
Synthesis of Randomly Substituted Anionic Cyclodextrins in Ball Milling



 ,
,
Abstract
:1. Introduction
2. Results and Discussion
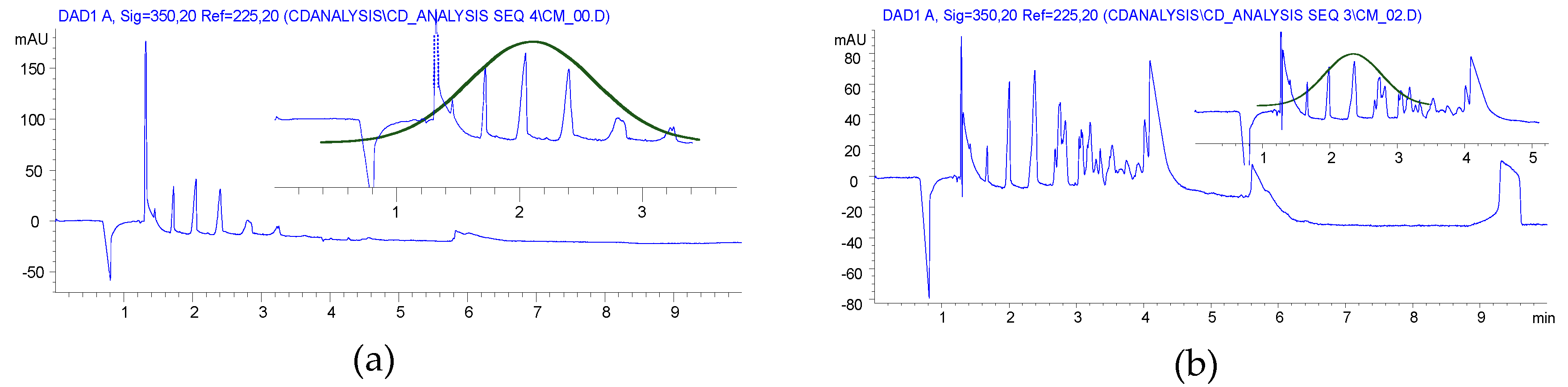
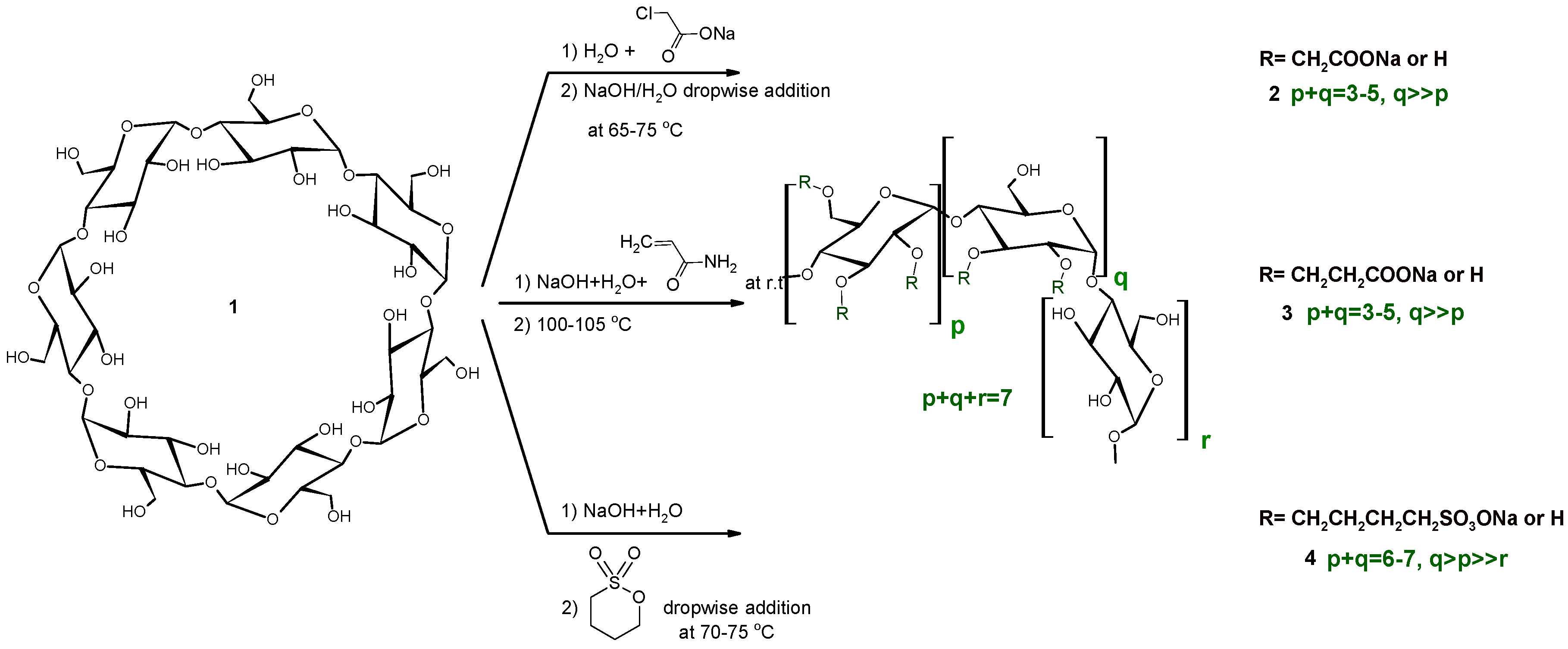
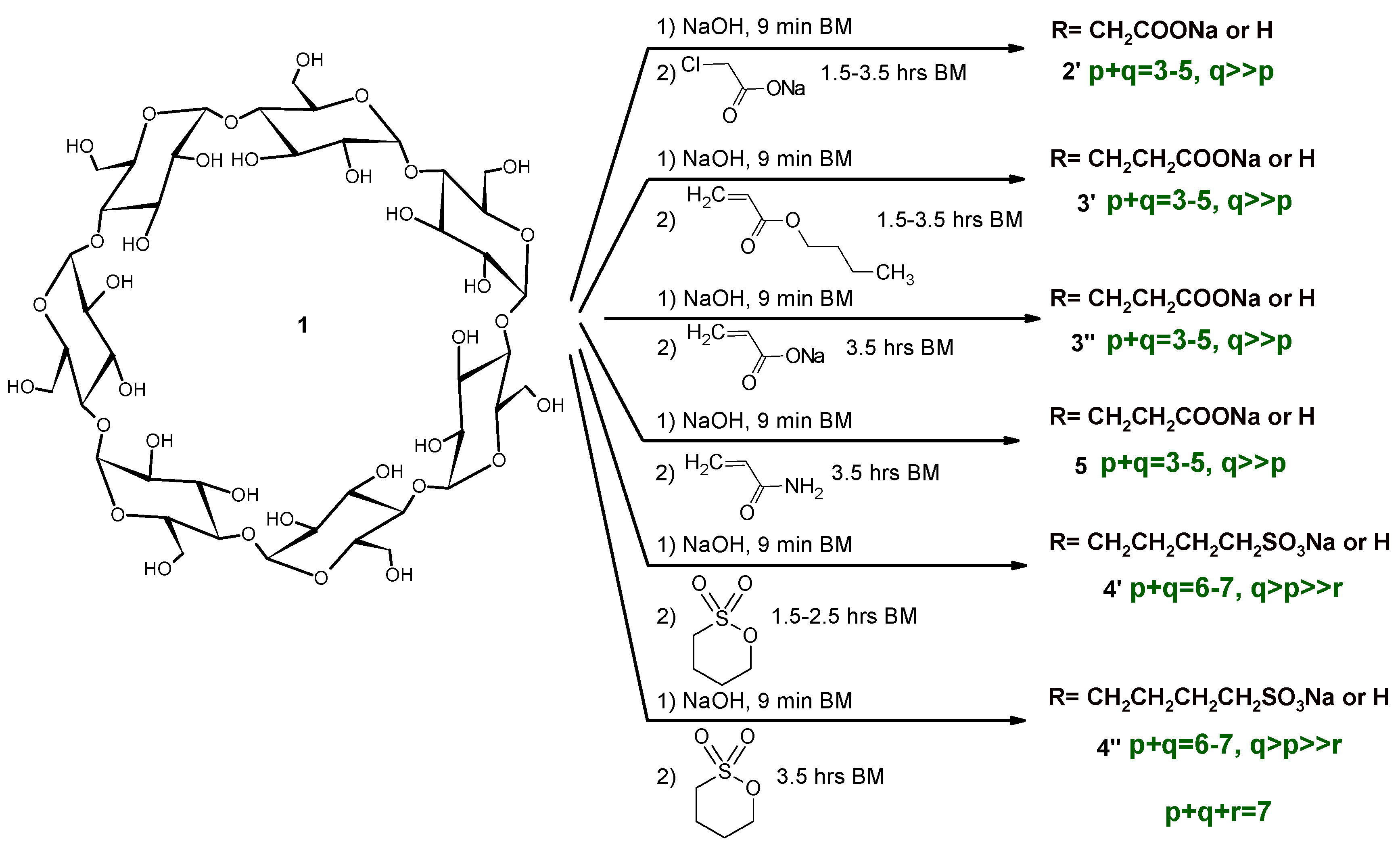
2.1. Carboxymethylation (Compounds 2 and 2’)
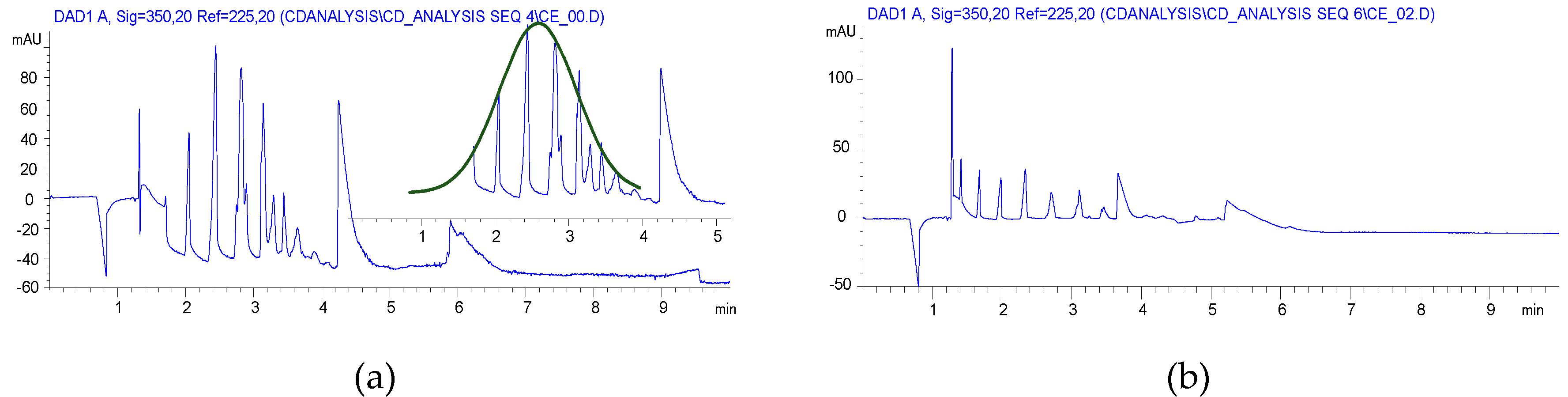
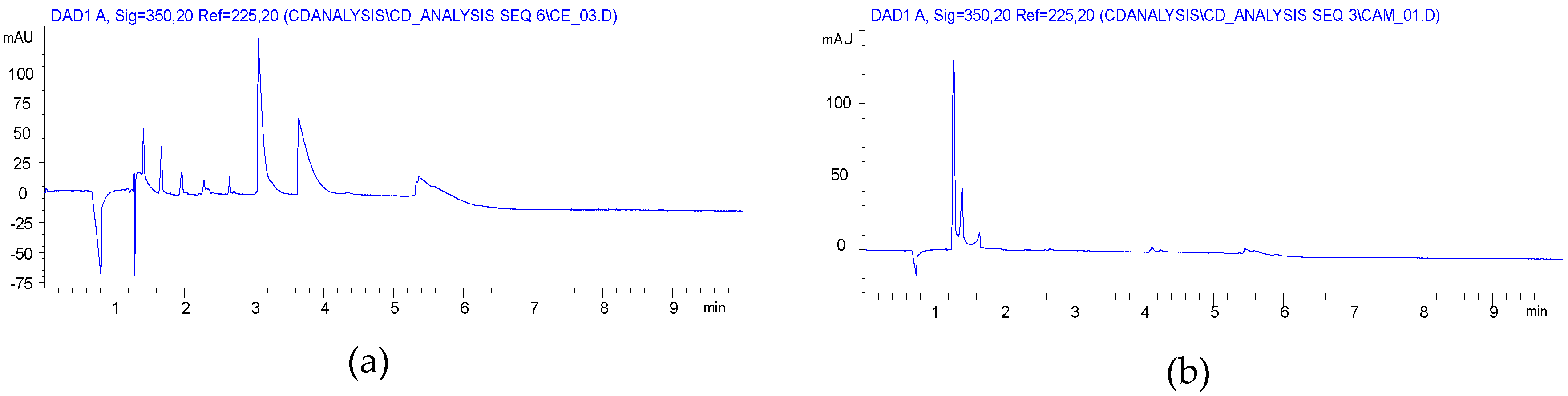
2.2. Carboxyethylation (Compounds 3 and 3′)
2.3. Carbamoylethylation (Compound 5)
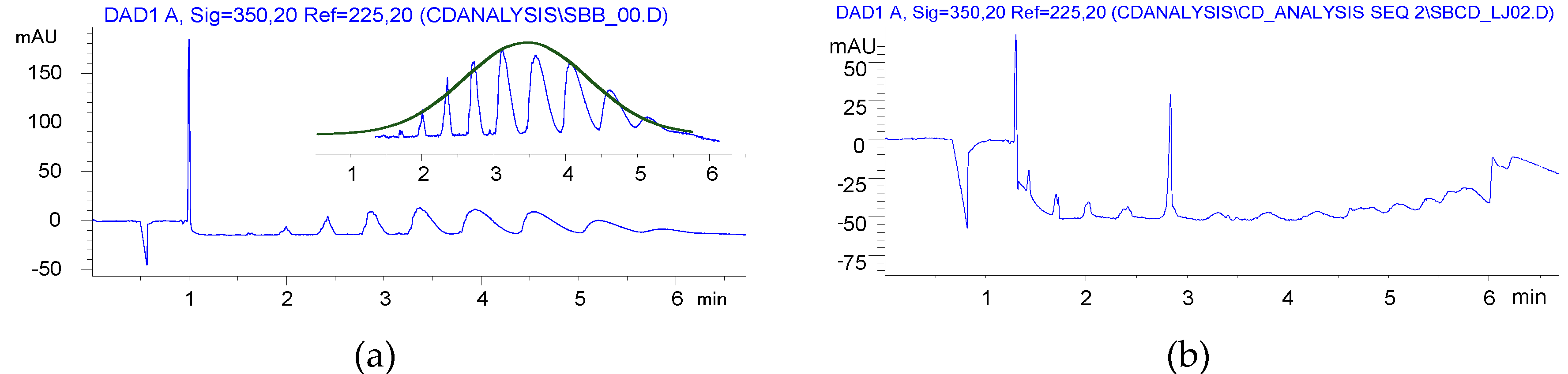
2.4. Sulfobutylation (Compounds 4 and 4′)
2.5. Other Reactions (Compounds 6 and 7)
3. Materials and Methods
3.1. General Information
3.2. Solution Reactions
3.2.1. Synthesis of Randomly Carboxymethylated βCD (2)
3.2.2. Synthesis of Randomly Carboxyethylated βCD (3)
3.2.3. Synthesis of Randomly Sulfobutylated βCD (4)
4-Hydroxybutane-1-Sulfonic Acid Sodium Salt (Dominant Impurity of Compounds 4 and 4′)
Di(1,1’-sulfonatobutyl)ether Disodium Salt(Potential Impurity of Compounds 4 and 4′)
3.3. Reactions in Ball Mill
3.3.1. Drying of βCD
3.3.2. General Method of the HEBM Reactions
3.3.3. Synthesis of Randomly Carboxymethylated βCD (2’)
3.3.4. Synthesis of Randomly Carboxyethylated βCD (3′)
3.3.5. Direct Synthesis of Randomly Carboxyethylated βCD (3′′)
3.3.6. Synthesis of Randomly Carbamoylethylated βCD (5)
3.3.7. Synthesis of Randomly Sulfobutylated βCD (4′)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Szejtli, J.; Osa, T. Cyclodextrins. In Comprehensive Supramolecular Chemistry; Atwood, J.L., Lehn, J.M., Eds.; Elsevier: Oxford, UK (aka Pergamon Press), 1996; Volume 3, pp. 1–693. [Google Scholar]
- Jicsinszky, L.; Hashimoto, H.; Fenyvesi, E.; Ueno, A. Cyclodextrin derivatives. In Comprehensive Supramolecular Chemistry; Szejtli, J., Osa, T., Eds.; Elsevier: Oxford, UK (aka Pergamon Press), 1996; Volume 3, pp. 57–188. [Google Scholar]
- Jicsinszky, L. Regioselective substitution of cyclodextrins without using protecting groups. In Proc. Ninth Int. Symp. Cyclodextrins; Labandeira, J.J.T., Vila-Jato, J.L., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1999; pp. 513–516. [Google Scholar]
- Jicsinszky, L.; Szejtli, J. Effect of catalysts on the acylation of cyclodextrins. In Minutes of Sixth Int. Symp. Cyclodextrins; Hedges, A.R., Ed.; Éditions de Santé: Paris, France, 1992; pp. 96–100. [Google Scholar]
- Rinaldi, L.; Binello, A.; Stolle, A.; Curini, M.; Cravotto, G. Efficient mechanochemical complexation of various steroid compounds with α-, β- and γ-cyclodextrin. Steroids 2015, 98, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Rogmann, N.; Seidel, J.; Mischnick, P. Formation of unexpected substitution patterns in sulfonylbutylation of cyclomaltoheptaose promoted by host-guest interaction. Carbohydr. Res. 2000, 327, 269–274. [Google Scholar] [CrossRef]
- Liversidge, G.G.; Cundy, K.C. Particle size reduction for improvement of oral bioavailability of hydrophobic drugs: I. Absolute oral bioavailability of nanocrystalline danazol in beagle dogs. Int. J. Pharm. 1995, 125, 91–97. [Google Scholar] [CrossRef]
- Beyer, M.K.; Clausen-Schaumann, H. Mechanochemistry: The mechanical activation of covalent bonds. Chem. Rev. 2005, 105, 2921–2948. [Google Scholar] [CrossRef] [PubMed]
- Hernández, J.; Avila-Ortiz, C.; Juaristi, E. Useful Chemical Activation Alternatives in Solvent-Free Organic Reactions. In Comprehensive Organic Synthesis II, 2nd ed.; Molander, G.A., Knochel, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 9, pp. 287–314. [Google Scholar]
- Stolle, A.; Szuppa, T.; Leonhardt, S.E.S.; Ondruschka, B. Ball milling in organic synthesis: Solutions and challenges. Chem. Soc. Rev. 2011, 40, 2317–2329. [Google Scholar] [CrossRef] [PubMed]
- Ranu, B.C.; Chatterjee, T.; Mukherjee, N. Carbon-heteroatom bond forming reactions and heterocycle synthesis under ball milling. In Ball Milling towards Green Synthesis; Stolle, A., Ranu, B., Eds.; Royal Society of Chemistry (RSC): London, UK, 2014; pp. 1–33. [Google Scholar]
- Jicsinszky, L.; Caporaso, M.; Tuza, K.; Martina, K.; Calcio Gaudino, E.; Cravotto, G. Nucleophilic substitutions of 6I-O-Monotosyl-β-cyclodextrin in a planetary ball mill. ACS Sustain. Chem. Eng. 2016, 4, 919–929. [Google Scholar] [CrossRef]
- Menuel, S.; Doumert, B.; Saitzek, S.; Ponchel, A.; Delevoye, L.; Monflier, E.; Hapiot, F. Selective secondary face modification of cyclodextrins by mechanosynthesis. J. Org. Chem. 2015, 80, 6259–6266. [Google Scholar] [CrossRef] [PubMed]
- Jicsinszky, L.; Caporaso, M.; Martina, K.; Calcio Gaudino, E.; Cravotto, G. Efficient mechanochemical synthesis of regioselective persubstituted cyclodextrin. Beilstein J. Org. Chem. 2016, 12, 2364–2371. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Rathish, I.G.; Sankaran, R. A review on current industrial trends for synthesis of medicinal compounds. Int. J. Pharm. Pharm. Sci. 2013, 5, 33–45. [Google Scholar]
- Tyagi, M.; Khurana, D.; Kartha, K.P.R. Solvent-free mechanochemical glycosylation in ball mill. Carbohydr. Res. 2013, 379, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Patil, P.R.; Kartha, K.P.R. Solvent-free synthesis of thioglycosides by ball milling. Green Chem. 2009, 11, 953–956. [Google Scholar] [CrossRef]
- Beak, P. Determinations of transition-state geometries by the endocyclic restriction test: Mechanisms of Substitution at Nonstereogenic Atoms. Acc. Chem. Res. 1992, 25, 215–222. [Google Scholar] [CrossRef]
- Berhenke, L.F.; Britton, E.C. Effect of pH on Hydrolysis Rate of Chloroacetic Acid. Ind. Eng. Chem. 1946, 38, 544–546. [Google Scholar] [CrossRef]
- Jortner, J.; Rabani, J. The Decomposition of Chloroacetic Acid in Aqueous Solutions by Atomic Hydrogen. I. Comparison with Radiation Chemical Data. J. Phys. Chem. 1962, 66, 2078–2081. [Google Scholar] [CrossRef]
- Jortner, J.; Rabani, J. The Decomposition of Chloroacetic Acid in Aqueous Solutions by Atomic Hydrogen. II. Reaction Mechanism in Alkaline Solutions. J. Phys. Chem. 1962, 66, 2081–2084. [Google Scholar] [CrossRef]
- Ivanyi, R.; Jicsinszky, L.; Hirschbergne Szejtli, G.; Vadasz, Zs.; Szente, L. Process for Preparing of Sulphobutylated Cyclodextrins of Pharmaceutical Additive Quality. HU228817 15 April 2009. [Google Scholar]
- Bicchi, C.; Artuffo, G.; D’Amato, A.; Manzin, V.; Galli, A.; Galli, M. Cyclodextrin derivatives in the GC separation of racemic mixtures of volatile compounds, part V: Heptakis 2,6-dimethyl-3-pentyl-β-cyclodextrins. J. High Res. Chrom. 1992, 15, 710–714. [Google Scholar] [CrossRef]
- Strohalm, M.; Kavan, D.; Novák, P.; Volný, M.; Havlíček, V. mMass 3: A Cross-Platform Software Environment for Precise Analysis of Mass Spectrometric Data. Anal. Chem. 2010, 82, 4648–4651. [Google Scholar] [CrossRef] [PubMed]
- Jicsinszky, L.; Szeman, J.; Szente, L.; Tuza, K.; Varga, E. Process for the Preparation of Anionic Cyclodextrins. HU1000534 29 May 2012. [Google Scholar]
- Sample Availability: Not Available.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Compound | Reagent 1 | NaOH/Reagent/CD (mole:mole:mole) | Reaction Time (h) | Yield (%) | DS | Residual Reagent 2 (%) |
|---|---|---|---|---|---|---|---|
| 1 | 2 | ClAcONa 3 | 6:5:1 | 3 | 89 | 4.3–4.5 | <2.5 |
| 2 | 2’ | ClAcONa | 6:5:1 | 1.5 | 96 | 4.3–4.6 | <2.5 |
| 3 | 2’ | ClAcONa | 6:5:1 | 2.5 | 98 | 4.4–4.6 | <0.5 |
| 4 | 2’ | ClAcONa 4 | 6:5:1 | 2.5 | 77 | 4.4–4.7 | <1.0 |
| 5 | 3’ | BuOAcr | 6:5:1 | 1.5 | 50 | 3.0–3.3 | ≈1.0 |
| 6 | 3’ | BuOAcr 4 | 6:5:1 | 2.5 | 30 | 2.8–3.2 | <1.0 |
| 7 | 3” | AcrONa | 6:5:1 | 3.5 | 52 | 2.8–3.1 | <11 |
| 8 | 3 | AcrAm 3 | 6:5:1 | 24 + 8 | 56 | 3.0–3.4 | <0.05 |
| 9 | 5 | AcrAm | 6:5:1 | 5.5 | 73 | 3.8–4.3 | <0.9 |
| 10 | 5 | AcrAm | 6:5:1 | 3.5 | 83 | 3.3–3.5 | <0.5 |
| 11 | 4 | BS 3 | 9.3:8.6:1 | 8 | 66 | 6.2–6.5 | <0.2 5 |
| 12 | 4’ | BS | 9:8:1 | 1.5 | 56 | 3.1–3.3 | ≈17 5 |
| 13 | 4’ | BS | 9:8:1 | 2.5 | 57 | 5.2–5.8 | ≈9.5 5 |
| 14 | 4’ | BS | 16:8:1 | 2.5 | 67 | 6.2–6.6 | <2 5 |
| 15 | 4’ | BS 4 | 16:8:1 | 2.5 | 72 | 7.5–7.8 | <0.6 5 |
| 16 | 4’ | BS 6 | 16:8:1 | 3 (1.5 + 1.5) | 71 | 4.4–4.9 | ≈9 5 |
| 17 | 4’ | BS | 16:8:1 | 3.5 | 79 | 6.9–7.5 | ≈0.6 5 |
| No | NaOH (g (mole)) | ClAcONa (g (mole)) | Milling Time (h) | Final Temp. (°C) | Yield (g (%)) | DS | Residual Reagent 1 (%) |
|---|---|---|---|---|---|---|---|
| 2 | 0.24 (0.006) | 0.61 (0.005) | 1.5 | 84 | 1.42 (95.9) | 4.3–4.6 | <2.5 |
| 3 | 0.24 (0.006) | 0.61 (0.005) | 2.5 | 87 | 1.48 (98.4) | 4.4–4.6 | <0.5 |
| 4 | 0.24 (0.006) | 0.61 (0.005) 2 | 2.5 | 87 | 1.15 (76.7) | 4.4–4.6 | <1.0 |
| No | NaOH (g (mole)) | Reagent (g (mole)) | Milling Time (h) | Final Temp. (°C) | Yield (g (%)) | DS | Residual Reagent 1 (%) |
|---|---|---|---|---|---|---|---|
| 5 | 0.24 (0.006) | 0.64 (0.005) 2 | 2.5 | 67 | 0.72 (50.3) | 3.0–3.3 | ≈1.0 |
| 6 | 0.24 (0.006) | 0.64 (0.005) 2,3 | 3.5 | 70 | 0.42 (29.6) | 2.8–3.2 | <1.0 |
| 7 | 0.24 (0.006) | 0.54 (0.005) 4 | 3.5 | 93 | 0.74 5 (52.1) | 2.8–3.1 | <11 |
| No | NaOH (g (mole)) | Reagent (g (mole)) | Milling Time (h) | Final Temp. (°C) | Yield (g (%)) | DS | Residual Reagent 1 (%) |
|---|---|---|---|---|---|---|---|
| 9 | 0.24 (0.006) | 0.36 (0.005) | 5.5 | 97 | 1.05 (73.4) | 3.8–4.3 | <0.9 |
| 10 | 0.24 (0.0051) | 0.36 (0.005) | 3.5 | 88 | 1.15 (83.3) | 3.3–3.5 | <0.5 |
| No | NaOH (g (mole)) | BS (g (mole)) | Milling Time (h) | Final Temp. (°C) | Yield (g (%)) 1 | DS | Residual Reagent 2 (%) |
|---|---|---|---|---|---|---|---|
| 12 | 0.36 (0.009) | 1.09 (0.008) | 1.5 | 67 | 1.17 (55.6) | 3.1–3.4 | ≈22 |
| 13 | 0.36 (0.009) | 1.09 (0.005) | 2.5 | 70 | 1.24 (54.6) | 5.2–5.8 | ≈12 |
| 14 | 0.64 (0.018) | 1.09 (0.005) | 2.5 | 82 | 1.59 (66.6) | 6.2–6.6 | <10 |
| 15 | 0.64 (0.006) | 1.09 (0.005) 3 | 2.5 | 82 | 1.78 (72.2) | 7.5–7.8 | ≈5 |
| 16 | 0.64 (0.018) | 1.09 (0.005) 4 | 3 (1.5 + 1.5) | 82/92 | 1.52 (70.7) | 4.4–4.9 | ≈13 |
| 17 | 0.64 (0.006) | 1.09 (0.005) | 3.5 | 88 | 1.89 (79.1) | 6.9–7.5 | ≈5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jicsinszky, L.; Caporaso, M.; Calcio Gaudino, E.; Giovannoli, C.; Cravotto, G. Synthesis of Randomly Substituted Anionic Cyclodextrins in Ball Milling. Molecules 2017, 22, 485. https://doi.org/10.3390/molecules22030485
Jicsinszky L, Caporaso M, Calcio Gaudino E, Giovannoli C, Cravotto G. Synthesis of Randomly Substituted Anionic Cyclodextrins in Ball Milling. Molecules. 2017; 22(3):485. https://doi.org/10.3390/molecules22030485
Chicago/Turabian StyleJicsinszky, László, Marina Caporaso, Emanuela Calcio Gaudino, Cristina Giovannoli, and Giancarlo Cravotto. 2017. "Synthesis of Randomly Substituted Anionic Cyclodextrins in Ball Milling" Molecules 22, no. 3: 485. https://doi.org/10.3390/molecules22030485
APA StyleJicsinszky, L., Caporaso, M., Calcio Gaudino, E., Giovannoli, C., & Cravotto, G. (2017). Synthesis of Randomly Substituted Anionic Cyclodextrins in Ball Milling. Molecules, 22(3), 485. https://doi.org/10.3390/molecules22030485