All chemical reagents used were purchased from commercial chemical suppliers. The NMR experiments were performed at 500 MHz (1H) on a Varian VNMR SYSTEM spectrometer. Chemical shifts are referenced to the residual solvent signals, 2.50 ppm for 1H in DMSO-d6 and 7.28 ppm for 1H in CDCl3. The MS measurements were performed on Shimadzu LCMS2020 LC/MS system. Flash chromatography was performed using Teledyne ISCO CombiFlash Lumen+ Rf. Purifications by preparative-HPLC were performed with Hanbon NS4205 Binary high pressure semi-preparative HPLC. Thin-layer chromatography was performed on TLC Silica Gel 60 F254. High resolution mass spectrometric measurements were performed using a Q-TOF Premier mass spectrometer (Waters Corporation, Milford, MA, USA) in positive electrospray ionization mode. Compounds 7–16 were purchased from Mcule Inc.
4.1. General Procedures for the Synthesis of Spiro[pyrrolidine-3,3′-oxindole] and Spiro[pyrrolidine-3,3′-indoline] Derivatives
4.1.1. Procedures to Afford 1-(Phenylsulfonyl)spiro[indoline-3,3′-pyrrolidin]-2-one (17)
Synthesis of Benzyl 2-Oxospiro[indoline-3,3′-pyrrolidine]-1′-carboxylate (27)
To the solution of benzyl 3,4-dihydro-1H-pyrido[3,4-b]indole-2(9H)-carboxylate (26) (918 mg, 3 mmol) in 10 mL tetrahydrofuran was added 10 mL distilled water and triethylamine (0.436 mL, 3.15 mmol, 1.05 equiv.) at room temperature. Afterwards N-chlorosuccinimide (431 mg, 3.24 mmol, 1.08 equiv.) was added at 0 °C in portions. The reaction mixture was stirred for 1.5 h at room temperature. The reaction mixture was diluted with 10 mL brine and was extracted using 2 × 10 mL ethyl acetate. The combined organic phases were dried over sodium-sulfate, filtered and evaporated under reduced pressure. The crude material was purified using flash chromatography (gradient: dichloromethane: methanol 0% to 5% methanol) to give 27 as a yellow solid. Yield: 399 mg (41%). 1H-NMR (500 MHz, DMSO-d6) δ ppm 7.42–7.28 (m, 8H, ArH-benzyl, ArH-4, ArH-5, ArH-6), 7.14 (d, J = 7.8 Hz, 1H, ArH-7), 5.11 (s, 2H, benzyl CH2), 3.70–3.53 (m, 4H, pyrrolidine H-2′, pyrrolidine H-5′), 2.19–2.05 (m, 2H, H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 176.9 (C-2), 154.8 (C=O), 142.8 (C-3a), 141.5 (C-7a), 135.9 (benzyl C-1), 128.5 (benzyl C-3, benzyl C-5), 127.9 (benzyl C-4, benzyl C-2, benzyl C-6), 123.8 (C-4), 122.1 (C-5), 109.3 (C-7), 67.9 (benzyl CH2), 57.7 (C-3), 45.1 (pyrrolidine C-2′), 43.3 (pyrrolidine C-5′), 34.2 (pyrrolidine C-4′). MS (DUIS+) m/z 323 [M + H]+.
Synthesis of Benzyl 2-oxo-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine]-1′-carboxylate (28)
(1) Procedure Using Sodium-Hydride
To the solution of benzyl 2-oxospiro[indoline-3,3′-pyrrolidine]-1′-carboxylate (27) (175 mg, 0.542 mmol) in 5 mL anhydrous tetrahydrofuran was added sodium-hydride (60%, 28 mg, 0.705 mmol; 1.3 equiv.) at 0 °C. After stirring the reaction mixture at room temperature for 0.5 h, benzenesulfonyl chloride (0.076 mL, 0.596 mmol, 1.1 equiv.) was added dropwise at 0 °C. The reaction mixture was stirred overnight at room temperature, then it was quenched by 10 mL brine and was extracted using ethyl acetate (2 × 10 mL). The collected organic phases were dried using sodium sulfate, filtered and evaporated under reduced pressure. The crude product was purified using flash chromatography (gradient: hexane: ethyl acetate 0% to 20%) to afford 28 as a colorless solid. Yield: 94 mg (38%).
(2) Procedure Using Lithium-Hexamethyl-Disilazane
To the solution of benzyl 2-oxospiro[indoline-3,3′-pyrrolidine]-1′-carboxylate (27) (161 mg, 0.5 mmol) and benzenesulfonyl chloride (0.076 mL, 0.6 mmol, 1.2 equiv.) in 5 mL anhydrous tetrahydrofuran was added lithiumhexamethyl-disilazane (1M in THF, 0.85 mL, 0.85 mmol, 1.7 equiv.) at 0 °C and the reaction mixture was stirred at this temperature for 0.5 h. After quenching the reaction by 10 mL brine, the reaction mixture was extracted using ethyl acetate (2 × 10 mL) and the collected organic phases were dried over sodium sulfate, filtered and evaporated under reduced pressure. The crude product was purified using flash chromatography (gradient: hexane: ethyl acetate 0% to 20%) to give 28 as a white solid. Yield: 230 mg (99%). 1H-NMR (500 MHz, DMSO-d6) δ ppm 8.02 (d, J = 7.7 Hz, 2H, ArH-3″, ArH-5″), 7.77 (m, 2H, ArH-4″, ArH-7), 7.67 (m, 2H, phenyl-sulfonyl ArH-2″, phenyl-sulfonyl ArH-6″), 7.43–7.30 (m, 7H, ArH-4, ArH-6, benzyl ArH-2,3,4,5,6), 7.23 (t, J = 7.4 Hz, 1H, ArH-5), 5.09 (m, 2H, benzyl CH2), 3.65–3.50 (m, 4H, pyrrolidine H-2′, pyrrolidine H-5′), 2.25–2.09 (m, 2H, pyrrolidine H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 170.1 (C-2), 154.3 (C=O), 136.2 (benzyl C-1), 135.5 (C-7a), 134.8 (phenyl-sulfonyl C-1″), 133.9 (C-3a), 133.1 (phenyl-sulfonyl C-4″), 129.2 (C-3″, C-5″), 128.5 (benzyl C-3, benzyl C-5), 128.0 (C-6, benzyl C-4), 127.5 (benzyl C-2, benzyl C-6), 127.4 (C-2″, C-6″), 123.3 (C-4), 121.8 (C-5), 113.8 (C-7), 67.5 (benzyl CH2), 66.1 (C-3), 46.4 (C-2′), 46.1 (C-5′), 30.3 (C-4′). MS (DUIS+) m/z 463 [M + H]+.
Synthesis of 1-(Phenylsulfonyl)spiro[indoline-3,3′-pyrrolidin]-2-one (17)
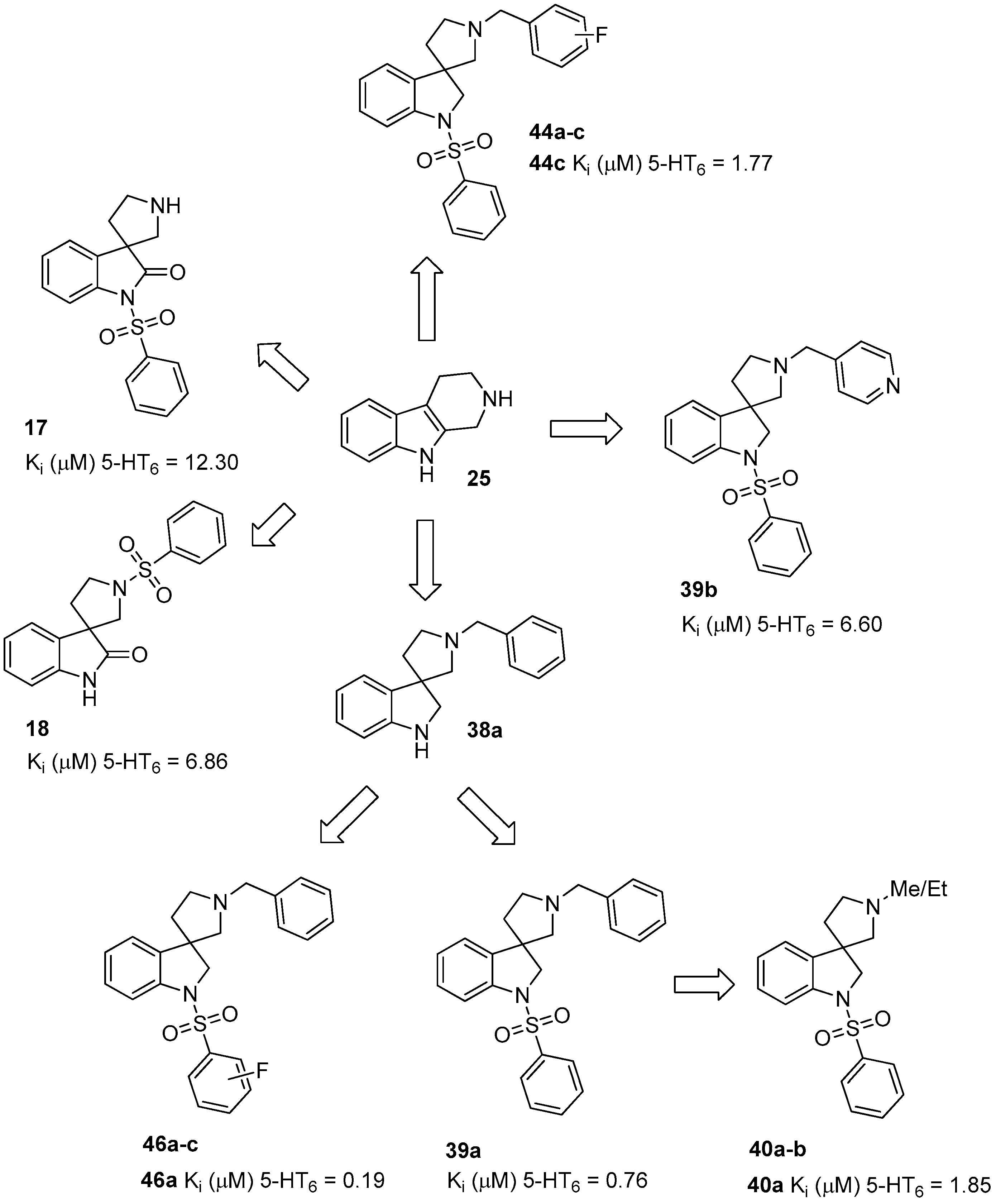
To the solution of benzyl 2-oxo-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine]-1′-carboxylate (28) (180 mg, 0.389 mmol) in methanol was added palladium (10%) on charcoal (90 mg, 5 w/w %) in an autoclave. The reactor was filled with 5 atm hydrogen gas and was stirred for 1 h at room temperature. The reaction mixture was then filtered on Celite and was evaporated under reduced pressure. The crude material was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 5% methanol) to give 17 as an orange solid. Yield: 108 mg (85%). 1H-NMR (500 MHz, CDCl3) δ ppm 8.10 (d, J = 8.0 Hz, 2H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-5″), 7.91 (d, J = 8.4 Hz, 1H, phenyl-sulfonyl ArH-4″), 7.65 (t, J = 7.2 Hz, 1H, ArH-7), 7.53 (t, J = 7.2 Hz, 2H, phenyl-sulfonyl ArH-2″, phenyl-sulfonyl ArH-6″), 7.47 (d, J = 7.2 Hz, 1H, ArH-4), 7.32 (t, J = 8.0 Hz, 1H, ArH-6), 7.19 (t, J = 8.0 Hz, 1H, ArH-5), 3.08 (m, 1H, H-2′), 2.93 (m, 1H, pyrrolidine H-2′), 2.74–2.69 (m, 2H, pyrrolidine H-5′), 2.28–2.23 (m, 1H, pyrrolidine H-4′), 2.11–2.04 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 175.2 (C-2), 166.8 (C-7a), 140.4 (phenyl-sulfonyl C-1″), 135.2 (C-3a), 133.0 (phenyl-sulfonyl C-4″), 129.5 (phenyl-sulfonyl C-3″, phenyl-sulfonyl C-5″), 128.4 (C-6), 127.1 (phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6″), 122.5 (C-4), 121.7 (C-5), 109.6 (C-7), 55.8 (C-3), 48.3 (pyrrolidine C-2′), 47.2 (pyrrolidine C-5′), 33.0 (pyrrolidine C-4′). MS (DUIS+) m/z 329 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C17H17N2O3S: 329.0960; found: 329.0949.
4.1.2. Synthesis of 1′-(Phenylsulfonyl)spiro[indoline-3,3′-pyrrolidin]-2-one (18)
To the solution of 2-(phenylsulfonyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole (29) (312 mg, 1 mmol) in tetrahydrofuran: water (10:10 mL) was added 10 mL glacial acetic acid at 0 °C. N-bromosuccinimide (178 mg, 1 mmol, 1 equiv.) was added slowly in portions at 0 °C. The reaction mixture was stirred for 1.5 h at 0 °C. Afterwards the reaction was quenched at 0 °C by 10 mL concentrated sodium-carbonate solution and was stirred for 0.5 h, allowing to warm up to room temperature. The mixture was extracted using ethyl acetate (2 × 10 mL). The combined organic phases were washed with concentrated sodium-hydrogen carbonate (2 × 10 mL) and by brine (2 × 10 mL). The organic phases were dried over sodium-sulfate, filtered and evaporated under reduced pressure. The crude product was purified using flash chromatography (gradient: hexane: ethyl acetate 0% to 20% ethyl acetate) to afford 18 as a colorless solid. Yield: 180 mg (55%). 1H-NMR (500 MHz, DMSO-d6) δ ppm 10.47 (s, 1H, NH), 7.87 (d, J = 7.3 Hz, 2H, phenyl-sulfonyl ArH-3″, ArH-5″), 7.76 (t, J = 7.3 Hz, 1H, phenyl-sulfonyl ArH-4″), 7.67 (t, J = 7.3 Hz, 2H, phenyl-sulfonyl ArH-2″, phenyl-sulfonyl ArH-6″), 7.15 (t, J = 7.3 Hz, 1H, ArH-4), 6.82 (t, J = 8.5 Hz, 2H, ArH-5, ArH-6), 6.69 (d, J = 7.3 Hz, 1H, ArH-7), 3.56 (t, J = 6.7 Hz, 1H, pyrrolidine H-2′), 3.35 (t, J = 7.3 Hz, 1H, pyrrolidine H-2′), 3.35 (d, J = 3.5 Hz, 2H, pyrrolidine H-5′), 2.10–2.05 (m, 1H, pyrrolidine H-4′), 1.94–1.88 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 181.4 (C-2), 167.0 (C-3a), 141.3 (C-7a), 135.8 (phenyl-sulfonyl C-1″), 133.3 (phenyl-sulfonyl C-4″), 129.5 (phenyl-sulfonyl C-3″, phenyl-sulfonyl C-5″), 128.3 (C-6), 127.4 (phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6″), 122.4 (C-4), 121.8 (C-5), 109.6 (C-7), 55.6 (C-3), 47.4 (pyrrolidine C-2′, pyrrolidine C-5′), 35.8 (pyrrolidine C-4′). MS (DUIS+) m/z 329 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C17H17N2O3S: 329.0960; found: 329.0957.
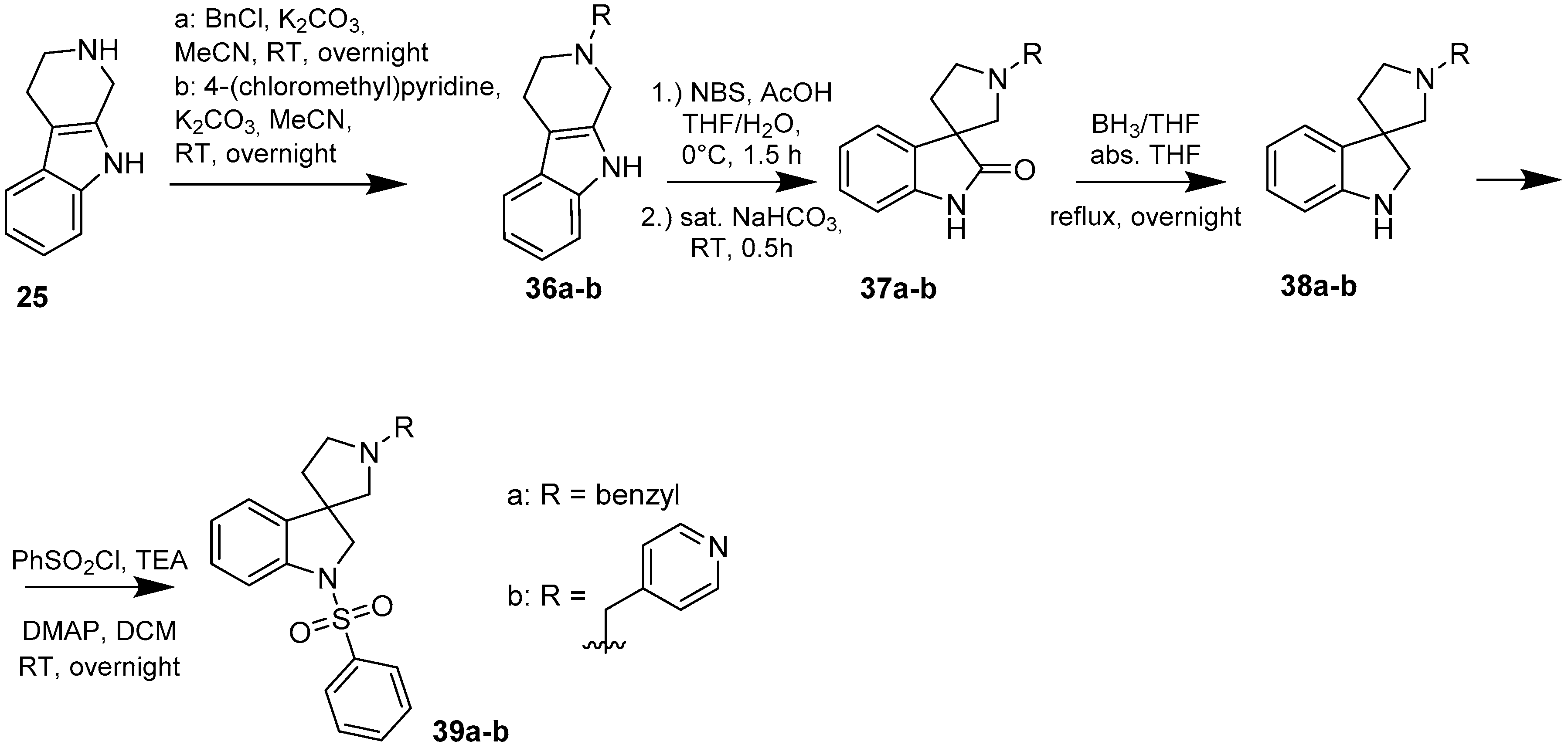
4.1.3. Procedures to Afford 1′-Benzyl-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (39a)
To the solution of 1′-benzylspiro[indoline-3,3′-pyrrolidin]-2-one (37a) (500 mg, 1.7985 mmol) in 15 mL absolutized tetrahydrofuran was added borane/tetrahydrofuran (1M) complex solution (4.495 mL, 4.495 mmol, 2.5 equiv.) and the reaction mixture was refluxed overnight. The reaction was allowed to cool down to room temperature and was quenched using 30 mL concentrated ammonium-chloride solution and was stirred for 20 min. Afterwards, 20 mL ethyl acetate was used for extraction. The combined organic phases were dried over sodium-sulfate, filtered and evaporated under reduced pressure to afford 400 mg 38a, used as a crude product in the further steps without purification. To the solution of crude 1′-benzylspiro[indoline-3,3′-pyrrolidine] (38a) (400 mg 1.513 mmol) (in anhydrous dichloromethane was added triethylamine (0.419 mL, 3.03 mmol, 2 equiv.) and N,N-dimethylpyridin-4-amine (10 mg, 0.076 mmol, 0.5 equiv.) at room temperature. Afterwards benzenesulfonyl chloride (0.203 mL, 1.59 mmol, 1.05 equiv.) was added to the solution at 5 °C. The reaction mixture was stirred at room temperature overnight. The reaction was quenched using 10 mL 10% HCl solution and was extracted by 10 mL dichloromethane. The dichloromethane phase was collected and dried over sodium-sulfate. The drying agent was filtered-off and the solution was evaporated under reduced pressure. The crude product was purified by flash chromatography to afford 39a as yellow oil. Yield: 621 mg (85% calculated to 37a). 1H-NMR (500 MHz, DMSO-d6) δ ppm 7.73 (d, J = 7.6 Hz, 2H, phenyl-sulfonyl H-2″, phenyl-sulfonyl H-6″), 7.58 (t, J = 7.6 Hz, 1H, H-6), 7.46 (t, J = 7.9 Hz, 3H, phenyl-sulfonyl H-3″, phenyl-sulfonyl H-4″, phenyl-sulfonyl H-5″), 7.31 (t, J = 7.6 Hz, 2H, phenyl-sulfonyl H-3″, phenyl-sulfonyl H-5″), 7.27–7.21 (m, 5H, benzyl H-1-5), 7.05 (t, J = 7.6 Hz, 1H, H-5), 3.89 (d, J = 10.9 Hz, 1H, H-2), 3.78 (d, J = 10.9 Hz, 1H, H-2), 3.52 (s, 2H, benzyl CH2), 2.69 (m, 1H, pyrrolidine H-2′), 2.57 (m, 1H, pyrrolidine H-2′), 2.24 (d, J = 9.1 Hz, 1H, pyrrolidine H-5′), 2.15 (d, J = 9.2 Hz, 1H, pyrrolidine H-5′), 1.91–1.85 (m, 1H, pyrrolidine H-4′), 1.78–1.73 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 141.0 (C-3a), 139.0 (C-7a), 134.2 (benzyl C-1), 129.8 (phenyl-sulfonyl C-1″), 129.8 (phenyl-sulfonyl C-4″), 128.7 (phenyl-sulfonyl C-3″, phenyl-sulfonyl C-5″), 128.7 (benzyl C-3, benzyl C-5), 127.4 (benzyl C-2, benzyl C-6), 127.4 (C-6), 124.9 (phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6″, benzyl C-4), 124.2 (C-4), 121.8 (C-5), 114.6 (C-7), 66.7 (pyrrolidine C-2′), 63.5 (benzyl CH2), 59.3 (C-3), 53.3 (pyrrolidine C-5′), 49.8 (C-4′). MS (DUIS+) m/z 405 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C24H25N2O2S: 405.1637; found: 405.1620.
4.1.4. Procedures to Afford 1-(Phenylsulfonyl)-1′-(pyridin-4-ylmethyl)spiro[indoline-3,3′-pyrrolidine] (39b)
Synthesis of 1′-(Pyridin-4-ylmethyl)spiro[indoline-3,3′-pyrrolidin]-2-one (37b)
To the solution of the crude 2-(pyridin-4-ylmethyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole (36b) (812 mg) in tetrahydrofuran:water (10:10 mL) was added 10 mL glacial acetic acid at 0 °C. N-bromosuccinimide (549 mg, 3.0874 mmol) was added slowly in portions at 0 °C. The reaction mixture was stirred for 1.5 h at 0 °C. Afterwards the reaction was quenched at 0 °C by 20 mL concentrated sodium-carbonate solution and was stirred for 0.5 h, allowing to warm up to room temperature. The mixture was extracted using ethyl-acetateethyl acetate (2 × 10 mL). The combined organic phases were washed with concentrated sodium-hydrogen carbonate (2 × 10 mL) and by brine (2 × 10 mL). The organic phases were dried over sodium-sulfate, filtered and evaporated under reduced pressure. The crude product was purified using flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) to afford 37b as a yellow solid. Yield: 293 mg (35% calculated to 25). 1H-NMR (500 MHz, DMSO-d6) δ ppm 11.63 (s, 1H, NH), 8.52 (d, J = 5.6 Hz, 2H, pyridine ArH-2, pyridine ArH-6), 7.59 (d, J = 7.2 Hz, 1H, ArH-4), 7.40 (d, J = 8.0 Hz, 1H, ArH-7), 7.32 (d, J = 5.5 Hz, 2H, pyridine ArH-3, pyridine ArH-5), 7.23 (t, J = 7.6 Hz, 1H, ArH-6), 7.05 (t, J = 7.3 Hz, 1H, ArH-5), 4.72 (s, 2H, pyridyl-methyl CH2), 3.65 (t, J = 6.7 Hz, 2H, pyrrolidine H-2′), 3.31 (m, 2H, pyrrolidine H-5′), 3.02 (t, J = 6.7 Hz, 2H, pyrrolidine H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 179.3 (C=O), 148.5 (pyridine C-2, pyridine C-6), 147.2 (pyridine C-1), 141.6 (C-7a), 134.7 (C-3a), 128.1 (C-6), 123.5 (C-4), 122.4 (pyridine C-3, pyridine C-5), 109.7 (C-7), 61.2 (pyridyl-methylene CH2), 60.1 (C-3), 54.4 (pyrrolidine C-2′), 53.6 (pyrrolidine C-5′), 34.7 (pyrrolidine C-4′). MS (DUIS+) m/z 280 [M + H]+.
Synthesis of 1-(Phenylsulfonyl)-1′-(pyridin-4-ylmethyl)spiro[indoline-3,3′-pyrrolidine] (39b)
To the solution of 1′-(pyridin-4-ylmethyl)spiro[indoline-3,3′-pyrrolidin]-2-one (37b) (289 mg, 1.05 mmol) in 10 mL anhydrousdry tetrahydrofuran was added borane/tetrahydrofuran (1 M) complex solution (2.63 mL, 2.63 mmol, 2.5 equiv.) and the reaction mixture was refluxed overnight. The reaction was allowed to cool down to room temperature and was quenched using 15 mL concentrated ammonium-chloride solution and was stirred for 20 min. Afterwards, 10 mL ethyl acetate was used for extraction. The combined organic phases were dried over sodium-sulfate, filtered and evaporated under reduced pressure to afford 292 mg 38b, used as a crude product in the further steps without purification. To the solution of crude 1′-(pyridin-4-ylmethyl)spiro[indoline-3,3′-pyrrolidine] (38b) (0.292 mg) in 5 mL anhydrous dichloromethane was added triethylamine (0.307 mL, 2.22 mmol) and N,N-dimethylpyridin-4-amine (6.8 mg, 0.0555 mmol) at room temperature. Afterwards benzenesulfonyl chloride (204 mg, 1.161 mmol) was added to the solution at 5 °C. The reaction mixture was stirred at room temperature overnight. The reaction was quenched using 10 mL 10% HCl solution and was extracted by 5 mL dichloromethane. The dichloromethane phase was collected and dried over sodium-sulfate. The drying agent was filtered-off and the solution was evaporated under reduced pressure. The crude product was purified by flash chromatography to afford 39b as yellow solid. Yield: 0.003 mg. 1H-NMR (500 MHz, DMSO-d6) δ ppm 8.50 (d, J = 6.2 Hz, 2H, pyridine ArH-2, pyridine ArH-6), 7.72 (d, J = 7.9 Hz, 2H, phenyl-sulfonyl ArH-2″, phenyl-sulfonyl ArH-6″), 7.58–7.53 (m, 3H, pyridine ArH-3, pyridine ArH-5, phenyl-sulfonyl ArH-4″), 7.49 (d, J = 7.8 Hz, 1H, ArH-7), 7.43 (t, J = 8.0 Hz, 2H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-5″), 7.28–7.23 (m, 2H, ArH-4, ArH-6), 7.07 (t, J = 7.1 Hz, 1H, ArH-5), 3.90 (d, J = 11.1 Hz, 1H, pyridyl methyl CH2), 3.83 (d, J = 11.1 Hz, 1H, pyridyl methyl CH2), 3.74 (d, J = 15.2 Hz, 1H, H-2), 3.67 (d, J = 15.2 Hz, 1H, H-2), 2.77 (m, 1H, pyrrolidine H-2′), 2.62 (m, 1H, pyrrolidine H-2′), 2.17 (m, 2H, pyrrolidine H-5′), 1.95–1.89 (m, 1H, pyrrolidine H-4′), 1.82–1.78 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 148.6 (pyridine C-2, pyridine C-6), 141.0 (pyridine C-1), 143.9 (C-3a), 143.1 (C-7a), 139.5 (phenyl-sulfonyl C-1″), 134.1 (phenyl-sulfonyl C-4″), 130.0 (phenyl-sulfonyl C-3,” phenyl-sulfonyl C-5″), 128.1 (C-6), 127.3 (phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6”), 127.6 (C-4), 124.2 (pyridine C-3, pyridine C-5), 124.8 (C-5), 114.1 (C-7), 65.6 (pyrrolidine C-2′), 61.5 (benzyl CH2), 60.1 (C-2), 53.0 (C-3), 51.3 (pyrrolidine C-5′), 32.9 (pyrrolidine C-4′). MS (DUIS+) m/z 406 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C23H24N3O2S: 406.1589; found: 406.1586.
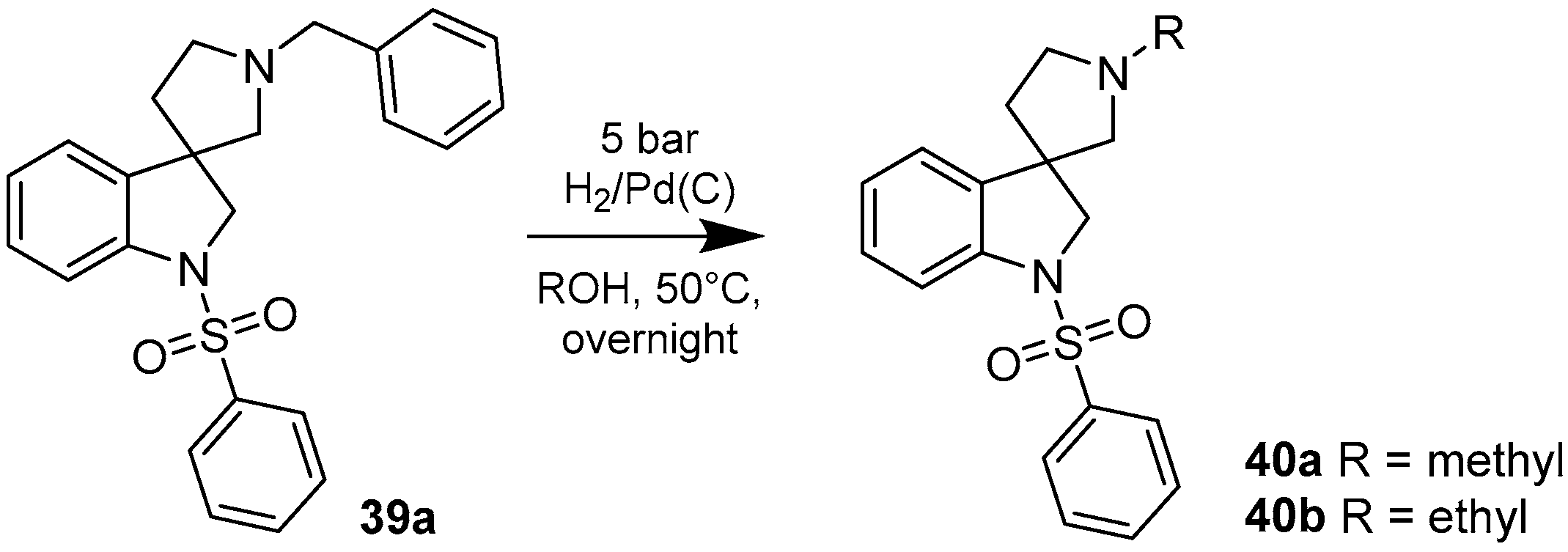
4.1.5. Synthesis of 1′-Methyl-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (40a)
The solution of 1′-benzyl-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (39a) (2 mg, 0.495 mmol) in 5 mL methanol was filled into an autoclave. Palladium (10%) on charcoal (100 mg, 5 w/w %) was added to the solution and reaction was stirred overnight under 5 bar hydrogen atmosphere at 50 °C. The reaction mixture was filtered through a pad of Celite and was evaporated under reduced pressure. The crude residual was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) to afford 40a as a colorless solid. Yield: 37 mg (23%). 1H-NMR (500 MHz, DMSO-d6) δ ppm 7.79 (d, J = 7.5 Hz, 2H, phenyl-sulfonyl ArH-2″, phenyl-sulfonyl ArH-6″), 7.66 (t, J = 7.2 Hz, 1H, phenyl-sulfonyl ArH-4″), 7.56 (t, J = 7.9 Hz, 2H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-5″), 7.48 (d, J = 7.9 Hz, 1H, ArH-7), 7.22 (m, 2H, ArH-4, H-6), 7.03 (t, J = 7.9 Hz, 1H, ArH-5), 3.87 (d, J = 11.8 Hz, 1H, H-2), 3.75 (d, J = 11.8 Hz, 1H, H-2), 2.62 (m, 1H, pyrrolidine H-2′), 2.44 (m, 1H, pyrrolidine H-2′), 2.24 (d, J = 9.1 Hz, 1H, pyrrolidine H-5′), 2.17 (s, 3H, methyl CH3), 2.15 (d, J = 9.1 Hz, 1H, pyrrolidine H-5′), 1.85–1.80 (m, 1H, pyrrolidine H-4′), 1.74–1.69 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 143.1 (C-3a, C-7a), 133.1 (phenyl-sulfonyl C-1″, phenyl-sulfonyl C-4″), 129.2 (phenyl-sulfonyl C-3″, phenyl-sulfonyl C-5″), 127.5 (C-6, phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6″), 124.1 (C-4), 114.5 (C-5), 110.0 (C-7), 69.1 (pyrrolidine C-2′), 60.2 (pyrrolidine C-5′), 55.5 (C-3), 52.8 (C-2), 42.3 (pyrrolidine N(1′)-methyl), 32.5 (pyrrolidine C-4′). MS (DUIS+) m/z 329 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C18H21N2O2S: 343.1324; found: 343.1320.
4.1.6. Synthesis of 1′-Ethyl-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (40b)
The solution of 1′-benzyl-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (39a) (100 mg, 0.2475 mmol) in 5 mL methanol was filled into an autoclave. Palladium (10%) on charcoal (500 mg, 5 w/w %) was added to the solution and reaction was stirred overnight under 5 bar hydrogen atmosphere at 50 °C. The reaction mixture was filtered through a pad of Celite and was evaporated under reduced pressure. The crude residual was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) to afford 40b as a colorless solid. Yield: 19 mg (22%). 1H-NMR (500 MHz, DMSO-d6) δ ppm 7.78 (m, 2H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-5″), 7.66 (m, 1H, phenyl-sulfonyl ArH-4″), 7.56 (m, 2H, phenyl-sulfonyl ArH-2″, phenyl-sulfonyl ArH-6″), 7.50–7.47 (m, 1H, ArH-7), 7.24–7.21 (m, 2H, ArH-4, ArH-6), 7.04 (t, J = 7.6 Hz, 1H, ArH-5), 3.86 (d, J = 10.8 Hz, 1H, H-2), 3.75 (d, J = 10.8 Hz, 1H, H-2), 2.68 (m, 1H, pyrrolidine H-2′), 2.44 (m, 1H, pyrrolidine H-2′), 2.34 (q, J = 6.4 Hz, 2H, ethyl CH2), 2.24 (m, 1H, pyrrolidine H-5′), 2.11 (q, J = 8.9 Hz, 1H, pyrrolidine H-5′), 1.86–180 (m, 1H, pyrrolidine H-4′), 1.73–167 (m, 1H, pyrrolidine H-4′), 0.93 (t, J = 7.6 Hz, 3H, ethyl CH3). 13C-NMR (125 MHz, DMSO-d6) δ ppm 143.3 (C-3a, C-7a), 133.1 (phenyl-sulfonyl C-1″, phenyl-sulfonyl C-4″), 129.5 (phenyl-sulfonyl C-3″, phenyl-sulfonyl C-5″), 127.7 (C-6, phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6″), 124.1 (C-4), 114.1 (C-5), 110.1 (C-7), 69.1 (pyrrolidine C-2′), 60.2 (pyrrolidine C-5′), 55.3 (C-3), 52.1 (C-2), 50.0 (pyrrolidine N(1′)-ethyl CH2), 32.5 (pyrrolidine C-4′), 13.6 (pyrrolidine N(1′)-ethyl CH3). MS (DUIS+) m/z 343 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C19H23N2O2S: 343.1480; found: 343.1496.
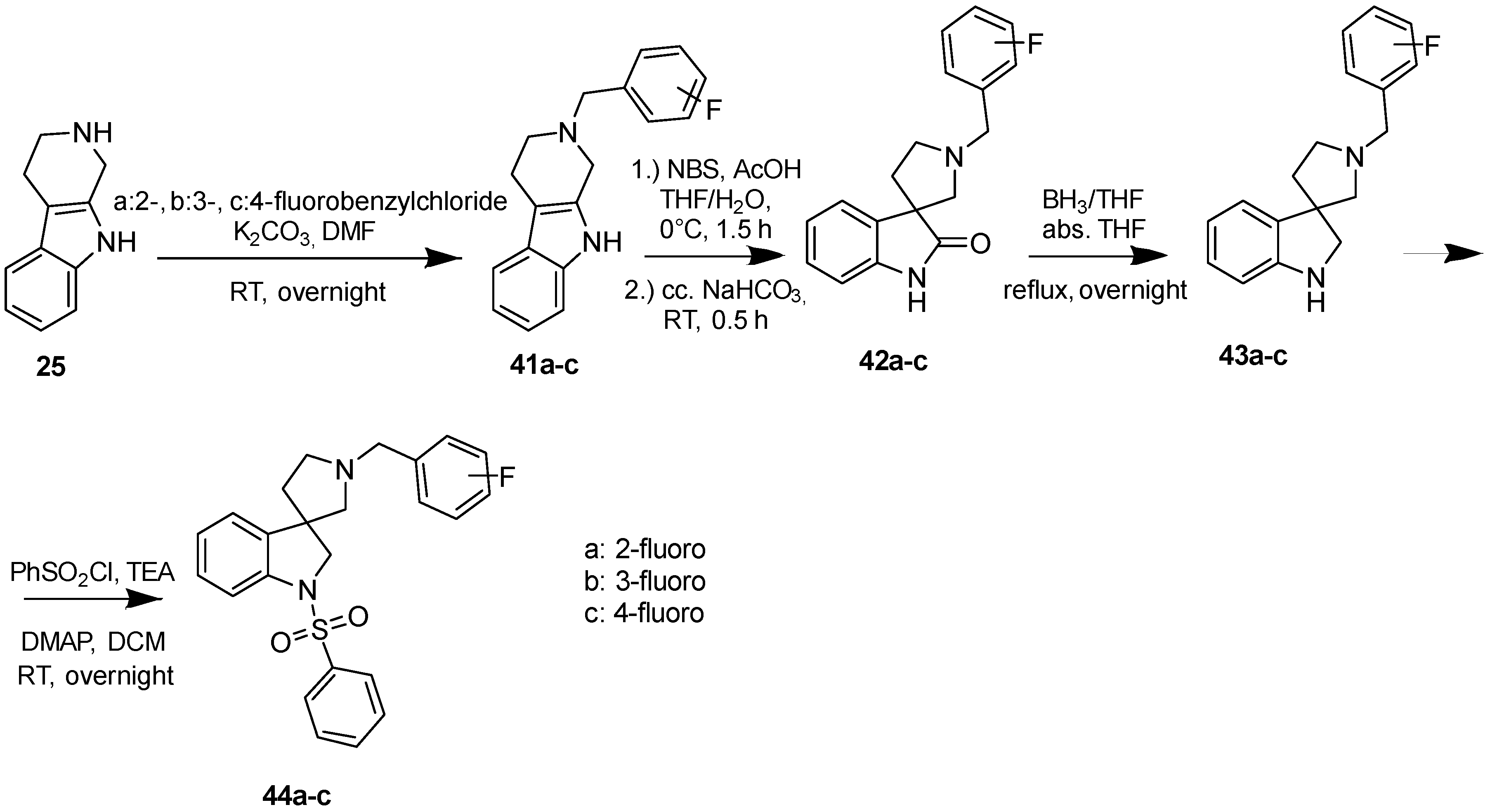
4.1.7. Procedures to Afford 1′-(2-Fluorobenzyl)-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (44a)
Synthesis of 1′-(2-Fluorobenzyl)spiro[indoline-3,3′-pyrrolidin]-2-one (42a)
To the solution of 41a (1.54 g, 5.5 mmol) in tetrahydrofuran: water (30:30 mL) was added 30 mL glacial acetic acid at 0 °C. N-bromosuccinimide (979 mg, 5.5 mmol, 1 equiv.) was added slowly in portions at 0 °C. The reaction mixture was stirred for 1.5 h at 0 °C. Afterwards the reaction was quenched at 0 °C by 40 mL concentrated sodium-carbonate solution and was stirred for 0.5 h, allowing to warm up to room temperature. The mixture was extracted using ethyl acetate (2 × 20 mL). The combined organic phases were washed with concentrated sodium-hydrogen carbonate (2 × 20 mL) and by brine (2 × 20 mL). The organic phases were dried over sodium-sulfate, filtered and evaporated under reduced pressure. The product 42a was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) Yield: 970 mg (56%). 1H-NMR (500 MHz, DMSO-d6) δ ppm 10.31 (s, 1H, NH), 8.01 (d, J = 8.8 Hz, 1H, benzyl ArH-6), 7.77 (d, J = 7.9 Hz, 1H, ArH-4), 7.65 (t, J = 7.9 Hz, 1H, benzyl ArH-4), 7.47 (t, J = 6.2 Hz, 1H, benzyl ArH-5), 7.24 (t, J = 8.4 Hz, 1H, ArH-6), 7.16 (m, 1H, benzyl ArH-3), 6.94 (t, J = 7.5 Hz, 1H, ArH-5), 6.80 (d, J = 7.5 Hz, 1H, ArH-7), 3.75 (s, 2H, benzyl CH2), 3.06 (m, 1H, pyrrolidine H-2′), 2.76 (d, J = 8.9 Hz, 1H, pyrrolidine H-5′), 2.69 (d, J = 8.9 Hz, 1H, pyrrolidine H-5′), 2.61 (q, J = 7.9 Hz, 1H, pyrrolidine H-2’), 2.20–2.15 (m, 1H, pyrrolidine H-4′), 1.92–1.87 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 181.5 (C=O), 178.0 (benzyl C-2), 141.6 (C-7a), 137.0 (C-3a), 135.5 (benzyl C-6), 130.2 (benzyl C-4), 127.6 (C-6), 124.7 (benzyl C-1), 123.9 (benzyl C-5), 123.3 (C-4), 122.3 (C-5), 115.7 (benzyl C-3), 109.6 (C-7), 64.0 (C-3), 53.86 (benzyl CH2), 52.9 (pyrrolidine C-2′), 51.7 (pyrrolidine C-5′), 37.1 (pyrrolidine C-4′). MS (DUIS+) m/z 297 [M + H]+.
Synthesis of 1′-(2-Fluorobenzyl)-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (44a)
Compound 43a was prepared according to the same procedure as 38a, starting from 42a (100 mg, 0.3378 mmol), using borane-tetrahydrofuran (0.6757 mL, 0.6757 mmol, 2.5 equiv.), to afford 10 mg crude product used in the next step without further purification. Compound 44a was prepared according to the same procedure as 39a, starting from crude 43a (10 mg, 0.03545 mmol), using benzylsulfonyl chloride (50 mg, 0.07445 mmol, 1.05 equiv.), triethylamine (0.010 mL, 0.0709 mmol, 2 equiv.) and N,N-dimethylpyridin-4-amine (0.2 mg, 0.0017 mmol, 0.5 equiv.). The product was purified by preparative-HPLC (gradient: water: acetonitrile 0% to 100% acetonitrile) Yield: 2 mg. 1H-NMR (500 MHz, CDCl3) δ ppm 8.22 (t, J = 6.7 Hz, 2H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-5″), 7.88 (m, 1H, phenyl-sulfonyl ArH-4″), 7.69 (m, 2H, phenyl-sulfonyl ArH-2″, phenyl-sulfonyl ArH-6″), 7.53 (t, J = 6.2 Hz, 1H, benzyl ArH-4), 7.47 (m, 1H, ArH-7), 7.33 (t, J = 7.8 Hz, 1H, benzyl ArH-5), 7.23 (t, J = 7.8 Hz, 1H, benzyl ArH-6), 7.17–7.08 (m, 2H, ArH-5, ArH-6), 6.73 (d, J = 7.0 Hz, 2H, benzyl ArH-3), 4.22 (m, 2H, H-2), 3.98 (m, 2H, benzyl CH2), 3.75 (m, 1H, pyrrolidine H-2′), 3.40 (m, 1H, pyrrolidine H-2′), 3.14 (m, 1H, pyrrolidine H-5′), 2.98 (m, 1H, pyrrolidine H-5′), 2.28 (m, 1H, pyrrolidine H-4′), 1.51 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, CDCl3) δ ppm 159.3 (benzyl C-2), 144.1 (C-3a), 142.5 (C-7a), 137.3 (phenyl-sulfonyl C-1″), 134.2 (phenyl-sulfonyl C-4″), 130.3 (benzyl C-6), 129.5 (phenyl-sulfonyl C-3″, phenyl-sulfonyl C-5″), 129.2 (benzyl C-4), 128.2 (C-6), 127.6 (phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6″), 125.8 (benzyl C-1), 125.3 (benzyl C-5), 125.0 (C-4), 121.6 (C-5), 115.7 (benzyl C-3), 113.0 (C-7), 62.3 (C-2′), 59.6 (benzyl CH2), 55.8 (C-3), 54.9 (C-5′), 53.6 (C-2), 33.1 (C-4′). MS (DUIS+) m/z 423 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C24H24N2O2FS: 423.1543; found: 423.1531.
4.1.8. Procedures to Afford 1′-(3-Fluorobenzyl)-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (44b)
Synthesis of 1′-(3-Fluorobenzyl)spiro[indoline-3,3′-pyrrolidin]-2-one (42b)
To the solution of 41b (1.681 g, 6 mmol) in tetrahydrofuran: water (30:30 mL) was added 30 mL glacial acetic acid at 0 °C. N-bromosuccinimide (1.069 g, 6 mmol, 1 equiv.) was added slowly in portions at 0 °C. The reaction mixture was stirred for 1.5 h at 0 °C. Afterwards the reaction was quenched at 0 °C by 40 mL concentrated sodium-carbonate solution and was stirred for 0.5 h, allowing to warm up to room temperature. The mixture was extracted using ethyl acetate (2 × 20 mL). The combined organic phases were washed with concentrated sodium-hydrogen carbonate (2 × 20 mL) and by brine (2 × 20 mL). The organic phases were dried over sodium-sulfate, filtered and evaporated under reduced pressure. The product 42b was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) Yield: 1.31 g (76% calculated to 29). 1H-NMR (500 MHz, DMSO-d6) δ ppm 10.31 (s, 1H, NH), 7.38–7.33 (m, 2H, benzyl H-5, benzyl ArH-6), 7.21–7.16 (m, 3H, ArH-4, benzyl H-2, benzyl ArH-4), 7.04 (m, 1H, ArH-6), 6.98 (t, J = 7.0 Hz, 1H, ArH-5), 6.81 (d, J = 8.0 Hz, 1H, ArH-7), 3.72 (s, 2H, benzyl CH2), 3.07 (m, 1H, pyrrolidine H-2′), 2.74 (d, J = 8.8 Hz, 1H, pyrrolidine H-5′), 2.66 (d, J = 8.8 Hz, 1H, pyrrolidine H-2′), 2.56 (m, 1H, pyrrolidine H-5′), 2.20 (m, 1H, pyrrolidine H-4′), 1.90 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, CDCl3) δ ppm 181.96 (C=O), 161.2 (benzyl C-3), 142.3 (C-7a), 141.4 (benzyl C-1), 130.4 (benzyl C-5), 127.8 (benzyl-C6, C-6), 122.1 (benzyl C-2, benzyl C-4), 109.4 (C-7), 64.7 (benzyl CH2), 63.9 (C-3), 53.9 (pyrrolidine C-2′), 52.7 (C-5′), 36.8 (pyrrolidine C-4′). MS (DUIS+) m/z 297 [M + H]+.
Synthesis of 1′-(3-Fluorobenzyl)-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (44b)
Compound 43b was prepared according to the same procedure as 38a, starting from 42b (385 mg, 1.3 mmol), using borane-tetrahydrofuran (3.250 mL, 3.25 mmolm 2.5 equiv.), to afford 338 mg crude product used in the next time without further purification. Compound 44b was prepared according to the same procedure as 39a, starting from crude 43b (338 mg, 1.197 mmol), using benzylsulfonyl chloride (221 mg, 1.2579 mmol, 1.05 equiv.), triethylamine (0.331 mL, 2.396 mmol, 2 equiv.) and N,N-dimethylpyridin-4-amine (7 mg, 0.0599 mmol, 0.5 equiv.). The product was purified by preparative HPLC (gradient: dichloromethane: methanol 0% to 10% methanol) Yield: 11 mg. 1H-NMR (500 MHz, CDCl3) δ ppm 7.83 (d, J = 8.2 Hz, 1H, ArH-7), 7.77 (d, J = 7.6 Hz, 1H, benzyl ArH-6), 7.73 (d, J = 7.6 Hz, 1H, ArH-4), 7.64 (d, J = 8.2 Hz, 1H, benzyl ArH-4), 7.58–7.53 (m, 1H, benzyl ArH-5), 7.50–7.39 (m, 3H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-4″, phenyl-sulfonyl ArH-5″), 7.37–7.27 (m, 2H, phenyl-sulfonyl ArH-2″, phenyl-sulfonyl ArH-6″), 7.16–7.12 (m, 1H, phenyl-sulfonyl ArH-6), 7.09–7.02 (m, 1H, phenyl-sulfonyl ArH-5), 6.99 (m, 1H, benzyl H-2), 4.23 (m, 1H, H-2), 4.11 (m, 1H, H-2), 3.93–3.71 (m, 2H, benzyl CH2), 3.23 (m, 1H, pyrrolidine H-2′), 2.94 (m, 1H, pyrrolidine H-2′), 2.83 (m, 1H, pyrrolidine H-5′), 2.69 (m, 1H, pyrrolidine H-5′), 2.39–2.25 (m, 1H, pyrrolidine H-4′), 2.18 (m, 1H, pyrrolidine H-5′). 13C-NMR (125 MHz, CDCl3) δ ppm 163.0 (benzyl C-3), 143.6 (C-3a), 143.5 (C-7a), 137.6 (phenyl-sulfonyl C-1″), 134.2 (phenyl-sulfonyl C-4″), 130.4 (benzyl C-5), 129.5 (phenyl-sulfonyl C-3″, phenyl-sulfonyl C-5″), 129.1 (C-6), 128.0 (phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6″), 127.7 (benzyl C-6), 125.6 (benzyl C-1), 125.3 (C-4), 124.1 (C-5), 121.8, 115.8 (benzyl C-2), 113.4 (C-7), 113.3 (benzyl C-4), 61.2 (benzyl CH2), 59.9 (pyrrolidine C-2′), 55.3 (C-3), 54.8 (pyrrolidine C-5′), 53.6 (C-2), 33.1 (pyrrolidine C-4’). MS (DUIS+) m/z 423 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C24H24N2O2SF: 423.1543; found: 423.1558.
4.1.9. Procedures to Afford 1′-(4-Fluorobenzyl)-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (44c)
Synthesis of 1′-(4-Fluorobenzyl)spiro[indoline-3,3′-pyrrolidin]-2-one (42c)
To the solution of 41c (2.361 g, 8.4321 mmol) in tetrahydrofuran: water (30:30 mL) was added 30 mL glacial acetic acid at 0 °C. N-bromosuccinimide (1.501 g, 8.4321 mmol, 1 equiv.) was added slowly in portions at 0 °C. The reaction mixture was stirred for 1.5 h at 0 °C. Afterwards the reaction was quenched at 0 °C by 40 mL concentrated sodium-carbonate solution and was stirred for 0.5 h, allowing to warm up to room temperature. The mixture was extracted using ethyl acetate (2 × 20 mL). The combined organic phases were washed with concentrated sodium-hydrogen carbonate (2 × 20 mL) and by brine (2 × 20 mL). The organic phases were dried over sodium-sulfate, filtered and evaporated under reduced pressure. The product 42c was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) Yield: 320 mg (19% calculated to 25). 1H-NMR (500 MHz, DMSO-d6) δ ppm 10.30 (s, 1H, NH), 7.41–7.35 (m, 3H, benzyl H-2, benzyl ArH-6, ArH-4), 7.16 (m, 3H, benzyl ArH-3, benzyl ArH-5, ArH-6), 6.96 (t, J = 7.2 Hz, 1H, ArH-5), 6.81 (d, J = 7.4 Hz, 1H, ArH-7), 5.73 (s, 2H, benzyl CH2), 3.03 (m, 1H, pyrrolidine H-5′), 2.73 (d, J = 9.3 Hz, 1H, pyrrolidine H-2′), 2.64 (d, J = 9.3 Hz, 1H, pyrrolidine H-2′), 2.58–2.53 (m, 1H, pyrrolidine H-5′), 2.21–2.16 (m, 1H, pyrrolidine H-4′), 1.95–1.87 (m, 1H, pyrrolidine H-4′) 13C-NMR (125 MHz, DMSO-d6) δ ppm 179.1 (C=O), 161.9 (benzyl C-4), 141.6 (C-7a), 138.2 (benzyl C-1), 134.6 (C-3a), 130.7 (benzyl C-2, benzyl C-6), 128.0 (C-6), 123.3 (C-4), 122.3 (C-2), 115.4 (benzyl C-3, benzyl C-5), 109.6 (C-7), 64.0 (benzyl CH2), 58.3 (C-3), 54.0 (pyrrolidine C-2′), 52.9 (pyrrolidine C-5′), 37.0 (pyrrolidine C-4′). MS (DUIS+) m/z 297 [M + H]+.
Synthesis of 1′-(4-Fluorobenzyl)-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (44c)
Compound 43c was prepared according to the same procedure as 38a, starting from 42c (310 mg, 1.0473 mmol), using borane-tetrahydrofuran (2.620 mL, 2.620 mmol, 2.5 equiv.), to afford 461 mg crude product used in the next step without further purification. Compound 44c was prepared according to the same procedure as 39a, starting from crude 43c (100 mg, 0.354 mmol), using benzylsulfonyl chloride (131 mg, 0.745 mmol, 1.05 equiv.), triethylamine (0.099 mL, 0.7092 mmol) and N,N-dimethylpyridin-4-amine (2 mg, 0.01773 mmol). The product 44c was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) Yield: 10 mg. 1H-NMR (500 MHz, CDCl3) δ ppm 7.84–7.77 (m, 2H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-5″), 7.74–7.70 (m, 1H, phenyl-sulfonyl ArH-4″), 7.67–7.57 (m, 3H, phenyl-sulfonyl ArH-2″, phenyl-sulfonyl ArH-6″, ArH-7), 7.50–7.45 (m, 2H, benzyl ArH-2, benzyl ArH-6), 7.43–7.40 (m, 1H, ArH-4), 7.24–7.20 (m, 1H, ArH-6), 7.16–7.12 (m, 2H, benzyl ArH-3, benzyl ArH-5), 7.09 (m, 1H, ArH-5), 4.39 (m, 1H, H-2), 4.29 (m, 1H, H-2), 4.18 (m, 1H, benzyl CH2), 413.06 (m, 1H, benzyl CH2), 3.90 (m, 1H, pyrrolidine H-2′), 3.70 (m, 1H, pyrrolidine H-2′), 3.06 (m, 1H), 2.68 (m, 1H, pyrrolidine H5′), 2.33 (m, 1H, pyrrolidine H-5′), 2.33 (m, 1H, pyrrolidine H-4′), 1.75 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, CDCl3) δ ppm 159.8 (benzyl C-4), 144.1 (C-3a), 142.6 (C-7a), 137.5 (benzyl C-1), 134.2 (phenyl-sulfonyl C-1″, phenyl-sulfonyl C-4″), 130.7 (benzyl C-2, benzyl C-6), 129.2 (phenyl-sulfonyl C-3″, phenyl-sulfonyl C-5″), 128.3 (C-6), 127.7 (phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6″), 125.8 (C-4), 125.2 (C-5), 115.5 (benzyl C-3, benzyl C-5), 113.1 (C-7), 62.4 (pyrrolidine C-2′), 59.8 (benzyl CH2), 55.5 (C-3), 55.0 (pyrrolidine C-5′), 53.7 (C-2), 33.0 (pyrrolidine C-4′). MS (DUIS+) m/z 423 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C24H24N2O2SF: 423.1543; found: 423.1542.
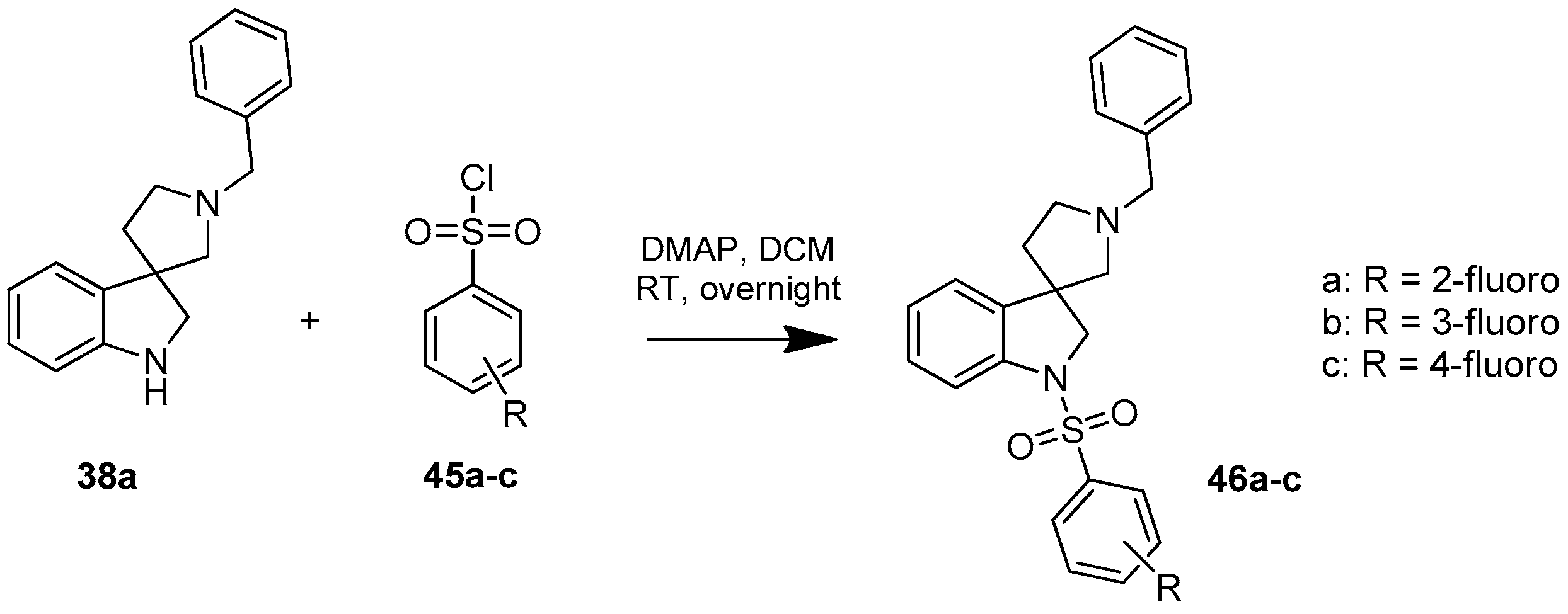
4.1.10. Synthesis of 1′-Benzyl-1-((2-fluorophenyl)sulfonyl)spiro[indoline-3,3′-pyrrolidine] (46a)
Compound 46a was prepared according to the same procedure as 39a, starting from crude 38a (15 mg, 0.0567 mmol), using 2-fluorobenzylsulfonyl chloride (23 mg, 0.11819 mmol), triethylamine (0.015 mL, 0.1188 mmol, 2.1 equiv.) and N,N-dimethylpyridin-4-amine (0.7 mg, 0.006 mmol, 0.1 equiv.). The product was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) Yield: 6.5 mg. 1H-NMR (500 MHz, CDCl3) δ ppm 8.00 (t, J = 6.7 Hz, 1H, phenyl-sulfonyl ArH-4″), 7.53 (m, 3H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-6″, ArH-7), 7.41–7.34 (m, 5H, benzyl ArH-2, benzyl ArH-3, benzyl ArH-5, benzyl ArH-6, ArH-4), 7.27 (t, J = 8.1 Hz, 1H, phenyl-sulfonyl ArH-5″), 7.17 (t, J = 8.1 Hz, 1H, benzyl ArH-4), 7.10 (t, J = 9.4 Hz, 1H, ArH-6), 7.04 (t, J = 7.6 Hz, 1H, ArH-5), 4.16–4.07 (m, 5H, benzyl CH2, H-2, pyrrolidine H-2′), 3.34 (m, 1H, pyrrolidine H-2′), 3.17 (m, 1H, pyrrolidine H-5′), 3.05 (m, 1H, pyrrolidine H-5′), 2.36 (m, 1H, pyrrolidine H-4′), 2.14 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 155.6 (phenyl-sulfonyl C-2″), 149.7 (C-3a), 142.8 (C-7a), 133.2 (benzyl C-1), 131.8 (phenyl-sulfonyl C-4″), 131.0 (phenyl-sulfonyl C-6″), 129.6 (benzyl C-3, benzyl C-5), 128.4 (benzyl C-2, benzyl C-6), 124.2 (phenyl-sulfonyl C-5″), 122.9 (benzyl C-4), 122.5 (C-4), 121.0 (C-5), 120.4 (phenyl-sulfonyl C-1″), 116.8 (phenyl-sulfonyl C-3″), 111.3 (C-7), 63.2 (pyrrolidine C-2′), 62.6 (benzyl CH2), 55.1 (C-3), 52.5 (pyrrolidine C-5′), 49.2 (C-2), 37.1 (pyrrolidine C-4′). MS (DUIS+) m/z 423 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C24H24N2O2SF: 423.1543; found: 423.1550.
4.1.11. Synthesis of 1′-Benzyl-1-((3-fluorophenyl)sulfonyl)spiro[indoline-3,3′-pyrrolidine] (46b)
Compound 45b was prepared according to the same procedure as 39a, starting from crude 38a (15 mg, 0.0567 mmol), using 3-fluorobenzylsulfonyl chloride (23 mg, 0.1188 mmol, 2.1 equiv.), triethylamine (0.015 mL, 0.1188 mmol, 2.1 equiv.) and N,N-dimethylpyridin-4-amine (0.7 mg, 0.006 mmol, 0.1 equiv.). The product was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) Yield: 5.5 mg. 1H-NMR (500 MHz, CDCl3) δ ppm 8.25 (m, 1H, phenyl-sulfonyl ArH-2″), 7.65–7.58 (m, 2H, phenyl-sulfonyl ArH-5″, phenyl-sulfonyl ArH-6″), 7.48–7.38 (m, 6H, ArH-7, benzyl ArH-2–6), 7.29–7.25 (m, 2H, ArH-4, ArH-6), 7.21 (m, 1H, phenyl-sulfonyl ArH-4″), 7.08 (t, J = 7.6 Hz, 1H, ArH-5), 4.16 (d, J = 12.7 Hz, 1H, benzyl CH2), 4.09 (d, J = 12.7 Hz, 1H, benzyl CH2), 3.99 (d, J = 10.9 Hz, 1H, H-2), 3.85 (d, J = 10.9 Hz, 1H, H-2), 3.33–3.24 (m, 2H, pyrrolidine H-2′), 3.12 (d, J = 11.6 Hz, 1H, pyrrolidine H-5′), 3.00 (d, J = 11.6 Hz, 1H, pyrrolidine H-5′), 2.26–2.20 (m, 1H, pyrrolidine H-4′), 2.05–2.00 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, CDCl3) δ ppm 158.6 (phenyl-sulfonyl C-3″), 146.8 (C-3a), 145.4 (C-7a), 137.5 (benzyl C-1), 130.5 (phenyl-sulfonyl C-1″), 129.7 (phenyl-sulfonyl C-5″), 129.0 (benzyl C-3, benzyl C-5), 128.7 (benzyl C-2, benzyl C-6), 124.5 (C-6), 122.8 (benzyl C-4), 122.7 (phenyl-sulfonyl C-6″), 121.7 (C-4), 120.2 (C-5), 117.7 (phenyl-sulfonyl C-4″), 114.3 (phenyl-sulfonyl C-2″), 109.5 (C-7), 63.0 (pyrrolidine C-2′), 62.6 (benzyl CH2), 55.4 (C-3), 52.7 (pyrrolidine C-5′), 49.2 (C-2), 37.9 (pyrrolidine C-4′). MS (DUIS+) m/z 423 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C24H24N2O2SF: 423.1543; found: 423.1547.
4.1.12. Synthesis of 1′-Benzyl-1-((4-fluorophenyl)sulfonyl)spiro[indoline-3,3′-pyrrolidine] (46c)
Compound 46c was prepared according to the same procedure as 39a, starting from crude 38a (15 mg, 0.0567 mmol), using 4-fluorobenzylsulfonyl chloride (23 mg, 0.1182 mmol, 2.1 equiv.), triethylamine (0.015 mL, 0.1188 mmol, 2.1 equiv.) and N,N-dimethylpyridin-4-amine (0.7 mg, 0.006 mmol, 0.1 equiv.). The product was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) Yield: 7.7 mg. 1H-NMR (500 MHz, CDCl3) δ ppm 7.80 (m, 2H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-5″), 7.63 (d, J = 7.9 Hz, 1H, ArH-7), 7.47 (m, 2H, benzyl H-3, benzyl ArH-5), 7.40 (m, 3H, benzyl ArH-2, benzyl ArH-4, benzyl ArH-6), 7.28 (m, 2H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-5″), 7.09–7.05 (m, 3H, ArH-4, ArH-5, ArH-6), 4.10 (d, J = 12.3 Hz, 1H, H-2), 4.02–3.96 (m, 2H, benzyl CH2), 3.84 (d, J = 11.3 Hz, 1H, H-2), 3.25 (m, 1H, pyrrolidine H-2′), 3.16 (m, 1H, pyrrolidine H-2′), 2.94 (m, 1H, pyrrolidine H-5′), 2.89 (m, 1H, pyrrolidine H-5′), 2.24–2.18 (m, 1H, pyrrolidine H-4′), 2.03–1.99 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, CDCl3) δ ppm 163.7 (phenyl-sulfonyl C-4″), 152.7 (C-3a), 150.6 (C-7a), 147.0 (benzyl C-1), 129.6 (phenyl-sulfonyl C-1″), 129.5 (phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6″), 129.2 (benzyl C-3, benzyl C-5), 129.1 (benzyl C-2, benzyl C-6), 128.8 (C-6), 128.6 (benzyl C-4), 125.6 (C-4), 124.4 (C-5), 122.8 (phenyl-sulfonyl C-3″, phenyl-sulfonyl C-5″), 116.1 (C-7), 63.1 (pyrrolidine C-2′), 62.4 (benzyl CH2), 54.1 (C-3), 52.8 (pyrrolidine C-5′), 49.1 (pyrrolidine C-2′), 36.6 (pyrrolidine C-4′). MS (DUIS+) m/z 423 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C24H24N2O2SF: 423.1543; found: 423.1563.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






















