4.1.2. Synthesis of Thiochroman-4-ones
2-Methylthiochroman-4-one (1a). To a mixture of crotonic acid (860 mg, 10 mmol) and thiophenol (1.650 g, 15 mmol) was added I2 (20 mol %, 255 mg, 1 mmol) and the mixture was stirred at room temperature for 12 h. After completion of the reaction (monitored by TLC), a cold saturated sodium thiosulfate solution (20 mL) was added and extracted with dichloromethane (2 × 50 mL); then, combined organic layers were mixed with a saturated solution of sodium bicarbonate and extracted to remove the unreacted starting material. The water layer was acidified with hydrochloric acid (10% aq) and extracted with dichloromethane (3 × 40 mL). The combined organic layers were dried over Na2SO4, evaporation of the solvent under reduced pressure afforded 1.962 g (86%) of the desired addition product. After, 200 mg (1.0 mmol) of this product were cooled down to 0 °C in an ice bath and 3.0 mL of concentrated sulfuric acid was added; the reaction mixture was stirred for 30 min, and, after that, the ice bath was removed allowing the reaction mixture to warm to room temperature for another 2 h under continuous stirring. The reaction was quenched with ice and the mixture was extracted with dichloromethane (3 × 25 mL). The combined organic layers were washed once with water, followed by addition of a saturated NaHCO3 solution. The combined organic layers were dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using hexane:EtOAc (9:1) as eluent, to give 137 mg (75%) of pure 1a as a yellowish oil. 1H-NMR (300 MHz, CDCl3) δ 8.08 (d, J = 8.0 Hz, 1H), 7.38 (t, J = 7.5 Hz, 1H), 7.25 (d, J = 7.8 Hz, 1H), 7.16 (t, J = 7.6 Hz, 1H), 3.74–3.53 (m, 1H), 2.98 (dd, J = 17.6, 8.8 Hz, 1H), 2.84–2.66 (m, 1H), 1.43 (d, J = 6.8 Hz, 3H). 13C-NMR (75 MHz, CDCl3) δ 194.9, 141.9, 133.7, 130.1, 129.1, 127.65, 125.1, 48.0, 36.5, 20.6. IR ν: 2964, 1679, 1587. HRMS (ESI) calculated for C10H11OS [M + H]+ 179.0525, found 179.0536.
6-Fluoro-2-methylthiochroman-4-one (1b). To a mixture of crotonic acid (172 mg, 2 mmol) and 4-fluorothiophenol (385 mg, 3.0 mmol) was added I2 (20% mol, 52 mg, 0.2 mmol) and the mixture was stirred at room temperature for 12 h. After completion of reaction (monitored by TLC), a cold saturated sodium thiosulfate solution (20 mL) was added and extracted with dichloromethane (2 × 25 mL); the combined organic layers were mixed with a saturated solution of sodium bicarbonate and extracted to remove the unreacted starting material. The water layer was acidified with hydrochloric acid (10% aq) and extracted with dichloromethane (3 × 25 mL). The combined organic layers were dried over Na2SO4, evaporation of the solvent under reduced pressure afforded 200 mg (94%) of the addition product. Thus, compounds were cooled down to 0 °C in an ice bath and 2.0 mL of concentrated sulfuric acid was added and the reaction mixture was allowed to warm to room temperature for 2 h with continuous stirring. The reaction was quenched with ice and the mixture was extracted with dichloromethane (3 × 25 mL). The combined organic layers were washed once with water, followed by saturated NaHCO3 solution. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using hexane:EtOAc (9:1) as eluent to give 128 mg (64%) of pure 1b as a yellowish oil. 1H-NMR (300 MHz, CDCl3) δ 7.84–7.72 (m, 1H), 7.39–6.97 (m, 2H), 3.78–3.47 (m, 1H), 3.07–2.95 (m, 1H), 2.80–2.68 (m, 1H), 1.43 (d, J = 6.9 Hz, 3H). 13C-NMR (75 MHz, CDCl3) δ 194.2, 160.7 (d, JC–F = 246 Hz), 137.6, 132.3, 129.7 (d, JC–F = 7.0 Hz), 121.8 (d, JC–F = 23.1 Hz), 115.4 (d, JC–F = 22.8 Hz), 47.9, 37.0, 20.7. IR ν: 2967, 1684, 1602. HRMS (ESI) calculated for C10H10FOS [M + H]+ 197.0431, found 197.0443.
6-Fluorothiochroman-4-one (1c). To a mixture of acrylic acid (700 μL, 720 mg, 10 mmol) and 4-fluorothiophenol (1985 mg, 15 mmol) was added I2 (20% mol, 760 mg, 3 mmol) and the mixture was stirred at 50 °C for 24 h. After completion of reaction (monitored by TLC), a cold saturated sodium thiosulfate solution (30 mL) was added and extracted with dichloromethane (2 × 25 mL); the combined organic layers were mixed with a saturated solution of sodium bicarbonate and extracted to remove the unreacted starting material. The water layer was acidified with hydrochloric acid (10% aq) and extracted with dichloromethane (3 × 50 mL). The combined organic layers were dried over Na2SO4, evaporation of the solvent under reduced pressure afforded 1150 mg (64%) of the desired addition product. The product was cooled down to 0 °C in an ice bath and 3 mL of concentrated sulfuric acid was added and the reaction mixture was allowed to warm to room temperature for 2 h with magnetic stirring. The reaction was quenched with ice and the mixture was extracted with dichloromethane (3 × 50 mL). The combined organic layers were washed once with water, followed by saturated NaHCO3 solution. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using hexane:EtOAc (9:1) as eluent to give 570 mg (90%) of pure 1c as a yellow solid. m.p.: 86–88 °C. 1H-NMR (300 MHz, CDCl3) δ 8.07 (dd, J = 8.0, 1.4 Hz, 1H), 7.16–7.11 (m, 2H), 3.25–3.23 (m, 2H), 2.99–2.96 (m, 2H). 13C-NMR (75 MHz, CDCl3) δ 193.5, 160.9 (d, JC–F = 245 Hz), 137.8, 132.7, 129.8 (d, JC–F = 7 Hz), 121.70 (d, JC–F = 23.0 Hz), 115.6 (d, JC–F = 22.6 Hz), 39.7, 27.1. IR ν: 1659, 1595, 1565. HRMS (ESI) calculated for C9H6FOS [M − H]− 181.0123, found 181.0165.
2,3,8,9-Tetrahydro-4H,10H-thiopyran[2,3-f]thiochromene-4,10-dione (1d). To a mixture of acrylic acid (288 mg, 280 μL, 4 mmol) and 1,3-benzenedithiol (350 μL, 3 mmol) was added I2 (240 mg, 1.0 mmol) and the mixture was stirred at 50 °C for 24 h. After completion of reaction (monitored by TLC), a cold saturated sodium thiosulfate solution (30 mL) was added and extracted with dichloromethane (2 × 30 mL); the combined organic layers were mixed with a saturated solution of sodium bicarbonate to form the diacid salt and extracted to remove the unreacted starting material. The water layer was acidified with hydrochloric acid (10% aq) and extracted with dichloromethane (3 × 50 mL). The combined organic layers were dried over Na2SO4, evaporation of the solvent under reduced pressure afforded 490 mg (57%); the desired addition product, 100 mg (0.35 mmol) of the later product was cooled down to 0 °C in an ice bath and 3 mL of concentrated sulfuric acid was added, the reaction mixture was allowed to warm to room temperature for 2 h with magnetic stirring and then heated to 60 °C. The reaction was quenched with ice and the mixture was extracted with dichloromethane (3 × 50 mL). The combined organic layers were washed once with water, followed by saturated NaHCO3 solution. The combined organic layers were dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using hexane:EtOAc (4:1) as eluent to give 22 mg of 1d, 25% of pure 2 as a white solid. m.p.: 145–147 °C. 1H-NMR (300 MHz, CDCl3) δ 8.15 (d, J = 8.0 Hz, 1H), 7.08 (d, J = 8.0 Hz, 1H), 3.29–3.25 (m, 2H), 3.10–3.05 (m, 4H), 2.99–2.94 (m, 2H). 13C-NMR (75 MHz, CDCl3) δ 194.3, 193.6, 151.4, 150.1, 132.7, 129.4, 127.6, 123.5, 40.3, 38.7, 27.3, 26.1. IR ν: 1648, 1559, 1351, 834. HRMS (ESI) calculated for C12H11O2S2 [M + H]+ 251.0200 found 251.0209.
4-Oxothiochroman-2-carboxylic acid (1e). Thiophenol (110 μL, 1.0 mmol) and furan-2,5-dione (maleic anhydride) (98 mg, 1.0 mmol) were mixed with triethylamine (10 μL). After stirring at 50 °C for 24 h, the mixture was cooled with an ice-cooling bath. AlCl3 (approx. 400 mg, 3.0 mmol) was added and the mixture was allowed to warm to room temperature and stirred for 1.5 h. The reaction mixture was quenched with 50 mL of 2.0 M HCl and then extracted with chloroform (4 × 50 mL). The combined organic layers were dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using hexane:EtOAc (1:4) as eluent to give 45 mg (22%) of 1e as a white solid. m.p.: 151–152 °C. 1H-NMR (300 MHz, CDCl3) δ 8.09 (d, J = 7.8 Hz, 1H), 7.4–7.38 (m, 1H), 7.25–7.21 (m, 2H) 4.13–4.11 (m, 1H), 3.19–3.17 (m, 2H). 13C-NMR (75 MHz, CDCl3) δ 192.4, 175.2, 138.4, 134.3, 130.6, 129.4, 127.7, 126.3, 42.2, 41.2. IR ν: 3300-2500, 2918, 1680, 1581, 7673. HRMS (ESI) calculated for C10H7O3S [M − H]− 207.0106 found 207.0107.
6-Nitrothiochroman-4-one (1f). Acrylic acid (510 μL, 504 mg, 7.0 mmol) and 4-nitrothiophenol (1240 mg, 8 mmol) were mixed with 75% aqueous solution of TBAF (512 μL, 1.4 mmol) and the mixture was stirred for 4h at 60 °C. A saturated solution of sodium bicarbonate was added and extracted with dichloromethane (3 × 25 mL) to remove the unreacted starting material. The water layer was acidified with hydrochloric acid (10% aq) and extracted with dichloromethane (3 × 30 mL). The combined organic layers were dried over Na2SO4, evaporation of the solvent under reduced pressure gave the crude addition product which was dissolved in anhydrous dichloromethane in an oven dried round bottomed flask under N2 in an ice cooling bath, oxalyl chloride (850 μL, 7.0 mmol) was added dropwise followed by two drops of DMF and the reaction mixture was allowed to warm to room temperature. After stirring 2.5 h, the solution was cooled to –10 °C, and a solution of 1M SnCl4 (8.4 mL, 8.4 mmol) in CH2Cl2 was added dropwise. The resulting mixture was stirred at 0 °C for 10 min and then allowed to warm to room temperature. After stirring at room temperature for 12 h, water (25 mL) was added and extracted with dichloromethane (3 × 25 mL). The combined organic layers dried over anhydrous Na2SO4, and concentrated under reduce pressure. The residue was purified by column chromatography over silica gel using hexane:EtOAc (2:1) as eluent to give 1.0 g (68%) of 1f as a white solid. m.p.: 153–154 °C. 1H-NMR (300 MHz, CDCl3) δ 8.91 (d, J = 2.6 Hz, 1H), 8.18 (dd, J = 8.8, 2.6 Hz, 1H), 7.43 (d, J = 8.8 Hz, 1H), 3.34 (t, J = 6.6 Hz, 2H), 3.04 (t, J = 6.6 Hz, 2H). 13C-NMR (75 MHz, CDCl3) δ 191.7, 150.6, 145.2, 130.8, 128.6, 126.8, 124.4, 38.4, 26.3. IR ν: 1921, 1676, 1594, 1505. HRMS (ESI) calculated for C9H6NO3S [M − H]− 208.0068 found 208.0081.
6-Aminothiochroman-4-one (1g). To a solution of the 6-nitrothiochroman-4-one 1f, (170 mg, 0.8 mmol) in 3:1 EtOH/H2O (3 mL) was added and iron wire (400 g, 7.3 mmol) and NH4Cl (100 mg, 1.8 mmol). The reaction mixture was stirred at 70 °C for 1 h. The mixture was filtered through a small pad of silica gel and then washed with EtOAc. The filtrate was concentrated and the resulting material was dissolved in (10% aq) hydrochloric acid and extracted with dichloromethane (3 × 25 mL) to remove unreacted staring material. The aqueous layer was basified with 2 M NaOH solution and extracted with dichloromethane (3 × 30 mL). The combined organic layers were dried over Na2SO4, evaporation of the solvent under reduced pressure. The resulting residue was purified by silica gel column chromatography using hexane:EtOAc (3:1) to provide 130 mg (75%) of 1g as a yellow solid. m.p.: 118–120 °C. 1H-NMR (300 MHz, CDCl3) δ 7.41 (d, J = 2.3 Hz, 1H), 7.06 (d, J = 8.4 Hz, 1H), 6.83 (dd, J = 8.4, 2.3 Hz, 1H), 3.77 (s, 2H), 3.20–3.06 (m, 2H), 2.95–2.81 (m, 2H). 13C-NMR (75 MHz, CDCl3) δ 195.3, 143.2, 131.5, 131.4, 128.7, 122.2, 114.9, 39.9, 26.8. IR ν: 3346, 3214, 1887, 1650, 1595. HRMS (ESI) calculated for C9H10NOS [M + H]+ 180.0483 found 180.0485.
4.1.3. Synthesis of Thioflavanones
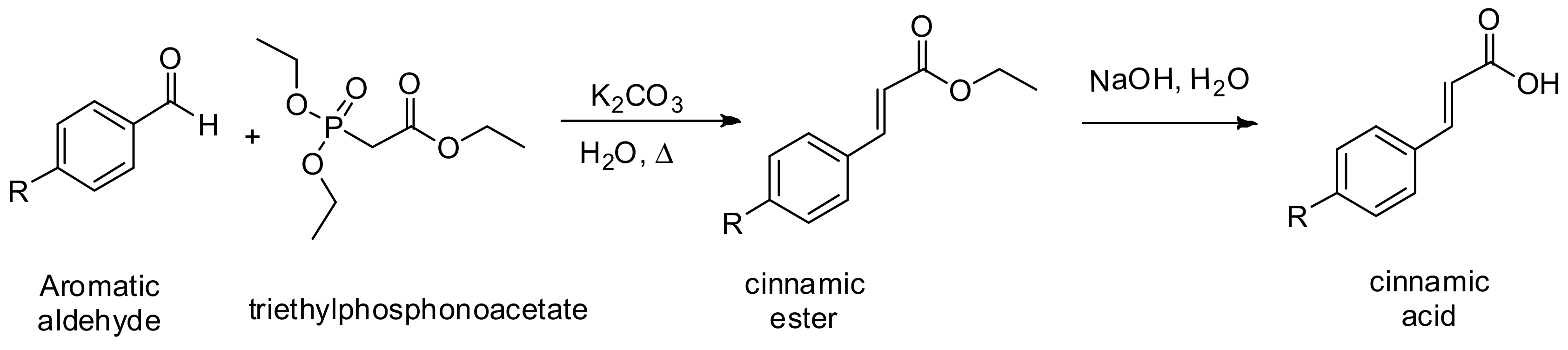
General Procedure I. Horner-Wadsworth-Emmons Reaction. Synthesis of Cinnamic Acids [30]
Cinnamic acids were prepared by the hydrolysis of the corresponding ethyl esters which were prepared by Horner-Wadsworth-Emmons reaction between aromatic aldehydes and triethylphosphonoacetate (
Scheme 2).
In a round bottomed flask equipped with a reflux condenser and an stirrer were mixed 12.0 mmol of the benzaldehyde, potassium carbonate, K2CO3 (32 mmol, 2.6 equiv.), 6.0 mL of triethylphosphonoacetate (30 mmol, 2.5 equiv.) and 2.0 mL of water and the reaction mixture was refluxed at 110 °C for 30 min. After completion of the reaction, it was quenched with water and, extracted with dichloromethane (3 × 50 mL). The combined organic layers were dried over Na2SO4 and concentrated under vacuum. The pure α,β-unsaturated ester was purified by column chromatography and then mixed with a 10% NaOH solution; then, reaction mixture was heated at 80 °C until the completion of the reaction and then 2.0 M HCl solution was added until acid pH, and extracted with chloroform (3 × 50 mL). Combined organic layer were dried over Na2SO4, and concentrated at reduced pressure, the α,β-unsaturated acid (cinnamic acid) obtained was used without further purification.
General Procedure II. Preparation of Thioflavanones
A cinnamic acid derivative (2 mmol) and thiophenol (3 mmol) were mixed with 75% aqueous solution of TBAF (140 μL sln, 0.4 mmol) and the mixture was stirred for 4 h at 60 °C. A saturated solution of sodium bicarbonate was added and extracted with dichloromethane (3 × 25 mL) to remove the unreacted starting material. The water layer was acidified with hydrochloric acid (10% aq) and extracted with dichloromethane (3 × 30 mL). The combined organic layers were dried over Na2SO4; evaporation of the solvent under reduced pressure gave the crude addition product which was dissolved in anhydrous dichloromethane and placed in an oven-dried round bottomed flask under N2 in an ice cooling bath. Consequently, oxalyl chloride (365 μL, 3.0 mmol) was added dropwise followed by two drops of DMF and the reaction mixture is left to warm to room temperature. After stirring for 2.5 h, the solution was cooled to −10 °C, and a solution of 1M SnCl4 (3.0 mL, 3.0 mmol) in CH2Cl2 was added dropwise. The resulting mixture was stirred at 0 °C for 10 min and then allowed to warm to room temperature. After stirring at room temperature for 12 h, water (25 mL) was added and extracted with dichloromethane (3 × 25 mL). The combined organic layers dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using hexane:EtOAc (2:1) as eluent to give the desired thioflavanone.
2-Phenylthiochroman-4-one (thioflavanone) (1h). The title compound was prepared from thiophenol (5.0 mmol) and cinnamic acid (310 mg, 3.0 mmol) according to the general procedure II. Yield 215 mg (45%) white solid. m.p.: 155–157 °C. 1H-NMR (300 MHz, CDCl3) δ 8.20 (dd, J = 7.9, 1.2 Hz, 1H), 7.52–7.36 (m, 6H), 7.35–7.30 (m, 1H), 7.30–7.21 (m, 1H), 4.77 (dd, J = 12.7, 3.3 Hz, 1H), 3.56–3.07 (m, 2H). IR ν: 1665, 1586, 1556, 1452, 1433. HRMS (ESI) calculated for C15H13OS [M + H]+ 241.0687, found 241.0694.
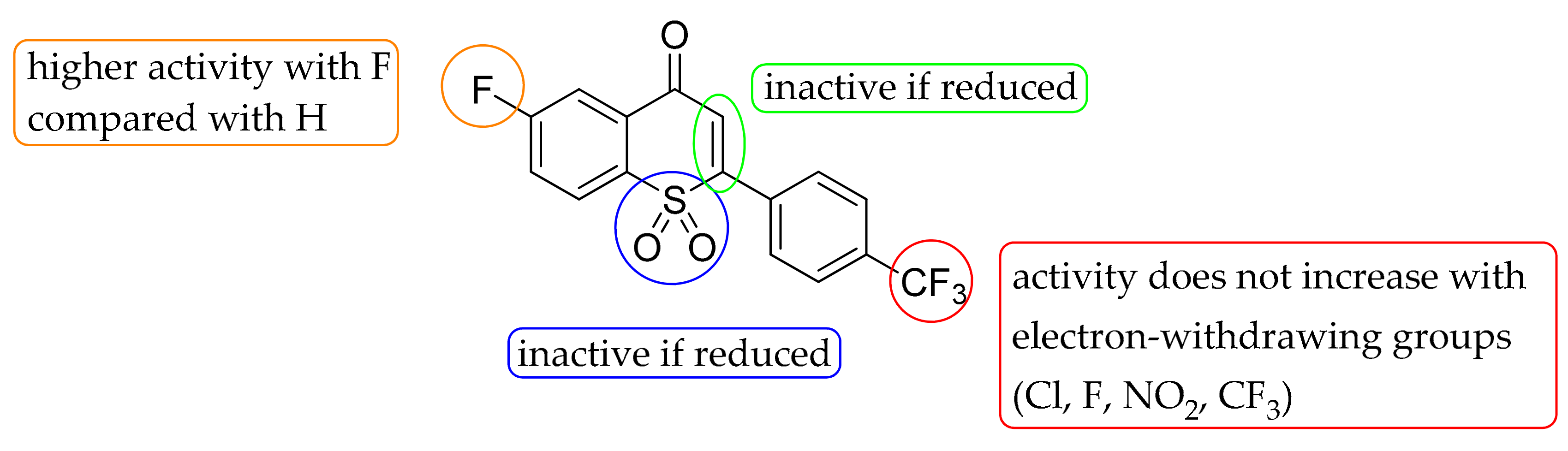
2-(4-(Trifluoromethyl)-phenyl)-thiochroman-4-one (1i). Trifluoromethylcinnamic acid (1070 mg, 5.0 mmol) and thiophenol (660 mg, 6.0 mmol) were mixed with 75% aqueous solution of TBAF (140 μL, 0.4 mmol) and the mixture was stirred for 24 h at 50 °C. A saturated solution of sodium bicarbonate was added and extracted with dichloromethane (3 × 25 mL) to remove the unreacted starting material. The water layer was acidified with hydrochloric acid (10% aq) and extracted with dichloromethane (3 × 50 mL). The combined organic layers were dried over Na2SO4, evaporation of the solvent under reduced pressure gave the crude addition product which was dissolved in methanesulfonic acid and heated at 60 °C for 2 h. Ice water was added to the mixture and the white precipitate was filtered off and washed with water. The residue was purified by column chromatography over silica gel using hexane:EtOAc (4:1) as eluent to give 650 mg (42%) of 1i as a white solid. m.p.: 108–110 °C. 1H-NMR (300 MHz, CDCl3) δ 8.18 (dd, J = 7.9, 1.4 Hz, 1H), 7.67 (d, J = 8.4 Hz, 2H), 7.58 (d, J = 8.4 Hz, 2H), 7.46 (ddd, J = 8.0, 7.3, 1.5 Hz, 1H), 7.36–7.22 (m, 3H), 4.79 (dd, J = 12.4, 3.3 Hz, 1H), 3.30 (ddd, J = 19.7, 16.4, 7.8 Hz, 2H). 13C-NMR (75 MHz, CDCl3) δ 193.7, 142.4, 141.3, 133.9, 130.8 (q, J = 32.4 Hz), 130.5, 129.3, 127.9, 127.3, 126.2 (q, J = 3.7 Hz), 125.6, 124.0 (q, J = 270.5 Hz), 46.3, 44.9. IR ν: 1677, 1323, 1106, 841, 757. HRMS (ESI) calculated for C16H12F3OS [M + H]+ 309.0561, found 309.0572.
6-Fluoro-2-(4-(trifluoromethyl)-phenyl)thiochroman-4-one (1j). The title compound was prepared from 4-fluorothiophenol (400 μL, 3.0 mmol) and trifluoromethylcinnamic acid (420 mg, 2.0 mmol) according to the general procedure II. Yield 346 mg (53%) of 1j as a white solid. m.p.: 55–57 °C. 1H-NMR (300 MHz, CDCl3) δ 7.82 (dd, J = 9.2, 2.8 Hz, 1H), 7.63 (d, J = 8.2 Hz, 2H), 7.53 (d, J = 8.2 Hz, 2H), 7.26 (dd, J = 8.9, 5.1 Hz, 1H), 7.17 (ddd, J = 8.7, 7.6, 2.9 Hz, 1H), 4.73 (dd, J = 11.6, 4.0 Hz, 1H), 3.37–3.15 (m, 2H). 13CNMR (75 MHz, CDCl3) δ 192.9 (d, J = 1.7 Hz), 161.0 (d, J = 247.2 Hz), 142.3, 136.6 (d, J = 3.0 Hz), 132.0 (d, J = 5.9 Hz), 131.0 (q, J = 32.7 Hz), 129.3 (d, J = 7.0 Hz), 128.1, 126.3 (q, J = 3.7 Hz), 124.0 (q, J = 272.7 Hz), 122.0 (d, J = 23.1 Hz), 115.6 (d, J = 22.8 Hz), 46.2, 45.3. IR ν: 1679, 1465, 1404, 1321, 1168, 1109. HRMS (ESI) calculated for C16H11F4OS [M + H]+ 327.0467, found 327.0478.
2-(4-Chlorophenyl)-thiochroman-4-one (1k). The title compound was prepared from thiophenol (440 μL, 4.0 mmol) and 4-chlorocinnamic acid (546 mg, 3.0 mmol) according to the general procedure II. Yield 537 mg (53%) of 1k as a yellowish solid. m.p.: 123–125 °C. 1H-NMR (300 MHz, CDCl3) δ 8.12 (dd, J = 7.9, 1.3 Hz, 1H), 7.41 (ddd, J = 8.1, 7.1, 1.6 Hz, 1H), 7.36–7.30 (m, 4H), 7.29–7.23 (m, 1H), 7.20 (ddd, J = 8.2, 7.1, 1.2 Hz, 1H), 4.67 (dd, J = 12.2, 3.6 Hz, 1H), 3.21 (m, 2H).13C-NMR (75 MHz, CDCl3) δ 194.0, 141.6, 137.0, 134.3, 133.8, 130.4, 129.3, 129.2, 128.8, 127.3, 125.5, 46.6, 44.8. IR ν: 1674, 1278, 1082, 851, 767. HRMS (ESI) calculated for C15H10ClOS [M − H]− 273.0141 found 273.0129.
2-(4-Fluorophenyl)-thiochroman-4-one (1l) The title compound was prepared from thiophenol (440 μL, 4.0 mmol) and 4-fluorocinnamic acid (500 mg, 3.0 mmol) according to the general procedure II. Yield 485 mg (63%) of 1l as a yellowish solid. m.p.: 100–102 °C. 1H-NMR (300 MHz, CDCl3) δ 8.19 (dd, J = 8.0, 1.4 Hz, 1H), 7.53–7.39 (m, 3H), 7.29 (ddd, J = 15.1, 8.9, 4.6 Hz, 2H), 7.18–7.06 (m, 2H), 4.75 (dd, J = 12.4, 3.5 Hz, 1H), 3.28 (qd, J = 16.4, 8.0 Hz, 2H). 13C-NMR (75 MHz, CDCl3) δ 194.2, 162.6 (d, J = 247.8 Hz), 141.8, 134.3 (d, J = 3.2 Hz), 133.8, 130.4, 129.3, 129.2 (d, J = 8.3 Hz), 127.2, 125.4, 116.0 (d, J = 21.7 Hz), 46.8, 44.74. IR ν: 2940, 1664, 1582, 1506, 1223, 842, 764. HRMS (ESI) calculated for C15H12FOS [M + H]+ 259.0593, found 259.0598.
2-(4-Nitrophenyl)-thiochroman-4-one (1m). The title compound was prepared from thiophenol (165 μL, 1.5 mmol) and 4-nitrocinnamic acid (192 mg, 1.0 mmol) according to the general procedure II. Yield 199 mg (70%) of 1m as a yellowish solid. m.p.: 158–160 °C. 1H-NMR (300 MHz, CDCl3) δ 8.29 (d, J = 8.7 Hz, 2H), 8.20 (dd, J = 7.9, 1.2 Hz, 1H), 7.66 (d, J = 8.7 Hz, 2H), 7.58–7.43 (m, 1H), 7.34 (d, J = 8.7 Hz, 1H), 7.29 (dd, J = 9.1, 1.9 Hz, 1H), 4.86 (dd, J = 11.3, 3.9 Hz, 1H), 3.46–3.24 (m, 2H). 13C-NMR (75 MHz, CDCl3) δ 193.2, 147.8, 145.6, 140.7, 134.1, 130.3, 129.4, 128.6, 127.3, 125.8, 124.3, 46.1, 44.7. IR ν: 2920, 1666, 1587, 1514, 1515, 1338, 761. HRMS (ESI) calculated for C15H12NO3S [M + H]+ 286.0538, found 286.0543.
4.1.5. General Procedure IV. Dehydrogenation with Iodine/DMSO [22]
A mixture of 0.5 mmol of substrate (thiochromanones or thioflavanones) in 2.0 mL of DMSO was added 125 μL of 0.1 M solution of iodine in (25% mol) was refluxed during 24 h, then cooled and poured into water. The mixture was extracted with dichloromethane (3 × 25 mL), and the combined organic layers were dried over sodium sulfate, and then concentrated at reduced pressure. The residue was purified in column chromatography using mixtures of ethyl acetate and hexane to give the desired dehydrogenation products in moderate yields.
6-Nitro-4H-thiochromen-4-one (2f). The title compound was prepared from 1f according to the general procedure III. Yield 50 mg (48%) of 2f as a yellowish solid. mp 184–185 °C. 1H-NMR (300 MHz, CDCl3) δ 9.32 (d, J = 2.5 Hz, 1H), 8.39 (dd, J = 8.9, 2.5 Hz, 1H), 7.85 (d, J = 10.5 Hz, 1H), 7.77 (d, J = 8.9 Hz, 1H), 7.06 (d, J = 10.5 Hz, 1H). 13C-NMR (75 MHz, CDCl3) δ 178.5, 147.4, 144.0, 137.9, 132.9, 128.5, 126.6, 125.4, 124.6. IR ν: 3096, 3031, 1602, 1505, 1339. HRMS (ESI) calculated for C9H6NO3S [M + H]+ 208.0068 found 208.0077.
2-(4-(Trifluoromethyl)-phenyl)-4H-thiochromen-4-one (2i). The title compound was prepared from 1i according to the general procedure III. Yield 69 mg (45%) of 21 as a yellowish solid. m.p.: 168–170 °C. 1H-NMR (300 MHz, CDCl3) δ 8.54 (d, J = 7.6 Hz, 1H), 7.85–7.71 (m, 4H), 7.69–7.51 (m, 3H), 7.24 (s, 1H). 13C-NMR (75 MHz, CDCl3) δ 180.6, 151.2, 140.0, 137.3, 132.6 (d, J = 32.9 Hz) *, 131.9, 130.8, 128.7, 128.1, 127.5, 126.5, 126.3 (q, J = 3.7 Hz), 124.4, 123.6 (d, J = 272.7 Hz). IR ν: 3033, 2930, 1615, 1438, 1113, 667. HRMS (ESI) calculated for C16H10F3OS [M + H]+ 307.0404 found 307.0446.
6-Fluoro-2-(4-(trifluoromethyl)-phenyl)-4H-thiochromen-4-one (2j). The title compound was prepared from 1j according to the general procedure III. Yield 90 mg (55%) of 2j as a yellowish solid. m.p.: 128–130 °C. 1H-NMR (300 MHz, CDCl3) δ 8.20 (dd, J = 9.3, 2.8 Hz, 1H), 7.82–7.72 (m, 4H), 7.67 (dd, J = 8.8, 4.7 Hz, 1H), 7.41 (ddd, J = 8.8, 7.6, 2.9 Hz, 1H), 7.22 (s, 1H). 13C-NMR (75 MHz, CDCl3) δ 180.0 (d, J = 2.2 Hz), 162.6 (d, J = 250.4 Hz), 151.8, 140.0, 133.1 (d, J = 7.5 Hz), 133.0 (q, J = 33.2 Hz), 132.8 (d, J = 3.7 Hz), 128.9 (d, J = 7.7 Hz), 127.7, 126.6 (q, J = 3.7 Hz), 123.8 (q, J = 272.4 Hz), 123.7, 121.0 (d, J = 24.2 Hz), 114.6 (d, J = 23.0 Hz). IR ν: 2923, 1713, 1623, 1605, 1118. HRMS (ESI) calculated for C16H9F4OS [M + H]+ 325.0310 found 325.0331.
2-(4-Chlorophenyl)-4H-thiochromen-4-one (2k). The title compound was prepared from 1k according to the general procedure III. Yield 82 mg (60%) of 2k as a yellowish solid. m.p.: 161–162 °C. 1H-NMR (300 MHz, CDCl3) δ 8.60–8.49 (m, 1H), 7.74–7.53 (m, 5H), 7.52–7.42 (m, 2H), 7.37 (s, 1H). 13C-NMR (75 MHz, CDCl3) δ 181.0, 151.9, 137.6, 137.4, 135.2, 132.0, 131.0, 129.8, 128.9, 128.4, 128.2, 126.7, 123.8. IR ν: 3014, 1627, 1588, 1087, 777. HRMS (ESI) calculated for C15H10ClOS [M + H]+ 273.0141 found 273.0172.
2-(4-Fluorophenyl)-4H-thiochromen-4-one (2l). The title compound was prepared from 1l according to the general procedure III. Yield 74 mg (58%) of 2l as a yellowish solid. m.p.: 160–161 °C. 1H-NMR (300 MHz, CDCl3) δ 8.62 (d, J = 7.7 Hz, 1H), 7.82–7.69 (m, 4H), 7.68–7.60 (m, 1H), 7.47 (s, 1H), 7.26 (t, J = 8.5 Hz, 2H). 13C-NMR (75 MHz, CDCl3) δ 180.8, 164.4 (d, J = 252.0 Hz), 151.9, 137.5, 132.8 (d, J = 3.3 Hz), 131.8, 130.8, 129.0 (d, J = 8.6 Hz), 128.7, 127.9, 126.5, 123.5, 116.5 (d, J = 22.1 Hz). IR ν: 2921, 1608, 1582, 1500, 1225, 845. HRMS (ESI) calculated for C15H10FOS [M + H]+ 257.0436 found 257.0463.
2-(4-nitrophenyl)-4H-thiochromen-4-one (2m). The title compound was prepared from 1m according to the general procedure IV. Yield 128 mg (91%) of 2m as a yellowish solid. mp 183–185 °C. 1H-NMR (300 MHz, CDCl3) δ 8.54 (d, J = 7.8 Hz, 1H), 8.35 (d, J = 8.7 Hz, 2H), 7.85 (d, J = 8.7 Hz, 2H), 7.72–7.62 (m, J = 3.0 Hz, 2H), 7.62–7.53 (m, 1H), 7.25 (s, 1H). 13C-NMR (75 MHz, CDCl3) δ 180.7, 150.3, 149.3, 142.7, 137.2, 132.3, 131.0, 129.0, 128.5, 128.3, 126.8, 125.2, 124.7. IR ν: 3015, 1738, 1628, 1513, 1354, 850. HRMS (ESI) calculated for C15H10NO3S [M + H]+ 284.0381 found 284.0407.
4.1.6. General Procedure V. Oxidation of Sulfides to Sulfones.
To a 25-mL glass tube, sulfide (1.0 mmol), oxone (0.9221 g, 1.5 mmol), and a water ethanol mixture (1:1) (3.0 mL) were added and the mixture was stirred at 60 °C for 12 h. The mixture was then cooled to room temperature and extracted with dichloromethane (3 × 25 mL). The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel using mixtures of hexane and EtOAc as eluent.
2-Methylthiochroman-4-one 1,1-dioxide (3a). The title compound was prepared from 1a according to the general procedure V. Yield 178 mg (82%) of 3a as a white solid. m.p.: 129–131 °C. 1H-NMR (300 MHz, CDCl3) δ 8.10 (dd, J = 7.7, 1.0 Hz, 1H), 8.04 (dd, J = 7.7, 0.8 Hz, 1H), 7.81 (td, J = 7.6, 1.3 Hz, 1H), 7.72 (td, J = 7.6, 1.2 Hz, 1H), 3.76 (dd, J = 14.2, 6.9 Hz, 1H), 3.33–3.19 (m, 2H), 1.54 (d, J = 6.9 Hz, 3H). 13C-NMR (75 MHz, CDCl3) δ 190.4, 140.7, 135.0, 133.3, 130.5, 128.5, 124.2, 54.6, 44.2, 11.7. IR ν: 3082, 2945, 1694, 1312, 1151, 728. HRMS (ESI) calculated for C10H9O3S [M − H]− 209.0272 found 209.0282.
6-Fluoro-2-methylthiochroman-4-one 1,1-dioxide (3b). The title compound was prepared from 1b according to the general procedure V. Yield 161 mg (71%) of 3b as a white solid. m.p.: 119–120 °C. 1H-NMR (300 MHz, CDCl3) δ 8.06 (dd, J = 8.7, 4.9 Hz, 1H), 7.75 (dd, J = 8.7, 2.6 Hz, 1H), 7.55–7.42 (m, 1H), 3.84–3.62 (m, 1H), 3.37–3.15 (m, 2H), 1.53 (2, J = 10.5 Hz, 3H). 13C-NMR (75 MHz, CDCl3) δ 189.4, 165.2 (d, J = 257.5 Hz), 136.9 (d, J = 3.6 Hz), 133.4 (d, J = 7.2 Hz), 127.5 (d, J = 8.6 Hz), 122.4 (d, J = 23.0 Hz), 115.4 (d, J = 23.8 Hz), 54.7, 44.3, 11.7. IR ν: 3100, 2939, 1697, 1580, 1310, 1273, 1147, 694. HRMS (ESI) calculated for C10H8FO3S [M − H]− 227.0178 found 227.0179.
6-Fluorothiochroman-4-one 1,1-dioxide (3c). The title compound was prepared from 1c according to the general procedure V. Yield 174 mg (81%) of 3c as a white solid. m.p.: 175–177 °C. 1H-NMR (300 MHz, CDCl3) δ 8.03 (dd, J = 8.7, 4.9 Hz, 1H), 7.76 (dd, J = 8.7, 2.6 Hz, 1H), 7.48 (ddd, J = 8.7, 7.7, 2.7 Hz, 1H), 3.74–3.62 (m, 2H), 3.48–3.37 (m, 2H). 13C-NMR (75 MHz, CDCl3) δ 189.1, 165.2 (d, J = 257.6 Hz), 137.6 (d, J = 3.6 Hz), 133.1 (d, J = 7.3 Hz), 126.9 (d, J = 8.7 Hz), 122.3 (d, J = 23.0 Hz), 115.7 (d, J = 23.8 Hz), 49.3, 36.9. IR ν: 3087, 2980, 1690, 1577, 1271, 1130, 749. HRMS (ESI) calculated for C9H6FO3S [M − H]− 213.0022 found 213.0025.
2-Phenylthiochroman-4-one 1,1-dioxide (3h). The title compound was prepared from 1h according to the general procedure V. Yield 217 mg (80%) of 3h as a white solid. m.p.: 154–155 °C. 1H-NMR (300 MHz, CDCl3) δ 8.22–8.12 (m, 1H), 8.07 (d, J = 7.4 Hz, 1H), 7.90–7.72 (m, 2H), 7.61–7.35 (m, 5H), 4.86 (dd, J = 12.8, 3.2 Hz, 1H), 3.96 (dd, J = 17.7, 12.8 Hz, 1H), 3.41 (dd, J = 17.7, 3.2 Hz, 1H). 13C-NMR (75 MHz, CDCl3) δ 190.9, 141.5, 135.1, 133.4, 130.5, 130.1, 129.9, 129.2, 128.8, 128.0, 124.5, 64.1, 43.1. IR ν: 2957, 1693, 1586, 1281, 1149, 1120, 764. HRMS (ESI) calculated for C15H11O3S [M − H]− 271.0429 found 271.0428.
2-(4-(Trifluoromethyl)-phenyl)-thiochroman-4-one 1,1-dioxide (3i). The title compound was prepared from 1i according to the general procedure V. Yield 146 mg (43%) of 3i as a white solid. m.p.: 155–157 °C. 1H-NMR (300 MHz, CDCl3) δ 8.18 (dd, J = 7.7, 1.2 Hz, 1H), 8.07 (dd, J = 7.7, 1.0 Hz, 1H), 7.85 (td, J = 7.6, 1.5 Hz, 1H), 7.78 (td, J = 7.6, 1.3 Hz, 1H), 7.72 (d, J = 8.2 Hz, 2H), 7.61 (d, J = 8.3 Hz, 2H), 4.93 (dd, J = 12.8, 3.2 Hz, 1H), 3.96 (dd, J = 17.7, 12.8 Hz, 1H), 3.41 (dd, J = 17.7, 3.3 Hz, 1H). 13C-NMR (75 MHz, CDCl3) δ 190.4, 141.3, 135.5, 133.87, 132.4 (q, J = 33Hz), 132.1, 130.6, 130.5, 129.1, 126.3 (dd, J = 7.5, 3.7 Hz), 124.7, 123.9 (q, J = 272.5 Hz), 63.8, 43.0. IR ν: 2927, 1691, 1294, 1153, 712. HRMS (ESI) calculated for C16H12F3O3S [M + H]+ 341.0459 found 341.0465.
6-Fluoro-2-(4-(trifluoromethyl)-phenyl)-thiochroman-4-one 1,1-dioxide (3j) The title compound was prepared from 1j according to the general procedure V. Yield 254 mg (62%) of 3j as a white solid. m.p.: 132–134 °C. 1H-NMR (300 MHz, CDCl3) δ 8.09 (dd, J = 8.7, 4.8 Hz, 1H), 7.83 (dd, J = 8.6, 2.6 Hz, 1H), 7.72 (d, J = 8.2 Hz, 2H), 7.60 (d, J = 8.2 Hz, 2H), 7.56–7.47 (m, 1H), 4.92 (dd, J = 12.7, 3.1 Hz, 1H), 3.97 (dd, J = 17.8, 12.8 Hz, 1H), 3.44 (dd, J = 17.8, 3.2 Hz, 1H). 13C-NMR (75 MHz, CDCl3) δ 189.1, 165.3 (d, J = 258.3 Hz), 137.1 (d, J = 3.6 Hz), 133.2 (d, J = 7.4 Hz), 132.3 (d, J = 33.0 Hz), 131.6, 130.3, 127.7 (d, J = 8.7 Hz), 126.1 (q, J = 3.5 Hz), 123.6 (q, J = 272.4 Hz), 122.5 (d, J = 23.0 Hz), 115.7 (d, J = 23.9 Hz), 63.6, 42.9. IR ν: 2918, 1697, 1579, 1298, 1158, 1114. HRMS (ESI) calculated for C16H9F4O3S [M − H]− 357.0209 found 357.0201.
2-(4-Chlorophenyl)-thiochroman-4-one 1,1-dioxide (3k). The title compound was prepared from 1k according to the general procedure V. Yield 199 mg (65%) of 3k as a white solid. m.p.: 160–161 °C. 1H-NMR (300 MHz, CDCl3) δ 8.16 (dd, J = 7.6, 1.1 Hz, 1H), 8.05 (dd, J = 7.7, 0.8 Hz, 1H), 7.83 (td, J = 7.6, 1.4 Hz, 1H), 7.76 (td, J = 7.6, 1.2 Hz, 1H), 7.47–7.36 (m, 4H), 4.83 (dd, J = 12.8, 3.2 Hz, 1H), 3.90 (dd, J = 17.7, 12.8 Hz, 1H), 3.37 (dd, J = 17.7, 3.2 Hz, 1H).13C-NMR (75 MHz, CDCl3) δ 190.7, 141.4, 136.5, 135.4, 133.7, 131.3, 130.6, 129.6, 129.0, 126.6, 124.7, 63.6, 43.1. IR ν: 2932, 1687, 1584, 1490 1314, 1280, 1147. HRMS (ESI) calculated for C15H10ClO3S [M − H]− 305.0039 found 305.0038.
2-(4-Fluorophenyl)thiochroman-4-one 1,1-dioxide (3l). The title compound was prepared from 1l according to the general procedure V. Yield 180 mg (62%) of 3l as a white solid. m.p.: 121–123 °C. 1H-NMR (300 MHz, CDCl3) δ 8.17 (d, J = 7.6 Hz, 1H), 8.06 (d, J = 7.8 Hz, 1H), 7.83 (t, J = 7.6, 1.2 Hz, 1H), 7.77 (t, J = 10.8, 4.3 Hz, 1H), 7.46 (dd, J = 8.6, 5.2 Hz, 2H), 7.14 (t, J = 8.6 Hz, 2H), 4.85 (dd, J = 12.9, 3.1 Hz, 1H), 3.91 (dd, J = 17.7, 12.9 Hz, 1H), 3.38 (dd, J = 17.7, 3.2 Hz, 1H). 13C-NMR (75 MHz, CDCl3) δ 190.8, 163.9 (d, J = 250.2 Hz), 141.5, 135.4, 133.7, 131.9 (d, J = 8.7 Hz), 130.6, 129.0, 124.7, 123.9 (d, J = 3.0 Hz), 116.5 (d, J = 21.8 Hz), 63.4, 43.3. IR ν: 2927, 1697, 1589, 1506, 1490, 1314, 1284, 1152. HRMS (ESI) calculated for C15H10FO3S [M − H]− 289.0335 found 289.0336.
2-(4-Nitrophenyl)-thiochroman-4-one 1,1-dioxide (3m). The title compound was prepared from 1m according to the general procedure V. Yield 237 mg (75%) of 3m as a white solid. m.p.: 180–182 °C. 1H-NMR (300 MHz, CDCl3) δ 8.31 (d, J = 8.8 Hz, 2H), 8.19 (dd, J = 7.6, 1.2 Hz, 1H), 8.06 (dd, J = 7.6, 0.9 Hz, 1H), 7.86 (td, J = 7.6, 1.5 Hz, 1H), 7.80 (td, J = 7.5, 1.3 Hz, 1H), 7.68 (d, J = 8.7 Hz, 2H), 4.98 (dd, J = 12.8, 3.2 Hz, 1H), 3.97 (dd, J = 17.6, 12.8 Hz, 1H), 3.43 (dd, J = 17.6, 3.2 Hz, 1H). 13CNMR (75 MHz, CDCl3) δ 189.9, 149.1, 141.1, 135.5, 135.1, 134.0, 131.2, 130.5, 129.2, 124.7, 124.4, 63.6, 42.9. IR ν: 3078, 2921, 1695, 1586, 1517, 1342, 1155. HRMS (ESI) calculated for C15H10NO5S [M − H]− 316.0280 found 316.0267.
2-Phenyl-4H-thiochromen-4-one 1,1-dioxide (4h). The title compound was prepared from 3h according to the general procedure IV. Yield 120 mg (89%) of 4h as a white solid. m.p.: 134–135 °C. 1H-NMR (300 MHz, CDCl3) δ 8.21 (dd, J = 7.8, 0.7 Hz, 1H), 8.14–8.05 (m, 1H), 7.87 (ddd, J = 8.3, 5.5, 1.2 Hz, 3H), 7.76 (t, J = 7.3 Hz, 1H), 7.60–7.43 (m, 3H), 6.82 (s, 1H). 13C-NMR (75 MHz, CDCl3) δ 178.3, 153.1, 141.7, 134.8, 133.0, 131.9, 129.3, 128.8, 128.7 (2CHs), 128.5, 128.1, 123.6. IR ν: 2920, 2849, 1964, 1650, 1588, 1287, 1242, 1149. HRMS (ESI) calculated for C15H11O3S [M + H]+ 271.0429 found 271.0434.
2-(4-(Trifluoromethyl)-phenyl)-4H-thiochromen-4-one 1,1-dioxide (4i). The title compound was prepared from 3i according to the general procedure IV. Yield 167 mg (99%) of 4i as a white solid. m.p.: 188–189 °C. 1H-NMR (300 MHz, CDCl3) δ 8.22 (dd, J = 7.9, 1.5 Hz, 1H), 8.10 (dd, J = 7.9, 1.4 Hz, 1H), 7.96 (d, J = 8.4 Hz, 2H), 7.90 (td, J = 7.6, 1.5 Hz, 1H), 7.79 (td, J = 7.5, 1.2 Hz, 1H), 7.77 (dd, J = 7.7, 1.2 Hz, 2H), 6.82 (s, J = 5.8 Hz, 1H). 13C-NMR (75 MHz, CDCl3) δ 178.2, 152.1, 141.6, 135.3, 133.6 (q, J = 33.1), 133.4, 132.4, 130.3, 129.6, 128.6, 128.5, 126.4 (q, J = 3.6 Hz), 123.9, 123.7 (q, J = 272.8). IR ν: 2917, 1651, 1587, 1295, 1119, 837. HRMS (ESI) calculated for C16H10F3O3S [M + H]+ 339.0303 found 339.0306.
6-Fluoro-2-(4-(trifluoromethyl)-phenyl)-4H-thiochromen-4-one 1,1-dioxide (4j). The title compound was prepared from 3j according to the general procedure IV. Yield 144 mg (81%) of 4j as a white solid. m.p.: 172–173°C. 1H-NMR (300 MHz, CDCl3) δ 8.12 (dd, J = 8.7, 4.7 Hz, 1H), 7.94 (d, J = 8.1 Hz, 2H), 7.86 (dd, J = 8.5, 2.6 Hz, 1H), 7.77 (d, J = 8.3 Hz, 2H), 7.71–7.46 (m, 1H), 6.83 (s, 1H). 13C-NMR (75 MHz, CDCl3) δ 177.2, 165.1 (d, J = 258.3 Hz), 152.5, 137.7 (d, J = 3.8 Hz), 133.9 (q, J = 33.3 Hz), 132.1, 131.4 (d, J = 7.6 Hz), 129.9, 129.7, 127.1 (d, J = 8.8 Hz), 126.5 (q, J = 3.6 Hz), 123.7 (q, J = 272.6 Hz), 122.9 (d, J = 23.3 Hz), 115.3 (d, J = 24.0 Hz). IR ν: 3040, 2918, 1658, 1580, 1292, 1119. HRMS (ESI) calculated for C16H9F4O3S [M + H]+ 357.0209 found 357.0210.
2-(4-Chlorophenyl)-4H-thiochromen-4-one 1,1-dioxide (4k). The title compound was prepared from 3k according to the general procedure IV. Yield 144 mg (95%) of 4k as a white solid. m.p.: 173–174 °C. 1H-NMR (300 MHz, CDCl3) δ 8.21 (d, J = 7.9 Hz, 1H), 8.09 (d, J = 7.9 Hz, 1H), 7.88 (t, J = 7.7 Hz, 1H), 7.80 (d, J = 8.2 Hz, 2H), 7.77 (t, J = 7.5 Hz, 1H), 7.49 (d, J = 8.2 Hz, 2H), 6.80 (s, 1H).13C-NMR (75 MHz, CDCl3) δ 178.3, 152.3, 141.7, 138.7, 135.2, 133.3, 130.4, 129.9, 129.2, 128.7, 128.4, 127.3, 123.9. IR ν: 2916, 1644, 1297, 1090. HRMS (ESI) calculated for C15H10ClO3S [M + H]+ 305.0039 found 305.0036.
2-(4-Fluorophenyl)-4H-thiochromen-4-one 1,1-dioxide (4l). The title compound was prepared from 3l according to the general procedure IV. Yield 36 mg (25%) of 4l as a white solid. m.p.: 173–174 °C. 1H-NMR (300 MHz, CDCl3) δ 8.21 (dd, J = 7.9, 1.0 Hz, 1H), 8.09 (dd, J = 7.9, 0.7 Hz, 1H), 7.94–7.82 (m, 3H), 7.77 (td, J = 7.7, 1.1 Hz, 1H), 7.21 (t, J = 8.7 Hz, 2H), 6.78 (s, 1H). 13C-NMR (75 MHz, CDCl3) δ 178.4, 165.2 (d, J = 254.3 Hz), 152.3, 141.8, 135.1, 133.3, 131.4 (d, J = 8.9 Hz), 129.0, 128.7, 128.4, 125.0 (d, J = 3.3 Hz), 123.9, 116.9 (d, J = 22.1 Hz). IR ν: 2849, 1650, 1585, 1505, 1286, 1147. HRMS (ESI) calculated for C15H10FO3S [M + H]+ 289.0335 found 289.0335.
2-(4-Nitrophenyl)-4H-thiochromen-4-one 1,1-dioxide (4m). The title compound was prepared from 3m according to the general procedure IV. Yield 99 mg (63%) of 4m as a white solid. m.p.: 187–189 °C. 1H-NMR (300 MHz, CDCl3) δ 8.43 (d, J = 8.4 Hz, 2H), 8.30 (d, J = 7.8 Hz, 1H), 8.17 (d, J = 7.9 Hz, 1H), 8.09 (d, J = 8.4 Hz, 2H), 7.99 (t, J = 7.7 Hz, 1H), 7.87 (t, J = 7.6 Hz, 1H), 6.92 (s, 1H). 13C-NMR (75 MHz, CDCl3) δ 177.8, 151.2, 149.7, 141.2, 135.3, 134.8, 133.4, 130.6, 130.2, 128.42, 128.38, 124.3, 123.7. IR ν: 2917, 2849, 1651, 1586, 1512, 1286, 1147. HRMS (ESI) calculated for C15H10NO5S [M + H]+ 316.0280 found 316.0281.

 ,
, 



{kind=link}
{kind=link}
{kind=link}