0.41 g, 0.90 mmol), diBoc-TACN (0.14 g, 0.82 mmol), dry K2CO3 (0.23 g, 1.62 mmol) and dry MeCN (8 mL) was stirred for 24 h at RT. After filtration and washing the solid material with CH2Cl2, the filtrate was evaporated to dryness. The product was purified by silica gel column chromatography using EtOH/CH2Cl2 (from 1:99 to 3:97) as an eluent. Yield: 0.31 g (53%). 1H-NMR (DMSO-d6, δ ppm): 11.48 (1H, s), 7.97 (1 H, s), 7.78–7.85 (3H, m), 7.66 (2H, d, J = 8.3 Hz), 4.38 (2H, q, J = 7.1 Hz); 3.80–3.85 (2H, m), 3.10–3.45 (8H, m), 2.65–2.75 (2H, m), 2.65–2.55 (2H, m), 1.43 (3H, s), 1.42 (3H, s), 1.40 (6H, s), 1.39 (6H, s), 1.34 (3 H, t, J = 7.1 Hz). 13C-NMR (DMSO-d6, δ ppm): 164.09, 155.78, 154.96, 154.80, 154.70, 154.56, 154.37, 154.08, 147.22, 137.51, 132.74, 132.54, 129.33, 127.03, 124.69, 120.85, 118.97, 116.69, 114.39, 112.62, 93.89, 86.35, 78.71, 61.44, 61.29, 51.42, 50.18, 49,69, 28.03, 14.02. Both spectra indicate the existence of rigid compound having different structural isomers. MS(ESI-TOF) calculated for C35H44F3N5O7[M + H]+ 704.33; found 705.09.
Compound 46. A mixture of compound 45 (0.29 g, 0.41 mmol) and TFA (2 mL) was stirred for 2 h at RT, evaporated to dryness and triturated with Et2O (40 mL). The product (0.34 g, 89%) was centrifuged, washed with Et2O (2 × 15 mL) and dried. 1H-NMR (DMSO-d6, δ ppm): 11.54 (1H, s), 7.82 (2 H, d, J = 8.4 Hz), 7.81 (1 H, s), 7.80 (1H, s), 7.71 (2 H, d, J = 8.4 Hz), 4.44 (2H, q, J = 7.0 Hz), 4.17 (2H, s), 3.69 (4H, bs), 3,26 (4H, bs), 2.97 (4H, bs), 1.39 (3H, t, J = 7.0 Hz). 13C-NMR (DMSO-d6, δ ppm): 154.41, 160.60, 155.48, 155.18, 154.89, 154.59, 147.17, 138.30, 133.24, 133.06, 129.87, 128.40, 125.22, 121.41, 118,57, 116.19, 114.84, 112.54, 95.54, 86.36, 62.47, 57.68, 50.42, 45.90, 45.36, 14,45. MS(ESI-TOF): calculated C25H28F3N5O3 [M + 2H]+ 505.54; found 505.31.
General Procedure for the Synthesis of Ligand Esters 47 and 48. A mixture of compound 46 (0.12 g, 0.15 mmol), 43 or 44 (0.32 mmol), di-isopropylethylamine (0.40 mL) and dry MeCN (3.0 mL) was stirred for 5.5 h at RT and evaporated to dryness. The product was purified by silica gel column chromatography.
Ligand Ester 47. The product was purified by silica gel column chromatography using EtOH/CH2Cl2/TEA (from 10:90:0 to 10:89:1) as an eluent. Yield: 84%. As the product contains 2–3 rigid isomers, the NMR spectra were too complicated to assign the isomers. MS(ESI-TOF) calculated for C75H78F3N7O19 [M + H]+ 1438.54; found 1439.41.
Ligand Ester 48. The product was purified by silica gel column chromatography using EtOH/CH2Cl2/TEA (from 10:90:0 to 15:80:5) as an eluent. Yield: 79%. As the product contains 2–3 rigid isomers, the NMR spectra were too complicated to assign the isomers. MS(ESI-TOF): calculated [M + H]+1642.60; found 1643.57.
General Procedure for the Synthesis of Eu(III) Chelates 49 and 50. A mixture of the compound 47 or 48 (64 μmol) and 0.5 M KOH in EtOH (6.5 mL) was stirred for 1 h at RT and H2O (3 mL) was added. After stirring for 4 h at RT, EtOH was evaporated, some H2O (2 mL) added and the residue was stirred for 24 h at RT. After addition of citric acid (41 mg, 0.21 mmol) in H2O (0.25 mL), the pH was adjusted to ca. 6.5 with 6 M HCl. Europium(III) chloride (26 mg, 71 μmol) in H2O (0.25 mL) was added within 10 min and the pH was adjusted to ca. 9.5 with 1 M NaOH. The mixture was stirred for 4–6 weeks at 95 °C (after the analytical HPLC chromatogram showed completed complexation), the pH was adjusted to ca. 7.0 with 1 M HCl, evaporated to dryness, dissolved in 20 mmol Triethylammonium acetate (TEAA) buffer (1 mL) and purified with HPLC.
Eu(III) Chelate 49. Rf(HPLC) = 19.2 min. UV: 350 nm. MS(ESI-TOF) calculated for C59H48EuN7O18 [M + H]+ 1296.24; found 1295.85. Ligand isomers shown in the HPLC during the Eu(III) loading: (1) Rf(HPLC) = 21.7 min, UV: 347 nm; (2) Rf(HPLC) = 22.3 min, UV: 339 nm; (3) Rf(HPLC) = 23.6 min, UV: 343 nm. All of these peaks finally gave the product peak at Rf(HPLC) = 19.2 min. UV: 350 nm.
Eu(III) Chelate 50. Rf(HPLC) = 16.0 min. UV: 360 nm MS(ESI-TOF) calculated for C63H62EuN7O24 [M + H]+ 1444.24; found 1443.96. Ligand isomers shown in HPLC during the Eu(III) loading: (1) Rf(HPLC) = 18.5 min, UV: 345 nm; (2) Rf(HPLC) = 20.4 min, UV: 347 nm; (3) Rf(HPLC) = 21.7 min, UV: 347 nm. All of these peaks finally gave the product peak at Rf = 16.0 min, UV = 360 nm.
The Eu complex formation caused the observed bathochromic shift of 13–15 nm at UV. This was separately secured with the complex 50 by an additional HPLC purification of the ligand isomers and loadings of Eu(III) ion to each isomers. All of these tests gave finally the same product at Rf(HPLC) = 16.0 min.
General Procedure for the Synthesis of Eu(III) Chelates 7 and 8. An aqueous solution (1 mL) of the chelate 49 or 50 (21 μmol) was added within 5 min to a mixture of CSCl2 (22 µL, 0.29 mmol) and NaHCO3 (28 mg, 0.33 mmol) and CHCl3 (1 mL). After stirring for 40 min at RT, the two phases were separated and the aqueous phase was washed with CHCl3 (3 × 1 mL). The product was precipitated with acetone (ca. 45 mL), isolated by centrifugation, washed with acetone (2 × 10 mL), and dried overnight in a vacuum desiccator. The products were used as such for next phase or for labeling the antibodies.
Eu(III) Chelate 7. Rf(HPLC) = 19.9 min. UV: 350 nm.
Eu(III) Chelate 8. Rf(HPLC) = 17.6 min. UV: 356 nm.
General Procedure for the Synthesis of Eu(III) Chelates 7a and 8a. A mixture of chelate 7 or 8 (2 mg) and taurine (2 mg) in 50 mM Na2CO3 buffer (300 μL, pH 9.8) was stirred overnight at RT. The product was purified by using HPLC.
Eu(III) Chelate 7a. Rf(HPLC) = 17.2 min. UV = 348 nm.
Eu(III) Chelate 8a. Rf(HPLC) = 15.2 min. UV = 354 nm.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}