3. Materials and Methods
General remarks: All reagents were obtained from Aladdin Reagent Shanghai Co., Ltd. (Shanghai, China), Lagewell Technology Co., Ltd., Meyer Reagent Shanghai Co., Ltd., Macklin Reagent Shanghai Co., Ltd., Chongqing Chuandong Chemical Co., Ltd. etc. without further purification unless otherwise noted. High resolution mass spectra (South China Agricultural University, Guangzhou) were measured on commercial instruments. NMR spectra were recorded on commercial instruments (Bruker company, Karlsruhe, Germany) and operated at 600 MHz for 1H-NMR and 151 MHz for 13C-NMR. Chemical shifts were reported in ppm from tetramethylsilane with the solvent resonance as the internal standard ((CD3)2SO, δ = 2.50, δ = 3.33) in 1H-NMR spectra and chemical shifts were reported in ppm from the tetramethylsilane with the solvent resonance as internal standard ((CD3)2SO, δ = 39.5) in 13C-NMR spectra. Spectra are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constants (Hz), integration and assignment.
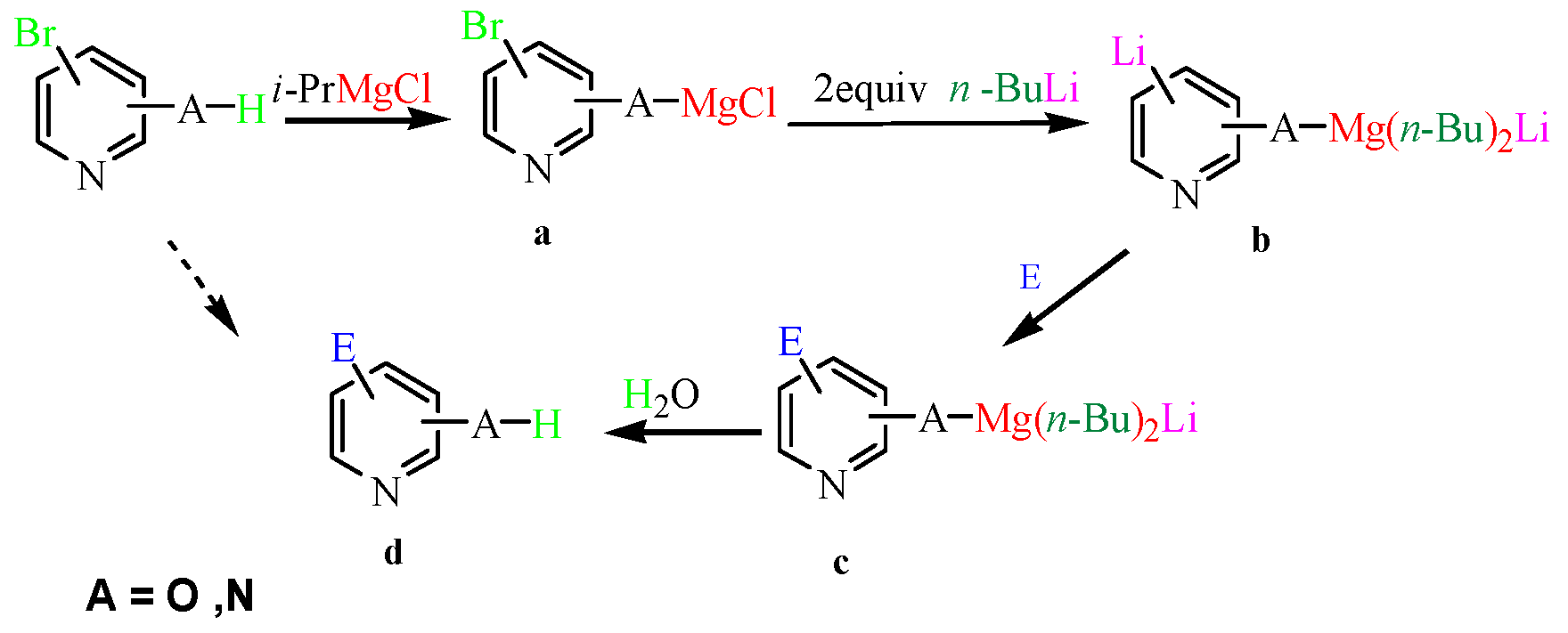
(5-Formyl-pyridin-2-yl)-carbamic acid tert-butyl ester (3a): To a solution of (5-Bromo-pyridin-2-yl)-carbamic acid tert-butyl ester (1.0 g, 3.7 mmol, 1.0 equiv.) in dry THF (12 mL) at 0 °C was added a 2 M solution of i-PrMgCl in THF (1.85 mL, 3.7 mmol, 1.0 equiv.) during 5 min. The clear solution was stirred at that temperature for an additional 5 min, and a 2.5 M solution of n-BuLi in hexanes (3 mL, 7.5 mmol, 2.0 equiv.) was added dropwise during 5 min, while maintaining the temperature below −20 °C. The resulting mixture was stirred at that temperature for 0.5 h, dry DMF (0.27 g, 3.7 mmol, 1.0 equiv.) in dry THF (5 mL) was added dropwise during 10 min. The resulting mixture was warmed to −20 °C in 0.5 h and quenched with water (6 mL). After stirring the mixture below −20 °C for 10 min, the phases were separated and the water phase was extracted one additional time with ethyl acetate. The resulting suspension was allowed to reach room temperature and fitered through a 0.5 × 1 cm pad of silica gel eluted with 10 mL of ethyl acetate. The filtrate was concentrated and the residue was purified by flash chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 10:1) to afford product 3a as white solid, 0.73 g (yield: 90%). 1H-NMR (600 MHz, DMSO) δ 10.42 (s, 1H), 9.94 (s, 1H), 8.93–8.60 (m, 1H), 8.17 (dd, J = 8.8, 2.3 Hz, 1H), 7.99 (d, J = 8.8 Hz, 1H), 1.48 (s, 9H). 13C-NMR (151 MHz, DMSO) δ 191.18, 157.08, 152.82, 152.40, 138.23, 127.30, 112.23, 80.97, 28.37.
1H-Indole-2-carboxylic acid (3b): To a solution of 2-Bromo-1H-indole (1.0 g, 5 mmol, 1.0 equiv.) in dry THF (20 mL) at 0 °C was added a 2 M solution of i-PrMgCl in THF (2.5 mL, 5 mmol, 1.0 equiv.) during 5 min. The clear solution was stirred at that temperature for an additional 5 min, and a 2.5 M solution of nBuLi in hexanes (4 mL, 10 mmol, 2.0 equiv.) was added dropwise during 5 min, while maintaining the temperature below −20 °C. The resulting mixture was stirred at that temperature for 0.5 h, dry CO2 (0.22 g, 5 mmol, 1.0 equiv.) was added to −20 °C. The resulting mixture was warmed to −20 °C in 0.5 h and quenched with water (6 mL). After stirring the mixture below −20 °C for 10 min, the phases were separated and the water phase was extracted one additional time with ethyl acetate. The resulting suspension was allowed to reach room temperature and fitered through a 0.5 × 1 cm pad of silica gel eluted with 10 mL of ethyl acetate. The filtrate was concentrated and the residue was purified by flash chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 3:1) to afford product 3b as white solid, 0.7 g (yield: 85%), m.p.: 203–204 °C. 1H-NMR (600 MHz, DMSO) δ 12.94 (s, 1H), 11.76 (s, 1H), 7.79–6.89 (m, 5H). 13C-NMR (151 MHz, DMSO) δ 163.29, 137.71, 128.88, 127.34, 124.73, 122.40, 120.41, 112.95, 107.77.
1H-Indole-3-carboxylic acid (3c): To a solution of 3-Bromo-1H-indole (0.86 g, 4.4 mmol, 1.0 equiv.) in dry THF (20 mL) at 0 °C was added a 2 M solution of i-PrMgCl in THF (2.2 mL, 4.4 mmol 1.0 equiv.) during 5 min. The clear solution was stirred at that temperature for an additional 5 min, and a 2.5 M solution of n-BuLi in hexanes (3.5 mL, 8.8 mmol, 2.0 equiv.) was added dropwise during 5 min, while maintaining the temperature below −20 °C. The resulting mixture was stirred at that temperature for 0.5 h, dry CO2 (0.2 g, 4.4 mmol, 1.0 equiv.) was added to −20 °C. The resulting mixture was warmed to −20 °C in 0.5 h and quenched with water (6 mL). After stirring the mixture below −20 °C for 10 min, the phases were separated and the water phase was extracted one additional time with ethyl acetate. The resulting suspension was allowed to reach room temperature and fitered through a 0.5 × 1 cm pad of silica gel eluted with 10 mL of ethyl acetate. The filtrate was concentrated and the residue was purified by flash chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 3:1) to afford product 3c as off-white solid, 0.63 g (yield: 89%), m.p.: 193–196 °C. 1H-NMR (600 MHz, DMSO) δ 11.82 (s, 1H), 8.02 (t, J = 5.7 Hz, 2H), 7.47 (d, J = 7.6 Hz, 1H), 7.32–6.99 (m, 2H). 13C-NMR (151 MHz, DMSO) δ 166.43, 136.89, 132.71, 126.48, 122.58, 121.42, 121.05, 112.65, 107.87.
1H-Indole-5-carboxylic acid (3d): To a solution of 5-Bromo-1H-indole (0.86 g, 4.4 mmol, 1.0 equiv.) in dry THF (20 mL) at 0 °C was added a 2 M solution of i-PrMgCl in THF (2.2 mL, 4.4 mmol, 1 equiv.) during 5 min. The clear solution was stirred at that temperature for an additional 5 min, and a 2.5 M solution of n-BuLi in hexanes (3.5 mL, 8.8 mmol, 2.0 equiv.) was added dropwise during 5 min, while maintaining the temperature below −20 °C. The resulting mixture was stirred at that temperature for 0.5 h, dry CO2 (0.2 g, 4.4 mmol, 1.0 equiv.) was added to −20 °C. The resulting mixture was warmed to −20 °C in 0.5 h and quenched with water (6 mL). After stirring the mixture below −20 °C for 10 min, the phases were separated and the water phase was extracted one additional time with ethyl acetate. The resulting suspension was allowed to reach room temperature and fitered through a 0.5 × 1 cm pad of silica gel eluted with 10 mL of ethyl acetate. The filtrate was concentrated and the residue was purified by flash chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 3:1) to afford product 3d as off-white solid, 0.46 g (yield: 65%), m.p.: 210–214 °C. 1H-NMR (600 MHz, DMSO) δ 12.39 (s, 1H), 11.46 (s, 1H), 8.25 (s, 1H), 7.72 (dd, J = 8.5, 1.5 Hz, 1H), 7.45 (dd, J = 8.4, 5.7 Hz, 2H), 6.57 (s, 1H). 13C-NMR (151 MHz, DMSO) δ 168.90, 138.80, 127.64, 127.35, 123.28, 122.67, 121.87, 111.57, 102.93.
5-Bromo-1H-indole-3-carbaldehyde (3e): To a solution of 3,5-dibromo-1H-indole (1.2 g, 4.4 mmol, 1.0 equiv.) in dry THF (20 mL) at 0 °C was added a 2 M solution of iPrMgCl in THF (2.2 mL, 4.4 mmol, 1.0 equiv.) during 5 min. The clear solution was stirred at that temperature for an additional 5 min, and a 2.5 M solution of n-BuLi in hexanes (3.5 mL, 8.8 mmol, 2.0 equiv.) was added dropwise during 5 min, while maintaining the temperature below −20 °C. The resulting mixture was stirred at that temperature for 0.5 h, dry DMF (0.32 g, 4.4 mmol, 1.0 equiv.) was added to −20 °C. The resulting mixture was warmed to −20 °C in 0.5 h and quenched with water (6 mL). After stirring the mixture below −20 °C for 10 min, the phases were separated and the water phase was extracted one additional time with ethyl acetate. The resulting suspension was allowed to reach room temperature and fitered through a 0.5 × 1 cm pad of silica gel eluted with 10 mL of ethyl acetate. The filtrate was concentrated and the residue was purified by flash chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 3:1) to afford product 3e as yellow solid, 0.78 g (yield: 80%), m.p.: 192–194 °C. 1H-NMR (600 MHz, DMSO) δ 12.35 (s, 1H), 9.93 (s, 1H), 8.47–8.05 (m, 2H), 7.57–7.21 (m, 2H). 13C-NMR (151 MHz, DMSO) δ 185.57, 139.67, 136.23, 126.49, 126.36, 123.39, 117.90, 115.27, 115.01.
5-Methoxy-1H-indole-2-carboxylic acid (3f): To a solution of 2-Bromo-5-methoxy-1H-indole (1.0 g, 4.4 mmol, 1.0 equiv.) in dry THF (20 mL) at 0 °C was added a 2 M solution of i-PrMgCl in THF (2.2 mL, 4.4 mmol, 1.0 equiv.) during 5 min. The clear solution was stirred at that temperature for an additional 5 min, and a 2.5 M solution of n-BuLi in hexanes (3.5 mL, 8.8 mmol, 2.0 equiv.) was added dropwise during 5 min, while maintaining the temperature below −20 °C. The resulting mixture was stirred at that temperature for 0.5 h, dry CO2 (0.20 g, 1.0 equiv.) was added to −20 °C. The resulting mixture was warmed to −20 °C in 0.5 h and quenched with water (6 mL). After stirring the mixture below −20 °C for 10 min, the phases were separated and the water phase was extracted one additional time with ethyl acetate. The resulting suspension was allowed to reach room temperature and fitered through a 0.5 × 1 cm pad of silica gel eluted with 10 mL of ethyl acetate. The filtrate was concentrated and the residue was purified by flash chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 3:1) to afford product 3f as brown solid, 0.68 g (yield: 80%), m.p.: 199–201 °C. 1H-NMR (600 MHz, DMSO) δ 7.36 (d, J = 8.9 Hz, 1H), 7.06 (d, J = 23.6 Hz, 2H), 6.90 (d, J = 8.8 Hz, 1H), 3.73 (s, 3H). 13C-NMR (151 MHz, DMSO) δ 163.25, 154.31, 133.07, 129.10, 127.65, 116.28, 113.83, 107.47, 102.44, 55.61.
1H-Benzoimidazole-5-carboxylic acid (3g): To a solution of 5-Bromo-1H-benzimidazole (0.87 g, 4.4 mmol, 1.0 equiv.) in dry THF (20 mL) at 0 °C was added a 2 M solution of i-PrMgCl in THF (2.2 mL, 1 equiv.) during 5 min. The clear solution was stirred at that temperature for an additional 5 min, and a 2.5 M solution of n-BuLi in hexanes (3.5 mL, 8.8 mmol, 2.0 equiv.) was added dropwise during 5 min, while maintaining the temperature below −20 °C. The resulting mixture was stirred at that temperature for 0.5 h, dry CO2 (0.20 g, 4.4mmol, 1.0 equiv.) was added to −20 °C. The resulting mixture was warmed to −20 °C in 0.5 h and quenched with water (6 mL). After stirring the mixture below −20 °C for 10 min, the phases were separated and the water phase was extracted one additional time with ethyl acetate. The resulting suspension was allowed to reach room temperature and fitered through a 0.5 × 1 cm pad of silica gel eluted with 10 mL of ethyl acetate. The filtrate was concentrated and the residue was purified by flash chromatography on silica gel (eluent:petroleum ether/ethyl acetate = 3:1) to afford product 3g as brown solid, 0.5 g (yield: 71%). 1H-NMR (600 MHz, DMSO) δ 8.43 (s, 1H), 8.26 (s, 1H), 7.87 (d, J = 8.4 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H). 13C-NMR (151 MHz, DMSO) δ 168.47, 144.83, 125.10, 123.74, 118.13, 115.12.
4-Hydroxy-benzaldehyde (3h): To a solution of 4-Bromo-phenol (1.5 g, 8.7 mmol, 1 equiv.) in dry THF (25 mL) at 0 °C was added a 2 M solution of i-PrMgCl in THF (4.3 mL, 8.7 mmol, 1.0 equiv.) during 5 min. The clear solution was stirred at that temperature for an additional 5 min, and a 2.5 M solution of n-BuLi in hexanes (7.0 mL, 17.4 mmol, 2.0 equiv.) was added dropwise during 5 min, while maintaining the temperature below −20 °C. The resulting mixture was stirred at that temperature for 0.5 h, dry DMF (0.63 g, 8.7 mmol, 1.0 equiv.) in dry THF (5 mL) was added dropwise during 10 min. The resulting mixture was warmed to −20 °C in 0.5 h and quenched with water (6 mL). After stirring the mixture below −20 °C for 10 min, the phases were separated and the water phase was extracted one additional time with ethyl acetate. The resulting suspension was allowed to reach room temperature and fitered through a 0.5 × 1 cm pad of silica gel eluted with 10 mL of ethyl acetate. The filtrate was concentrated and the residue was purified by flash chromatography on silica gel (eluent:petroleum ether/ethyl acetate = 5:1) to afford product 3h as white solid, 0.94 g (yield: 90%), m.p.: 114–116 °C. 1H-NMR (600 MHz, DMSO) δ 10.59 (s, 1H), 9.78 (s, 1H), 7.75 (d, J = 8.6 Hz, 2H), 6.93 (d, J = 8.6 Hz, 2H). 13C-NMR (151 MHz, DMSO) δ 191.35, 191.33, 163.76, 132.53, 128.89, 116.29.
N-(4-Formyl-phenyl)-acetamide (3i): To a solution of 4-Bromo-phen N-(4-Formyl -phenyl)-acetamide (1.5 g, 7.0 mmol, 1.0 equiv.) in dry THF (25 mL) at 0 °C was added a 2 M solution of iPrMgCl in THF (3.5 mL, 7.0 mmol, 1.0 equiv.) during 5 min. The clear solution was stirred at that temperature for an additional 5 min, and a 2.5 M solution of n-BuLi in hexanes (5.6 mL, 14.0 mmol, 2.0 equiv.) was added dropwise during 5 min, while maintaining the temperature below −20 °C. The resulting mixture was stirred at that temperature for 0.5 h, dry DMF (0.5 g, 1.0 equiv.) in dry THF (5 mL) was added dropwise during 10 min. The resulting mixture was warmed to −20 °C in 0.5 h and quenched with water (6 mL). After stirring the mixture below −20 °C for 10 min, the phases were separated and the water phase was extracted one additional time with ethyl acetate. The resulting suspension was allowed to reach room temperature and fitered through a 0.5 × 1 cm pad of silica gel eluted with 10 mL of ethyl acetate. The filtrate was concentrated and the residue was purified by flash chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 10:1) to afford product 3i as white solid, 1.0 g (yield: 94%) , m.p.: 157–158 °C. 1H-NMR (600 MHz, DMSO) δ 10.35 (s, 1H), 9.86 (s, 1H), 7.81 (dd, J = 32.9, 8.6 Hz, 4H), 2.10 (s, 3H). 13C-NMR (151 MHz, DMSO) δ 191.92, 169.55, 145.29, 131.56, 131.27, 119.01, 24.68.
1H-Pyrrole-2-carbaldehyde (3j): To a solution of 2-Bromo-1H-pyrrole (0.5 g, 3.4 mmol, 1.0 equiv.) in dry THF (15 mL) at 0 °C was added a 2 M solution of i-PrMgCl in THF (1.7 mL, 3.4 mmol, 1.0 equiv.) during 5 min. The clear solution was stirred at that temperature for an additional 5 min, and a 2.5 M solution of n-BuLi in hexanes (2.7 mL, 6.8 mmol, 2.0 equiv.) was added dropwise during 5 min, while maintaining the temperature below −20 °C. The resulting mixture was stirred at that temperature for 0.5 h, dry DMF (0.25 g, 1.0 equiv.) in dry THF (5 mL) was added dropwise during 10 min. The resulting mixture was warmed to −20 °C in 0.5 h and quenched with water (6 mL). After stirring the mixture below −20 °C for 10 min, the phases were separated and the water phase was extracted one additional time with ethyl acetate. The resulting suspension was allowed to reach room temperature and fitered through a 0.5 × 1 cm pad of silica gel eluted with 10 mL of ethyl acetate. The filtrate was concentrated and the residue was purified by flash chromatography on silica gel (eluent:petroleum ether/ethyl acetate = 10:1) to afford product 3j as white solid, 0.29 g (yield: 89%) , m.p.: 42–44 °C. 1H-NMR (600 MHz, DMSO) δ 12.19 (s, 1H), 11.69 (s, 1H), 7.04–6.85 (m, 1H), 6.72 (dd, J = 4.2, 2.9 Hz, 1H), 6.12 (dd, J = 5.8, 2.4 Hz, 1H). 13C-NMR (151 MHz, DMSO) δ 162.31, 123.82, 123.35, 115.11, 109.73.
1H-Imidazole-2-carbaldehyde (3k): To a solution of 2-bromo-1H-imidazole (0.65 g, 4.4 mmol, 1.0 equiv.) in dry THF (20 mL) at 0 °C was added a 2 M solution of i-PrMgCl in THF (2.2 mL, 4.4 mmol, 1.0 equiv.) during 5 min. The clear solution was stirred at that temperature for an additional 5 min, and a 2.5 M solution of n-BuLi in hexanes (3.5 mL, 8.8 mmol, 2.0 equiv.) was added dropwise during 5 min, while maintaining the temperature below −20 °C. The resulting mixture was stirred at that temperature for 0.5 h, dry DMF (0.32 g, 4.4 mmol, 1.0 equiv.) was added to −20 °C. The resulting mixture was warmed to −20 °C in 0.5 h and quenched with water (6 mL). After stirring the mixture below −20 °C for 10 min, the phases were separated and the water phase was extracted one additional time with ethyl acetate. The resulting suspension was allowed to reach room temperature and fitered through a 0.5 × 1cm pad of silica gel eluted with 10 mL of ethyl acetate. The filtrate was concentrated and the residue was purified by flash chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 10:1) to afford product 3k as pale yellow solid, 0.38 g (yield: 91%), m.p.: 205–206 °C. 1H-NMR (600 MHz, DMSO) δ 13.60 (s, 1H), 9.64 (s, 1H), 7.42 (s, 2H). 13C-NMR (151 MHz, DMSO) δ 181.66, 146.09.
1H-Imidazole-4-carbaldehyde (3l): To a solution of 4-bromo-1H-imidazole (0.65 g, 4.4 mmol, 1.0 equiv.) in dry THF (20 mL) at 0 °C was added a 2 M solution of i-PrMgCl in THF (2.2 mL, 4.4 mmol, 1.0 equiv.) during 5 min. The clear solution was stirred at that temperature for an additional 5 min, and a 2.5 M solution of n-BuLi in hexanes (3.5 mL, 8.8 mmol, 2.0 equiv.) was added dropwise during 5 min, while maintaining the temperature below −20 °C. The resulting mixture was stirred at that temperature for 0.5 h, dry DMF (0.32 g, 4.4 mmol, 1.0 equiv.) was added to −20 °C. The resulting mixture was warmed to −20 °C in 0.5 h and quenched with water (6 mL). After stirring the mixture below −20 °C for 10 min, the phases were separated and the water phase was extracted one additional time with ethyl acetate. The resulting suspension was allowed to reach room temperature and fitered through a 0.5 × 1 cm pad of silica gel eluted with 10 mL of ethyl acetate. The filtrate was concentrated and the residue was purified by flash chromatography on silica gel (eluent:petroleum ether/ethyl acetate = 10:1) to afford product 3l as off-white solid, 0.36 g (yield: 85%), m.p.: 175–177 °C. 1H-NMR (600 MHz, DMSO) δ 9.74 (s, 1H), 7.99 (s, 1H), 7.94 (s, 1H). 13C-NMR (151 MHz, DMSO) δ 184.46, 139.44, 134.9, 129.5.
6-Hydroxy-pyridine-2-carboxylic acid (3m): To a solution of 2-bromo-6-hydroxypyridine (0.76 g, 4.4 mmol, 1.0 equiv.) in dry THF (20 mL) at 0 °C was added a 2 M solution of -PrMgCl in THF (2.2 mL, 4.4 mmol, 1.0 equiv.) during 5 min. The clear solution was stirred at that temperature for an additional 5 min, and a 2.5 M solution of n-BuLi in hexanes (3.5 mL, 8.8 mmol, 2.0 equiv.) was added dropwise during 5min, while maintaining the temperature below −20 °C. The resulting mixture was stirred at that temperature for 0.5 h, dry CO2 (0.20 g, 1.0 equiv.) was added to −20 °C. The resulting mixture was warmed to −20 °C in 0.5 h and quenched with water (6 mL). After stirring the mixture below −20 °C for 10 min, the phases were separated and the water phase was extracted one additional time with ethyl acetate. The resulting suspension was allowed to reach room temperature and fitered through a 0.5 × 1 cm pad of silica gel eluted with 10 mL of ethyl acetate. The filtrate was concentrated and the residue was purified by flash chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 10:1) to afford product 3m as off-white solid, 0.56 g (yield: 93%) , m.p.: 275–277 °C. 1H-NMR (600 MHz, DMSO) δ 7.56 (dd, J = 8.9, 7.0 Hz, 1H), 6.97 (d, J = 6.8 Hz, 1H), 6.65 (d, J = 9.0 Hz, 1H). 13C-NMR (151 MHz, DMSO) δ 163.28, 162.67, 140.51, 137.97, 123.88, 110.42.
2-Hydroxy-nicotinic acid (3n): To a solution of 3-bromo-2-hydroxypyridine (0.76 g, 4.4 mmol, 1.0 equiv.) in dry THF (20 mL) at 0 °C was added a 2 M solution of i-PrMgCl in THF (2.2 mL, 4.4 mmol, 1.0 equiv.) during 5 min. The clear solution was stirred at that temperature for an additional 5 min, and a 2.5 M solution of n-BuLi in hexanes (3.5 mL, 8.8 mmol, 2.0 equiv.) was added dropwise during 5 min, while maintaining the temperature below −20 °C. The resulting mixture was stirred at that temperature for 0.5 h, dry CO2 (0.20 g, 1.0 equiv.) was added to −20 °C. The resulting mixture was warmed to −20 °C in 0.5 h and quenched with water (6 mL). After stirring the mixture below −20 °C for 10 min, the phases were separated and the water phase was extracted one additional time with ethyl acetate. The resulting suspension was allowed to reach room temperature and fitered through a 0.5 × 1 cm pad of silica gel eluted with 10 mL of ethyl acetate. The filtrate was concentrated and the residue was purified by flash chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 10:1) to afford product 3n as off-white solid, 0.48 g (yield: 79%), m.p.: 255–257 °C. 1H-NMR (600 MHz, DMSO) δ 14.76 (s, 1H), 13.38 (s, 1H), 8.38 (dd, J = 7.2, 2.0 Hz, 1H), 7.95 (dd, J = 6.3, 2.0 Hz, 1H), 6.68 (t, J = 6.7 Hz, 1H). 13C-NMR (151 MHz, DMSO) δ 165.46, 165.04, 146.61, 141.95, 117.12, 109.09.
5-Bromo-3-hydroxyethynyl-pyridin-2-ol (3o): To a solution of 3,5-dibromo-2-hydroxypyridine (1.1 g, 4.4 mmol, 1.0 equiv.) in dry THF (20 mL) at 0 °C was added a 2 M solution of i-PrMgCl in THF (2.2 mL, 4.4 mmol, 1.0 equiv.) during 5 min. The clear solution was stirred at that temperature for an additional 5 min, and a 2.5 M solution of n-BuLi in hexanes (3.5 mL, 8.8 mmol, 2.0 equiv.) was added dropwise during 5 min, while maintaining the temperature below −20 °C. The resulting mixture was stirred at that temperature for 0.5 h, dry CO2 (0.20 g, 4.4 mmol, 1.0 equiv.) was added to −20 °C. The resulting mixture was warmed to −20 °C in 0.5 h and quenched with water (6 mL). After stirring the mixture below −20 °C for 10 min, the phases were separated and the water phase was extracted one additional time with ethyl acetate. The resulting suspension was allowed to reach room temperature and fitered through a 0.5 × 1 cm pad of silica gel eluted with 10 mL of ethyl acetate. The filtrate was concentrated and the residue was purified by flash chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 10:1) to afford product 3o as Off-white solid, 1.0 g (yield: 80%), m.p.: 245–247 °C. 1H-NMR (600 MHz, DMSO) δ 13.82 (s, 2H), 8.31 (d, J = 2.8 Hz, 1H), 8.23 (d, J = 2.8 Hz, 1H). 13C-NMR (151 MHz, DMSO) δ 164.43, 163.72, 147.95, 142.51, 118.49, 99.96.
Experimental procedures and analytical data of all compounds (
1H-and
13C-NMR), copy of the
1H,
13C and data are available in the
Supplementary Materials.





{kind=link}
{kind=link}
{kind=link}
{kind=link}































