New Chiral Ebselen Analogues with Antioxidant and Cytotoxic Potential

 ,
, 

Abstract
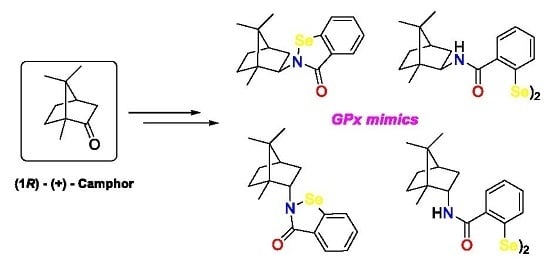
:
1. Introduction
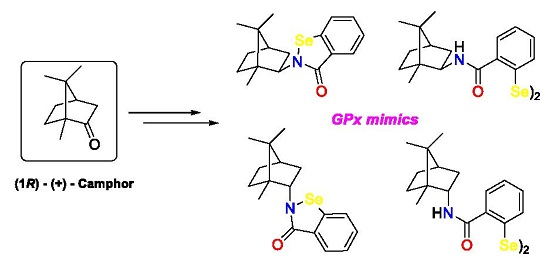
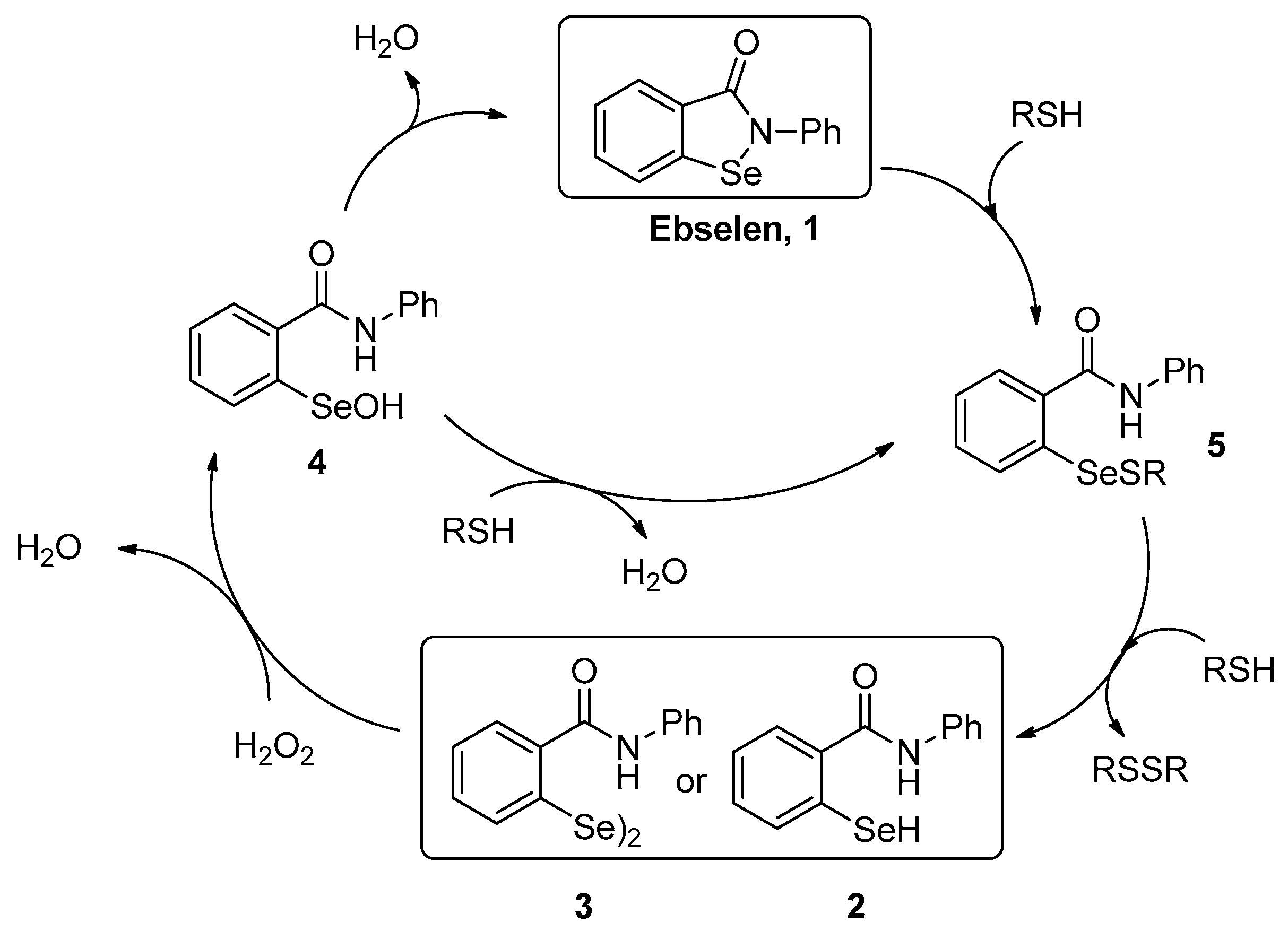
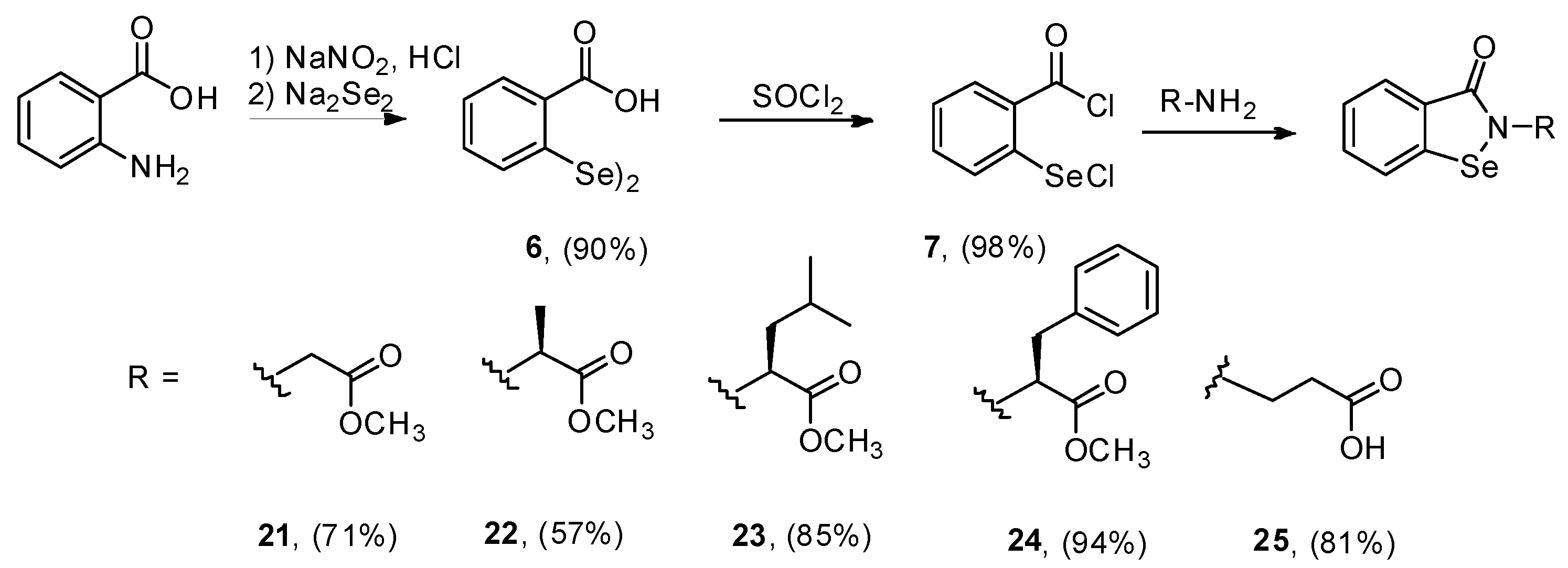
2. Results and Discussion
3. Experimental
3.1. General
3.2. Procedures and Analysis Data
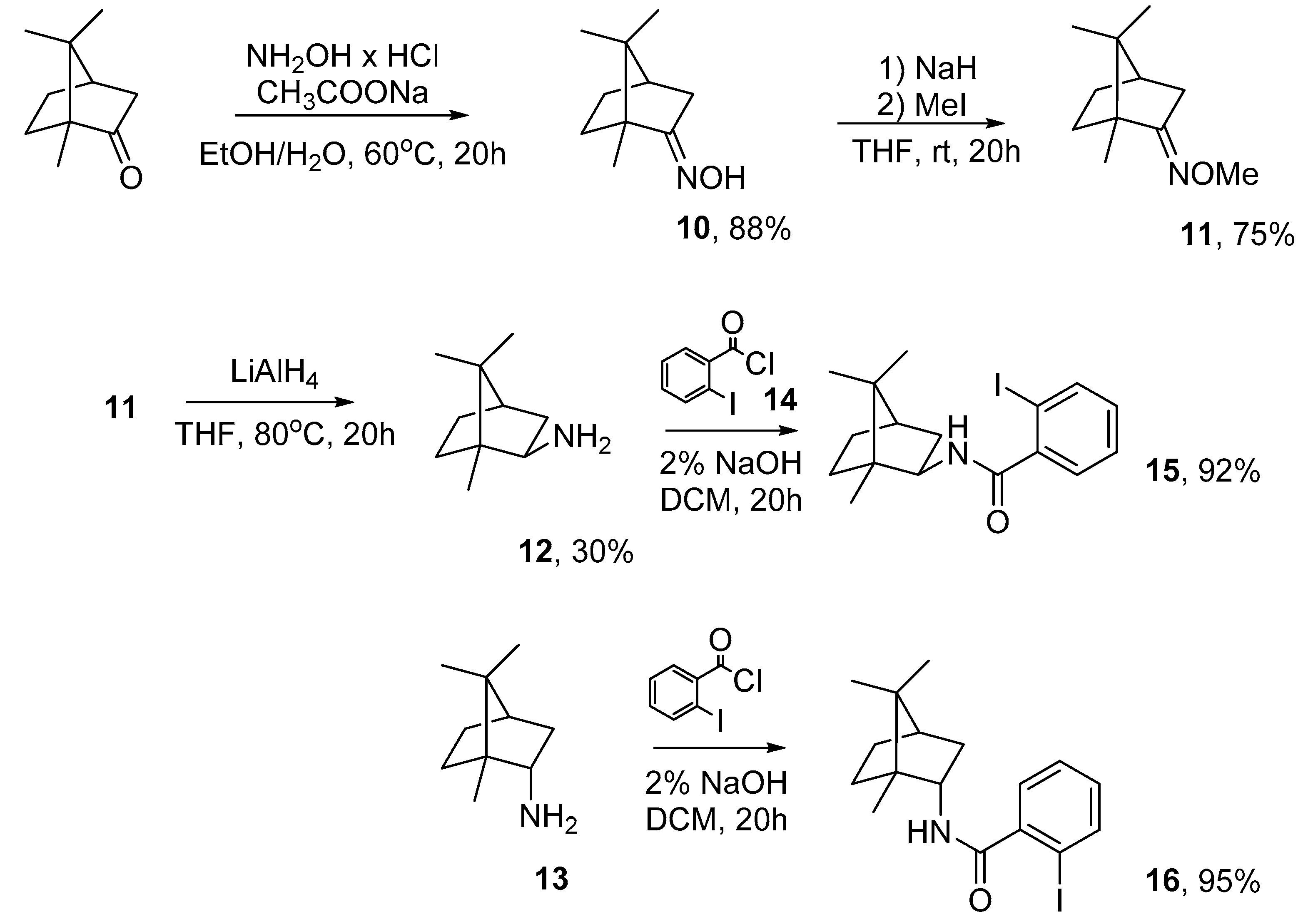
3.2.1. (1R)-Camphor Oxime (10)
3.2.2. (1R)-Camphor O-methyloxime (11)
3.2.3. (R)-(−)-Isobornylamine (12)
3.2.4. General Procedure for the Synthesis of Compounds 15 and 16
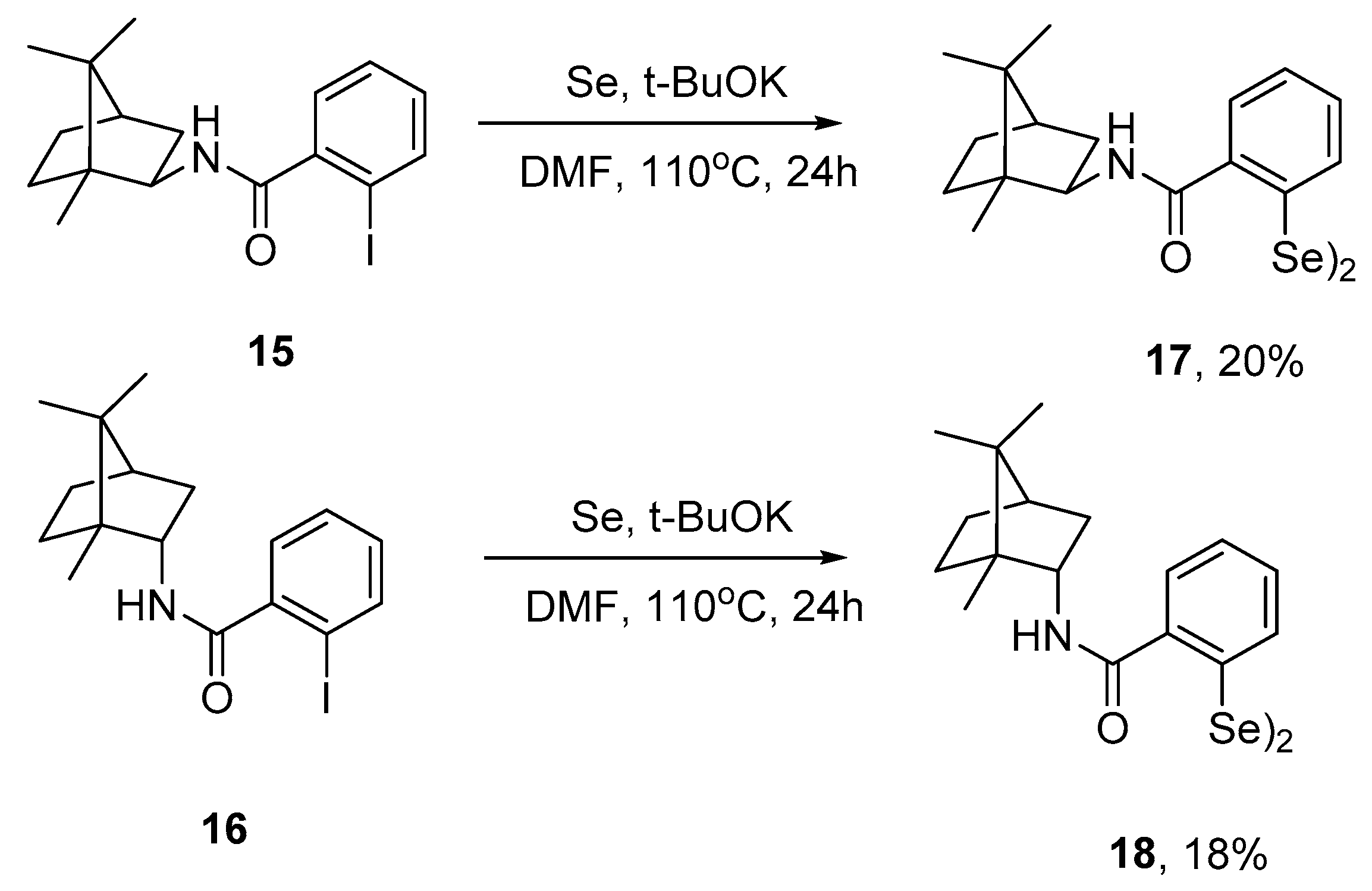
3.2.5. General Procedure for the Synthesis of Compounds 16 and 17
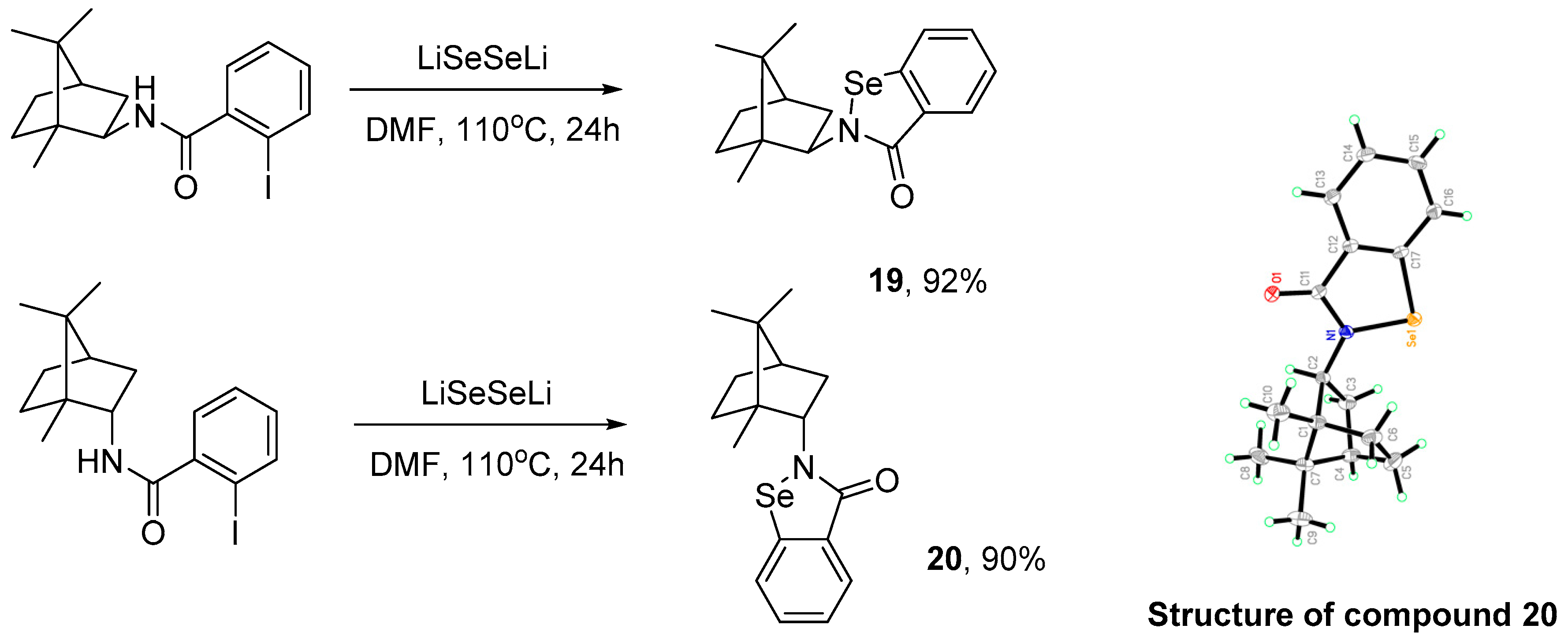
3.2.6. General Procedure for the Synthesis of Compounds 19 and 20
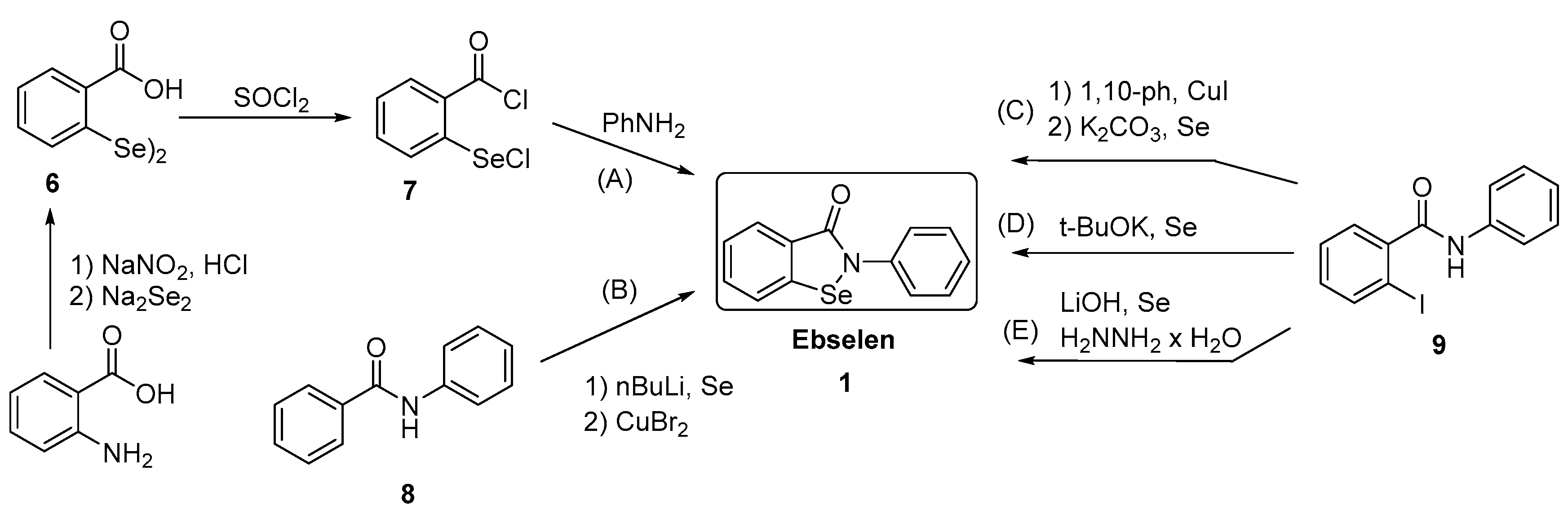
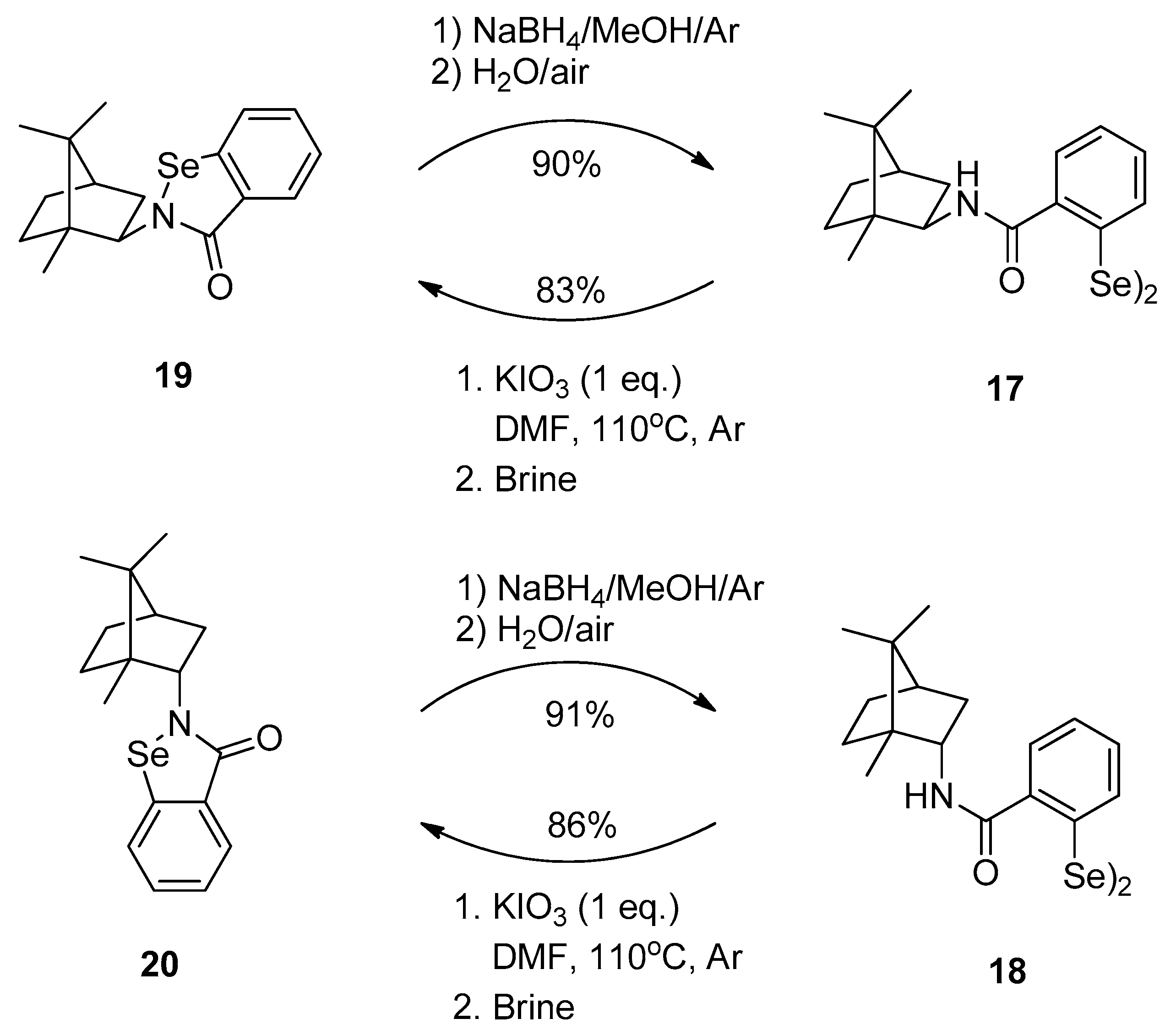
3.2.7. Conversion of Diselenides to Benzisoselenazolones
3.2.8. Synthesis of 2,2-Diselenobis(benzoic acid) 6 and 2-(Chloroseleno)benzoyl Chloride 7
3.2.9. General Procedure for the Synthesis of Compounds 21–25
3.3. Antioxidant Activity Assay
3.4. Cell Viability Assays
3.4.1. MTT Viability Assay
Cell Culture
MTT Assay
3.4.2. SRB Viability Assay
Cell Culture
SRB Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Parnham, J.M.; Sies, H. The early research and development of ebselen. Biochem. Pharm. 2013, 86, 1248–1253. [Google Scholar] [CrossRef] [PubMed]
- Bhujan, B.J.; Mugesh, G. Biological and Biochemical Aspects of Selenium Compounds. In Organoselenium Chemistry; Wirth, T., Ed.; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Soares, J.C.M.; Folmer, V.; da Rocha, J.B.T.; Nogueira, C.W. Ebselen exhibits glycation-inhibiting properties and protects against osmotic fragility of human erythrocytes in vitro. Cell. Biol. Int. 2014, 38, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kang, S.; Kim, D.S.; Shin, B.K.; Moon, N.R.; Daily, J.W., III. Ebselen pretreatment attenuates ischemia/reperfusion injury and prevents hyperglycemia by improving hepatic insulin signaling and β-cell survival in gerbils. Free Radic. Res. 2014, 48, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yun, J.-W.; Lei, X.G. Glutathione peroxidase mimic ebselen improves glucose-stimulated insulin secretion in murine islets. Antioxid. Redox Signal. 2014, 20, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.M.; Sharma, A.; Yuen, D.Y.C.; Stefanovic, N.; Krippner, G.; Mugesh, G.; Chai, Z.; de Haan, J.B. The Modified Selenenyl Amide, M-hydroxy Ebselen, Attenuates Diabetic Nephropathy and Diabetes-Associated Atherosclerosis in ApoE/GPx1 Double Knockout Mice. PLoS ONE 2013, 8, e69193. [Google Scholar] [CrossRef] [PubMed]
- Martini, F.; Bruning, C.A.; Soares, S.M.; Nogueira, C.W.; Zeni, G. Inhibitory Effect of Ebselen on Cerebral Acetylcholinesterase Activity In Vitro: Kinetics and Reversibility of Inhibition. Curr. Pharm. Des. 2015, 21, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Lynch, E.D.; Kill, J. Compounds for the prevention and treatment of noise-induced hearing loss. Drug Discov. Today 2005, 10, 1291–1298. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Sano, K.; Takakura, K.; Saito, I.; Shinohara, Y.; Asano, T.; Yasuhara, H. Ebselen in Acute Ischemic Stroke. A Placebo-Controlled, Double-blind Clinical Trial. Stroke 1998, 29, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, L.; Du, J.; Li, M.; Qian, C.; Cheng, Y.; Peng, Y.; Xie, J.; Wang, D. Induction of Apoptosis in Human Multiple Myeloma Cell Lines by Ebselen via Enhancing the Endogenous Reactive Oxygen Species Production. BioMed Res. Int. 2014, 2014, 696107. [Google Scholar] [CrossRef] [PubMed]
- Pacuła, A.J.; Mangiavacchi, F.; Sancineto, L.; Lenardao, E.J.; Ścianowski, J.; Santi, C. An Update on “Selenium Containing Compounds from Poison to Drug Candidates: A Review on the GPx-like Activity”. Curr. Chem. Biol. 2015, 9, 97–112. [Google Scholar] [CrossRef]
- Orian, L.; Toppo, S. Organochalcogen peroxidase mimetics as potential drugs: A long story of a promise still unfulfilled. Free Radic. Biol. Med. 2014, 66, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Mugesh, G. Glutathione Peroxidase Activity of Ebselen and its Analogues: Some Insights into the Complex Chemical Mechanisms Underlying the Antioxidant Activity. Curr. Med. Chem. 2013, 7, 47–56. [Google Scholar] [CrossRef]
- Pacuła, A.J.; Kaczor, K.B.; Wojtowicz, A.; Antosiewicz, J.; Janecka, A.; Długosz, A.; Janecki, T.; Ścianowski, J. New glutathione peroxidase mimetics—Insights into antioxidant and cytotoxic activity. Bioorg. Med. Chem. 2017, 25, 126–131. [Google Scholar]
- Welter, A.; Christiaens, L.; Ferdinand, W.P. New Benzisoselenazolones, Process for Producing the Same and Pharmaceutical Preparations Containing the Same. EP 0044453 A2, 27 January 1982. [Google Scholar]
- Engman, L.; Hallberg, A. Expedient synthesis of ebselen and related compounds. J. Org. Chem. 1989, 54, 2964–2966. [Google Scholar] [CrossRef]
- Balkrishna, S.J.; Bhakuni, B.S.; Chopra, D.; Kumar, S. Cu-catalyzed efficient synthetic methodology for ebselen and related Se-N heterocycles. Org. Lett. 2010, 12, 5394–5397. [Google Scholar] [CrossRef] [PubMed]
- Balkrishna, S.J.; Kumar, S.; Azad, K.G.; Bhakuni, B.S.; Panini, P.; Ahalawat, N.; Tomar, R.S.; Detty, R.M.; Kumar, S. An ebselen like catalyst with enhanced GPx activity via a selenol intermediate. Org. Biomol. Chem. 2014, 12, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Pacuła, A.J.; Ścianowski, J.; Aleksandrzak, K.B. Highly efficient synthesis and antioxidant capacity of N-substituted benzisoselenazol-3(2H)-ones. RSC Adv. 2014, 4, 48959–48962. [Google Scholar] [CrossRef]
- Elsherbini, M.; Hamama, W.S.; Zoorob, H.H.; Bhowmick, D.; Mugesh, G.; Wirth, T. Synthesis and antioxidant activities of novel chiral ebselen analogues. Heteroatom Chem. 2014, 25, 320–325. [Google Scholar] [CrossRef]
- Satheeshkumar, K.; Mugesh, G. Synthesis and Antioxidant Activity of Peptide-Based Ebselen Analogues. Chem. Eur. J. 2011, 17, 4849–4857. [Google Scholar] [CrossRef] [PubMed]
- Mlochowski, J.; Gryglewski, R.J.; Inglot, A.D.; Jakubowski, A.; Juchniewicz, L.; Kloc, K. Synthesis and Properties of 2-Carboxyalkyl-l,2-benzisoselenazol-3(2H)-ones and Related Organoselenium Compounds as Nitric Oxide Synthase Inhibitors and Cytokine Inducers. Liebigs Ann. 1996, 1996, 1751–1755. [Google Scholar] [CrossRef]
- Pacuła, A.J.; Ścianowski, J. Terpenes as Green Starting Materials for New Organoselenium and Organotellurium Compounds. Curr. Green Chem. 2016, 3, 36–50. [Google Scholar] [CrossRef]
- Ścianowski, J.; Rafalski, J.; Banach, A.; Czaplewska, J.; Komoszyńska, A. Synthesis and reactions of the optically active selenols derived from monoterpenes. Tetrahedron Asymmetry 2013, 24, 1089–1096. [Google Scholar] [CrossRef]
- Rafiński, Z.; Ścianowski, J. Synthesis and reactions of enantiomerically pure dialkyl diselenides from the p-menthane group. Tetrahedron Asymmetry 2008, 19, 1237–1244. [Google Scholar] [CrossRef]
- Ścianowski, J.; Rafinski, Z.; Wojtczak, A.; Burczynski, K. Syntheses and reactions of terpene β-hydroxyselenides and β-hydroxydiselenides. Tetrahedron Asymmetry 2009, 20, 2871–2879. [Google Scholar] [CrossRef]
- Ścianowski, J. Convenient route to dialkyl diselenides from alkyl tosylates. Synthesis of Di(cis-myrtanyl) diselenide. Tetrahedron Lett. 2005, 46, 3331–3334. [Google Scholar] [CrossRef]
- Ścianowski, J.; Rafiński, Z.; Wojtczak, A. Syntheses and Reactions of New Optically Active Terpene Dialkyl Diselenides. Eur. J. Org. Chem. 2006, 14, 3216–3225. [Google Scholar] [CrossRef]
- Ścianowski, J.; Banach, A.; Uzarewicz-Baig, M.; Wojtczak, A. Terpenyl Selenides: Synthesis and Application in Asymmetric Epoxidation. Eur. J. Org. Chem. 2015, 16, 3477–3485. [Google Scholar]
- Rafinski, Z.; Ścianowski, J.; Wojtczak, A. Synthesis and Reactions of the Optically Active Dialkyl Diselenides from the Pinane Group. Lett. Org. Chem. 2009, 6, 321–328. [Google Scholar] [CrossRef]
- Ścianowski, J.; Rafiński, Z.; Szuniewicz, A.; Wojtczak, A. New chiral selenium electrophiles derived from functionalized terpenes. Tetrahedron 2009, 65, 10162–10174. [Google Scholar] [CrossRef]
- Flack, H.D. On enantiomorph-polarity estimation. Acta Cryst. 1983, A39, 876–881. [Google Scholar] [CrossRef]
- Allen, H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Cryst. 2002, B58, 380–388. [Google Scholar] [CrossRef]
- Selvakumar, K.; Singh, H.B.; Butcher, R.J. Strained dimethyl 2-(bromoselanyl)-5-tert-butylisophthalate: A reactive precursor for the synthesis of ebselen analogs. Tetrahedron Lett. 2011, 52, 6831–6834. [Google Scholar] [CrossRef]
- Bhabak, K.P.; Vernekar, A.A.; Jakka, S.R.; Roy, G.; Mugesh, G. Mechanistic investigations on the efficient catalytic decomposition of peroxynitrite by ebselen analogues. Org. Biomol. Chem. 2011, 9, 5193–5200. [Google Scholar] [CrossRef] [PubMed]
- Bhabak, K.P.; Mugesh, G. Synthesis, characterization, and antioxidant activity of some ebselen analogues. Chem. Eur. J. 2007, 13, 4594–4601. [Google Scholar] [CrossRef]
- Piatek, M.; Oleksyn, B.; Sliwinski, J. 2-Methyl-2H-1,2-benzisoselenazol-3-one. Acta Crystallogr. Sect. 1995, C51, 298–301. [Google Scholar] [CrossRef]
- Zade, S.S.; Panda, S.; Tripathi, S.K.; Singh, H.B.; Wolmershäuser, G. Convenient Synthesis, Characterization and GPx-Like Catalytic Activity of Novel Ebselen Derivatives. Eur. J. Org. Chem. 2004, 2004, 3857–3864. [Google Scholar] [CrossRef]
- Syper, L.; Młochowski, J. The Convenient Syntheses of Organoselenium Reagents. Synthesis 1984, 1984, 439–442. [Google Scholar] [CrossRef]
- Bird, M.L.; Challenger, F. Potassium alkaneselenonates and other alkyl derivatives of selenium. J. Chem. Soc. 1942, 570–574. [Google Scholar] [CrossRef]
- Klayman, D.L.; Griffin, T.S. Reaction of selenium with sodium borohydride in protic solvents. A Facile Method for the introduction of selenium into organic molecules. J. Am. Chem. Soc. 1973, 95, 197–199. [Google Scholar] [CrossRef]
- Lou, Z.; Li, P.; Sun, X.; Yang, S.; Wang, B.; Han, K. A fluorescent probe for rapid detection of thiols and imaging of thiols reducing repair and hydrogen peroxide oxidative stress cycles in living cells. Chem. Commun. 2013, 49, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Kumakura, F.; Mishra, B.; Priyadarsini, K.I.; Iwaoka, M. A Water-Soluble Cyclic Selenide with Enhanced Glutathione Peroxidase-Like Catalytic Activities. Eur. J. Org. Chem. 2010, 3, 440–444. [Google Scholar] [CrossRef]
- Rao, M.N.; Haridas, M.; Gangwar, M.K.; Rajakannu, P.; Kalita, A.C.; Ghosh, P. Asymmetric Base-Free Michael Addition at Room Temperature with Nickel-Based Bifunctional Amido-Functionalized N-Heterocyclic Carbene Catalysts. Eur. J. Inorg. Chem. 2015, 9, 1604–1615. [Google Scholar] [CrossRef]
- Dickmu, G.C.; Stahl, L.; Smoliakovaet, I.P. A new enantiopure d-camphor-derived palladacycle. J. Organomet. Chem. 2014, 756, 27–33. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.








| Remaining Dithiotreitol (%) | |||||
|---|---|---|---|---|---|
| Catalyst | 3 min | 5 min | 15 min | 30 min | 60 min |
| 17 | 59 | 36 | 12 | 0 | 0 |
| 18 | 48 | 38 | 28 | 25 | 22 |
| 19 | 50 | 17 | 0 | 0 | 0 |
| 20 | 30 | 11 | 0 | 0 | 0 |
| 21 | 99 | 99 | 98 | 97 | 95 |
| 22 | 96 | 95 | 93 | 91 | 89 |
| 23 | 45 | 0 | 0 | 0 | 0 |
| 24 | 96 | 95 | 94 | 93 | 90 |
| 25 | 94 | 93 | 92 | 92 | 91 |
| 26 | 77 | 58 | 42 | 28 | 13 |
| Ebselen (1) | 84 | 75 | 64 | 58 | 52 |
| Diselenide (3) | 89 | 83 | 74 | 68 | 63 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Structure | MCF-7 (MTT) | HEP G2 (MTT) | HL60 (MTT) | DU-145 (SRB) | PNT1A (SRB) |
|---|---|---|---|---|---|---|
| IC50, µM | ||||||
| 20 |  | 220 ± 34 | 232 ± 28 | 190 ± 20 | >40 | >40 |
| 23 |  | 18 ± 1.1 | 23.5 ± 3.2 | 21.5 ± 1.8 | >40 | >40 |
| 26 |  | 29 ± 1.9 | 100 ± 12 | 43.5 ± 5.0 | 30.6 ± 0.2 | 30.35 ± 0.06 |
| Control | Carboplatin | 0.70 ± 0.30 | 3.19 ± 0.46 | 9.70 ± 1.20 | ----- | ------ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacuła, A.J.; Kaczor, K.B.; Antosiewicz, J.; Janecka, A.; Długosz, A.; Janecki, T.; Wojtczak, A.; Ścianowski, J. New Chiral Ebselen Analogues with Antioxidant and Cytotoxic Potential. Molecules 2017, 22, 492. https://doi.org/10.3390/molecules22030492
Pacuła AJ, Kaczor KB, Antosiewicz J, Janecka A, Długosz A, Janecki T, Wojtczak A, Ścianowski J. New Chiral Ebselen Analogues with Antioxidant and Cytotoxic Potential. Molecules. 2017; 22(3):492. https://doi.org/10.3390/molecules22030492
Chicago/Turabian StylePacuła, Agata J., Katarzyna B. Kaczor, Jędrzej Antosiewicz, Anna Janecka, Angelika Długosz, Tomasz Janecki, Andrzej Wojtczak, and Jacek Ścianowski. 2017. "New Chiral Ebselen Analogues with Antioxidant and Cytotoxic Potential" Molecules 22, no. 3: 492. https://doi.org/10.3390/molecules22030492
APA StylePacuła, A. J., Kaczor, K. B., Antosiewicz, J., Janecka, A., Długosz, A., Janecki, T., Wojtczak, A., & Ścianowski, J. (2017). New Chiral Ebselen Analogues with Antioxidant and Cytotoxic Potential. Molecules, 22(3), 492. https://doi.org/10.3390/molecules22030492