
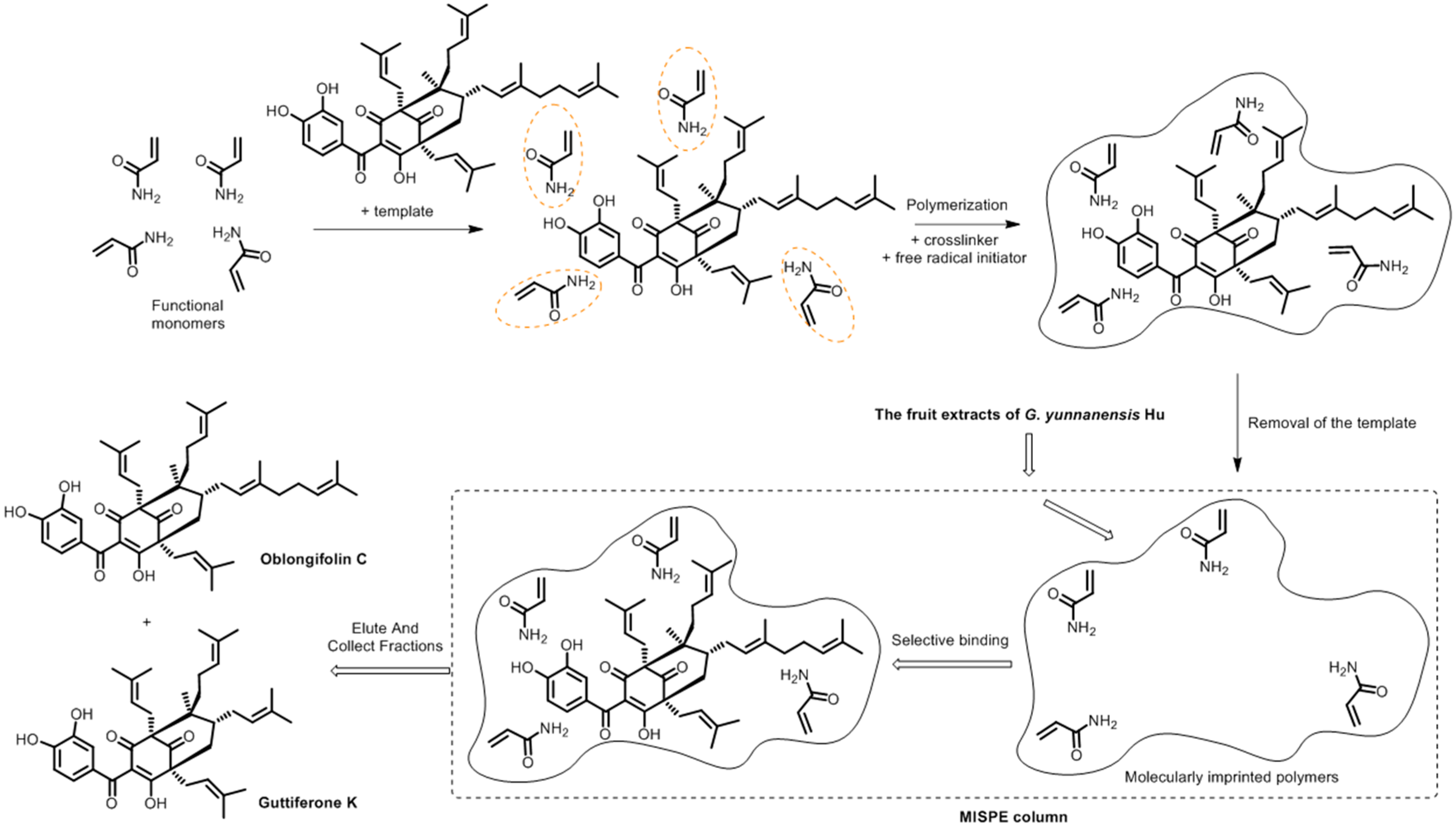
Molecularly Imprinted Polymers for Selective Extraction of Oblongifolin C from Garcinia yunnanensis Hu


Abstract
:1. Introduction
2. Results and Discussion
2.1. Comparative Study of Different Polymer Preparation Conditions
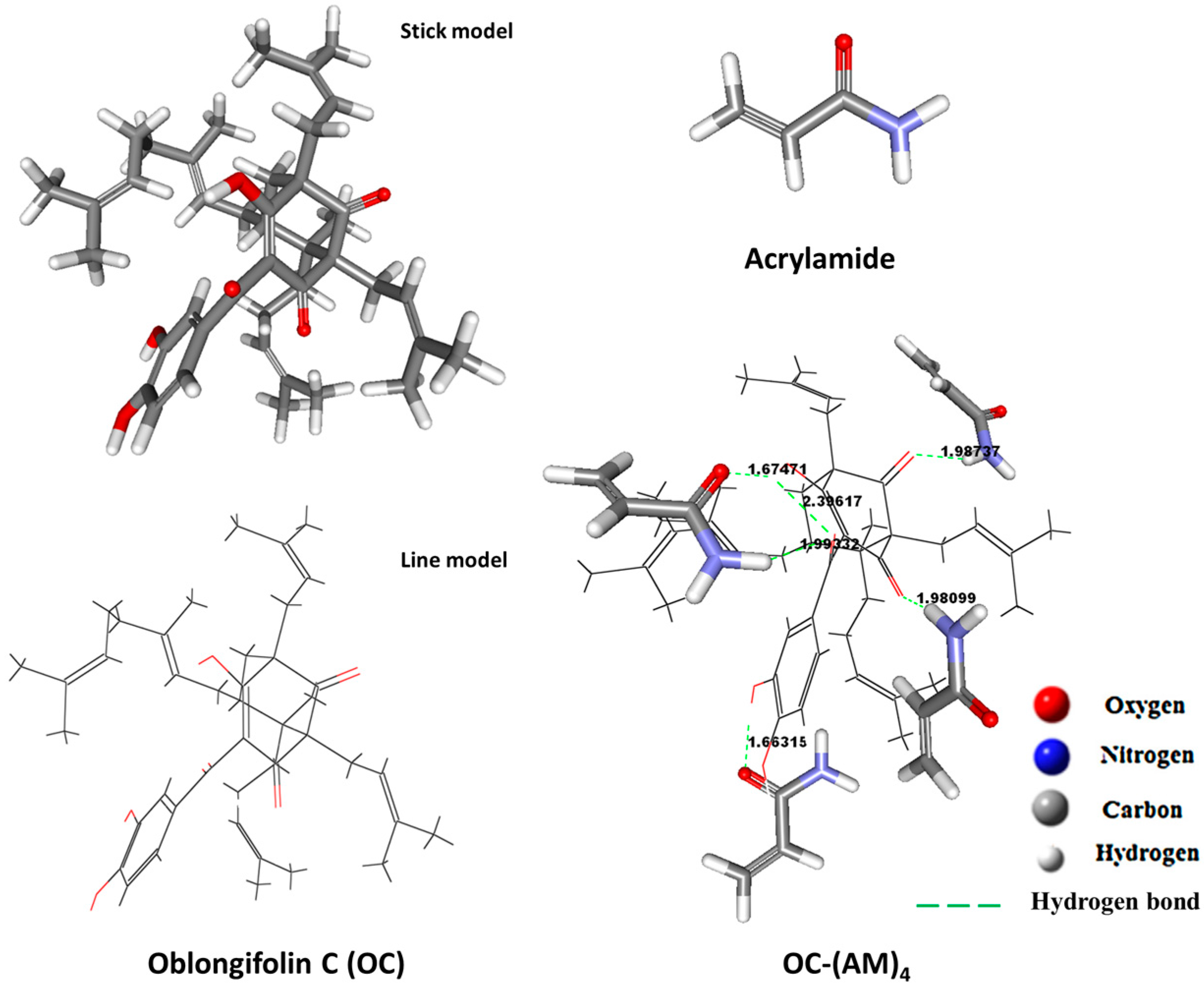
2.2. Molecular Modeling and Computational Optimization
2.3. Characterization of the Morphology
2.4. Evaluation of the Adsorption Properties
2.4.1. Adsorption Isotherm
2.4.2. Adsorption Kinetics
2.4.3. Selectivity Study of the Sorbents
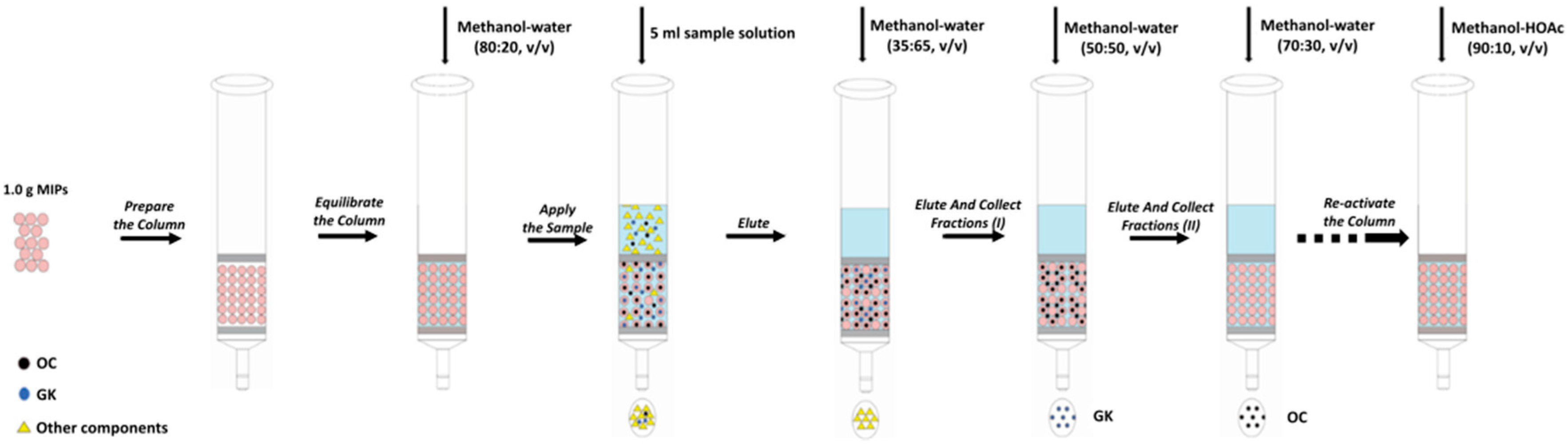
2.5. Optimization of the MISPE Procedures
2.5.1. The Type and Volume of the Loading Solvent
2.5.2. Washing and Eluting Solvent Selection
2.5.3. Reusability of MISPE
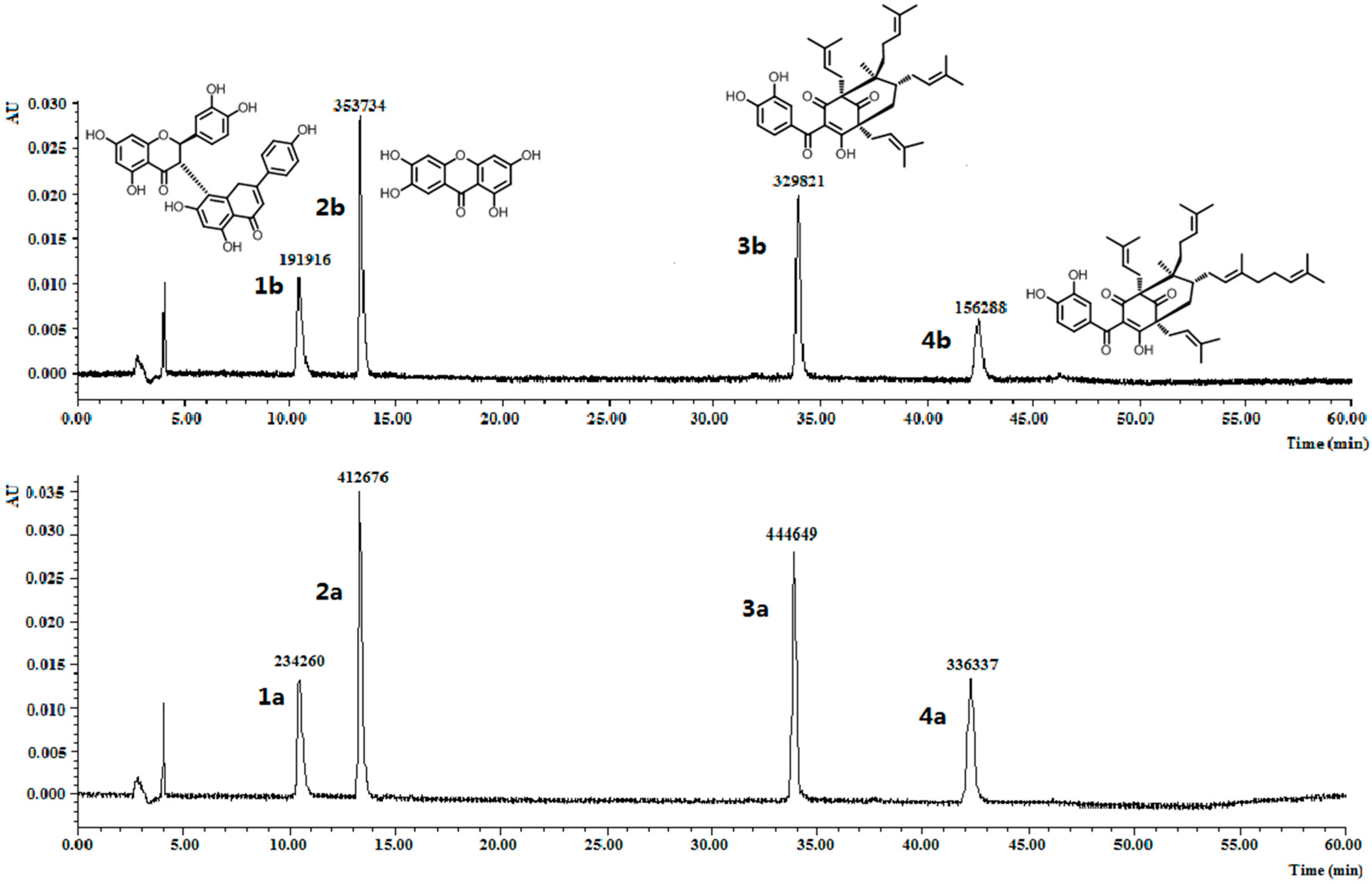
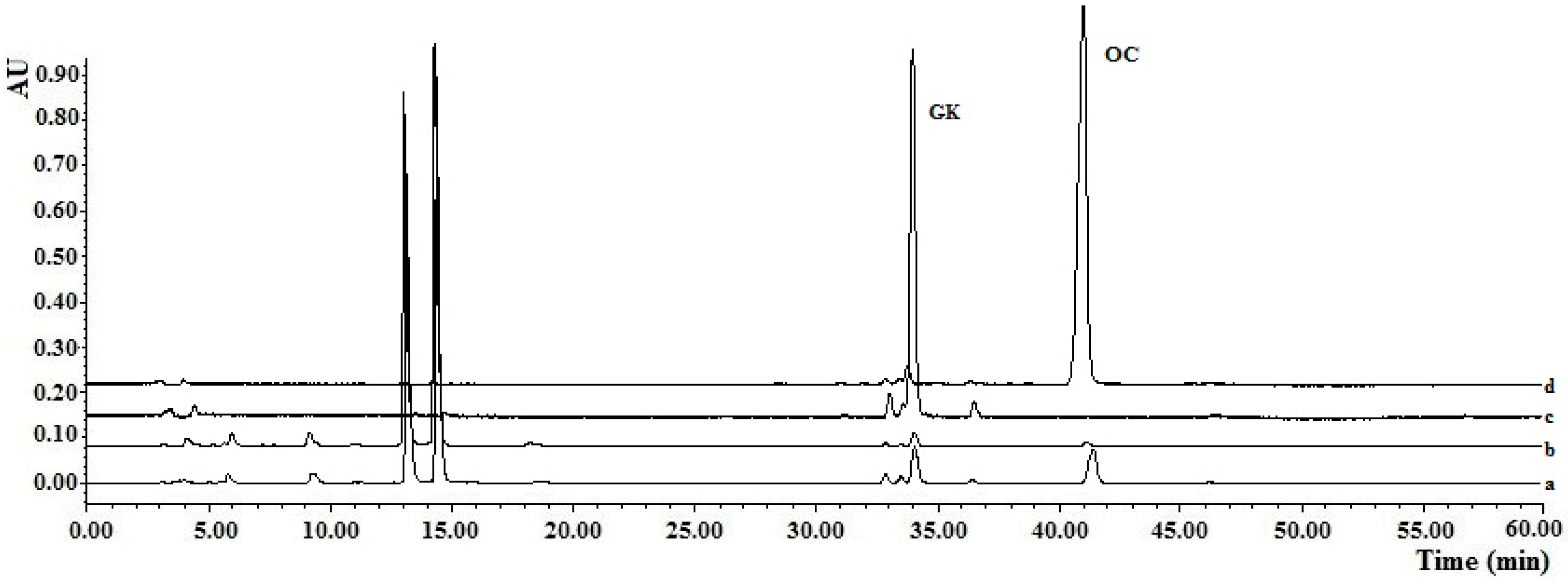
2.6. Application of MISPE to Fruit Extracts of G. yunnanensis Hu
3. Materials and Methods
3.1. Reagents and Materials
3.2. Apparatus
3.3. Computational Optimization: Monomers Molar Ratio Screening
3.4. Preparation of MIPs
3.5. Evaluation of MIPs Performance
3.5.1. Equilibrium Batch Rebinding Experiments
3.5.2. Adsorption Isotherms
3.5.3. Adsorption Kinetics
3.5.4. Selectivity Experiments
3.5.5. Characterization of the Molecularly Imprinted Polymer
3.6. Preparation of MISPE Column
3.7. Preparation of the Fruit Extracts of G. yunnanensis Hu
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Xu, G.; Feng, C.; Zhou, Y.; Han, Q.B.; Qiao, C.F.; Huang, S.X.; Chang, D.C.; Zhao, Q.S.; Luo, K.Q.; Xu, H.X. Bioassay and ultraperformance liquid chromatography/mass spectrometry guided isolation of apoptosis-inducing benzophenones and xanthone from the pericarp of Garcinia yunnanensis Hu. J. Agric. Food Chem. 2008, 56, 11144–11150. [Google Scholar] [PubMed]
- Li, X.W.; Li, J.; Stevens, P.F. Garcinia Linnaeus. In Flora of China; Wu, Z.Y., Raven, P.H., Eds.; Science Press and Missouri Botanical Garden Press: Beijing, China, 2007; Volume 13, pp. 40–47. [Google Scholar]
- Lu, Y.; Cai, S.; Tan, H.; Fu, W.; Zhang, H.; Xu, H. Inhibitory effect of oblongifolin C on allergic inflammation through the suppression of mast cell activation. Mol. Cell. Biochem. 2015, 406, 263–271. [Google Scholar] [PubMed]
- Feng, C.; Zhou, L.Y.; Yu, T.; Xu, G.; Tian, H.L.; Xu, J.J.; Xu, H.X.; Luo, K.Q. A new anticancer compound, oblongifolin C, inhibits tumor growth and promotes apoptosis in HeLa cells through Bax activation. Int. J. Cancer 2012, 131, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Lao, Y.; Wan, G.; Liu, Z.; Wang, X.; Ruan, P.; Xu, W.; Xu, D.; Xie, W.; Zhang, Y.; Xu, H.; et al. The natural compound oblongifolin C inhibits autophagic flux and enhances antitumor efficacy of nutrient deprivation. Autophagy 2014, 10, 736–749. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Cheng, M.; Lao, Y.; Wang, X.; Wu, J.; Zhou, L.; Zhang, Y.; Xu, H.; Xu, N. DNA damage and ER stress contribute to oblongifolin C-induced cell killing in Bax/Bak-deficient cells. Biochem. Biophys. Res. Commun. 2015, 457, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lao, Y.; Xu, N.; Xi, Z.; Wu, M.; Wang, H.; Li, X.; Tan, H.; Sun, M.; Xu, H. Oblongifolin C inhibits metastasis by up-regulating keratin 18 and tubulins. Sci. Rep. 2015, 5, 10293. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.X.; Feng, C.; Zhou, Y.; Xu, G.; Han, Q.B.; Qiao, C.F.; Chang, D.C.; Luo, K.Q.; Xu, H.X. Bioassay-guided isolation of xanthones and polycyclic prenylated acylphloroglucinols from Garcinia oblongifolia. J. Nat. Prod. 2009, 72, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Wulff, G.; Sarhan, A. The use of polymers with enzyme-analogous structures for resolution of racemates. Angew. Chem. Int. Ed. Engl. 1972, 11, 341–344. [Google Scholar]
- Whitcombe, M.J.; Kirsch, N.; Nicholls, I.A. Molecular imprinting science and technology: A survey of the literature for the years 2004–2011. J. Mol. Recognit. 2014, 27, 297–401. [Google Scholar] [PubMed]
- Chen, L.; Xu, S.; Li, J. Recent advances in molecular imprinting technology: Current status, challenges and highlighted applications. Chem. Soc. Rev. 2011, 40, 2922–2942. [Google Scholar] [PubMed]
- Liu, X.; Zong, H.Y.; Huang, Y.P.; Liu, Z.S. Liquid crystal-based molecularly imprinted nanoparticles with low crosslinking for capillary electrochromatography. J. Chromatogr. A 2013, 1309, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Cheong, W.J.; Yang, S.H.; Ali, F. Molecular imprinted polymers for seperation science: A review of reviews. J. Sep. Sci. 2013, 36, 609–628. [Google Scholar] [PubMed]
- Tse Sum Bui, B.; Haupt, K. Molecularly imprinted polymers: Synthetic receptors in bioanalysis. Anal. Bioanal. Chem. 2010, 398, 2481–2492. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, Y.; Dai, Q.; Zhou, Y. Magnetic deep eutectic solvents molecularly imprinted polymers for the selective recognition and separation of protein. Anal. Chim. Acta 2016, 936, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, M.P.; Prasad, A. Molecularly imprinted polymer based enantioselective sensing devices: A review. Anal. Chim. Acta 2015, 853, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Huynh, T.-P.; Kutner, W. Molecularly imprinted polymers as recognition materials for electronic tongues. Biosens. Bioelectron. 2015, 74, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Ashley, J.; Shahbazi, M.-A.; Kant, K.; Chidambara, V.A.; Wolff, A.; Bang, D.D.; Sun, Y. Molecularly imprinted polymers for sample preparation and biosensing in food analysis: Progress and perspectives. Biosens. Bioelectron. 2017, 91, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Soppera, O.; Haupt, K. Photopolymerization and photostructuring of molecularly imprinted polymers for sensor applications—A review. Anal. Chim. Acta 2012, 717, 7–20. [Google Scholar] [PubMed]
- Yang, B.; Lv, S.; Chen, F.; Liu, C.; Cai, C.; Chen, C.; Chen, X. A resonance light scattering sensor based on bioinspired molecularly imprinted polymers for selective detection of papain at trace levels. Anal. Chim. Acta 2016, 912, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Mirata, F.; Resmini, M. Molecularly Imprinted Polymers for Catalysis and Synthesis. Adv. Biochem. Eng. Biotechnol. 2015, 150, 107–129. [Google Scholar] [PubMed]
- Resmini, M. Molecularly imprinted polymers as biomimetic catalysts. Anal. Bioanal. Chem. 2012, 402, 3021–3026. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.M.; Zhang, X.; Xu, Z.X. The Applications of Molecularly Imprinted Polymer in Immunoassay, Biosensor and Enzyme Mimic Catalyst-A Critical Review. Adv. Mater. Res. 2012, 466–467, 84–87. [Google Scholar] [CrossRef]
- Alvarezlorenzo, C.; Concheiro, A. Molecularly imprinted polymers for drug delivery. J. Chromatogr. B 2004, 804, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Puoci, F.; Iemma, F.; Picci, N. Stimuli-responsive molecularly imprinted polymers for drug delivery: A review. Curr. Drug Deliv. 2008, 5, 85. [Google Scholar] [CrossRef] [PubMed]
- Dhanashree, S.; Priyanka, M.; Manisha, K.; Vilasrao, K. Molecularly imprinted polymers: Novel discovery for drug delivery. Curr. Drug Deliv. 2016, 13, 632–645. [Google Scholar] [CrossRef] [PubMed]
- Suedee, R. The Use of Molecularly Imprinted Polymers for Dermal Drug Delivery. Pharm. Anal. Acta 2013, 4. [Google Scholar] [CrossRef]
- Li, Z.Y.; Quan, H.J.; Gong, C.B.; Yang, Y.Z.; Tang, Q.; Wei, Y.B.; Ma, X.B.; Lam, H.W. Photocontrolled solid-phase extraction of guanine from complex samples using a novel photoresponsive molecularly imprinted polymer. Food Chem. 2015, 172, 56–62. [Google Scholar] [PubMed]
- Sellergren, B. Direct Drug Determination by Selective Sample Enrichment on an Imprinted Polymer. Anal. Chem. 1994, 66, 1678–1582. [Google Scholar] [CrossRef]
- Tsermentseli, S.K.; Manesiotis, P.; Assimopoulou, A.N.; Papageorgiou, V.P. Molecularly imprinted polymers for the isolation of bioactive naphthoquinones from plant extracts. J. Chromatogr. A 2013, 1315, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Chen, L.; Ma, X.; Wang, X.; Gao, Q.; Geng, Y.; Huang, L. Molecularly imprinted polymers with novel functional monomer for selective solid-phase extraction of gastrodin from the aqueous extract of Gastrodia elata. J. Chromatogr. A 2014, 1342, 1–7. [Google Scholar] [PubMed]
- Saad, E.M.; Madbouly, A.; Ayoub, N.; El Nashar, R.M. Preparation and application of molecularly imprinted polymer for isolation of chicoric acid from Chicorium intybus L. medicinal plant. Anal. Chim. Acta 2015, 877, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Roy, E.; Patra, S.; Madhuri, R.; Sharma, P.K. Gold nanoparticle mediated designing of non-hydrolytic sol-gel cross-linked metformin imprinted polymer network: A theoretical and experimental study. Talanta 2014, 120, 198–207. [Google Scholar] [PubMed]
- Meier, F.; Schott, B.; Riedel, D.; Mizaikoff, B. Computational and experimental study on the influence of the porogen on the selectivity of 4-nitrophenol molecularly imprinted polymers. Anal. Chim. Acta 2012, 744, 68–74. [Google Scholar] [PubMed]
- Yan, H.; Row, K. Characteristic and Synthetic Approach of Molecularly Imprinted Polymer. Int. J. Mol. Sci. 2006, 7, 155. [Google Scholar] [CrossRef]
- Umpleby, R.J., II; Baxter, S.C.; Bode, M.; Berch, J.K., Jr.; Shan, R.N.; Shimizu, K.D. Application of the Freundlich adsorption isotherm in the characterization of molecularly imprinted polymers. Anal. Chim. Acta 2001, 435, 35–42. [Google Scholar]
- Ji, W.H.; Ma, X.L.; Zhang, J.H.; Xie, H.K.; Liu, F.; Wang, X. Preparation of the high purity gingerols from ginger by dummy molecularly imprinted polymers. J. Chromatogr. A 2015, 1387, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A. The COSMO and COSMO-RS solvation models. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 699–709. [Google Scholar] [CrossRef]
- Dutra, J.D.; Filho, M.A.; Rocha, G.B.; Freire, R.O.; Simas, A.M.; Stewart, J.J. Sparkle/PM7 Lanthanide Parameters for the Modeling of Complexes and Materials. J. Chem. Theory Comput. 2013, 9, 3333–3341. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar]
- Baggiani, C.; Giraudi, G.; Giovannoli, C.; Tozzi, C.; Anfossi, L. Adsorption isotherms of a molecular imprinted polymer prepared in the presence of a polymerisable template: Indirect evidence of the formation of template clusters in the binding site. Anal. Chim. Acta 2004, 504, 43–52. [Google Scholar]
- Spivak, D.A. Optimization, evaluation, and characterization of molecularly imprinted polymers. Adv. Drug Deliv. Rev. 2005, 57, 1779–1794. [Google Scholar] [CrossRef] [PubMed]
- Manzoor, S.; Buffon, R.; Rossi, A.V. Molecularly imprinted solid phase extraction of fluconazole from pharmaceutical formulations. Talanta 2015, 134, 1–7. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds oblongifolin C and guttiferone K are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MIPs | Molecular Template 1 | Functional Monomer | Crosslinker | Porogen | Molar Ratio 2 | IF 3 | RSD 4 (%) |
|---|---|---|---|---|---|---|---|
| A | OC | 4-VP | EDGMA | DMSO (3 mL) | 1:4:10 | 1.26 | 6.67 |
| B | OC | MAA | EDGMA | DMSO (3 mL) | 1:4:10 | 1.37 | 1.25 |
| C | OC | AM | EDGMA | DMSO (3 mL) | 1:4:10 | 3.42 | 0.42 |
| D | OC | AM | EDGMA | MeCN (3 mL) | 1:4:10 | 0.87 | 0.76 |
| E | OC | AM | EDGMA | Tol. (3 mL) | 1:4:10 | 1.36 | 6.66 |
| F | OC | AM | EDGMA | CHCl3 (3 mL) | 1:4:10 | 0.93 | 4.98 |
| G | OC | AM | EDGMA | DMSO (3 mL) | 1:4:8 | 2.34 | 8.03 |
| H | OC | AM | EDGMA | DMSO (3 mL) | 1:4:20 | 1.10 | 3.41 |
| I | OC | AM | EDGMA | DMSO (3 mL) | 1:1:10 | 1.93 | 0.78 |
| J | OC | AM | EDGMA | DMSO (3 mL) | 1:2:10 | 1.95 | 1.17 |
| K | OC | AM | EDGMA | DMSO (3 mL) | 1:3:10 | 2.08 | 0.73 |
| Complexes | Energies (Hartree) 1 | Binding Energies (kcal·mol−1) 2 |
|---|---|---|
| OC (Template) | −2122.941 | / |
| AM | −247.118 | / |
| OC-AM | −2368.641 | −2.343 |
| OC-(AM)2 | −2609.601 | −8.594 |
| OC-(AM)3 | −2843.979 | −13.128 |
| OC-(AM)4 | −3110.504 | −15.465 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Fu, W.; Shen, Y.; Tan, H.; Xu, H. Molecularly Imprinted Polymers for Selective Extraction of Oblongifolin C from Garcinia yunnanensis Hu. Molecules 2017, 22, 508. https://doi.org/10.3390/molecules22040508
Wang L, Fu W, Shen Y, Tan H, Xu H. Molecularly Imprinted Polymers for Selective Extraction of Oblongifolin C from Garcinia yunnanensis Hu. Molecules. 2017; 22(4):508. https://doi.org/10.3390/molecules22040508
Chicago/Turabian StyleWang, Liping, Wenwei Fu, Yunhui Shen, Hongsheng Tan, and Hongxi Xu. 2017. "Molecularly Imprinted Polymers for Selective Extraction of Oblongifolin C from Garcinia yunnanensis Hu" Molecules 22, no. 4: 508. https://doi.org/10.3390/molecules22040508
APA StyleWang, L., Fu, W., Shen, Y., Tan, H., & Xu, H. (2017). Molecularly Imprinted Polymers for Selective Extraction of Oblongifolin C from Garcinia yunnanensis Hu. Molecules, 22(4), 508. https://doi.org/10.3390/molecules22040508