
Revision of the Structure of Acremine P from a Marine-Derived Strain of Acremonium persicinum

Abstract
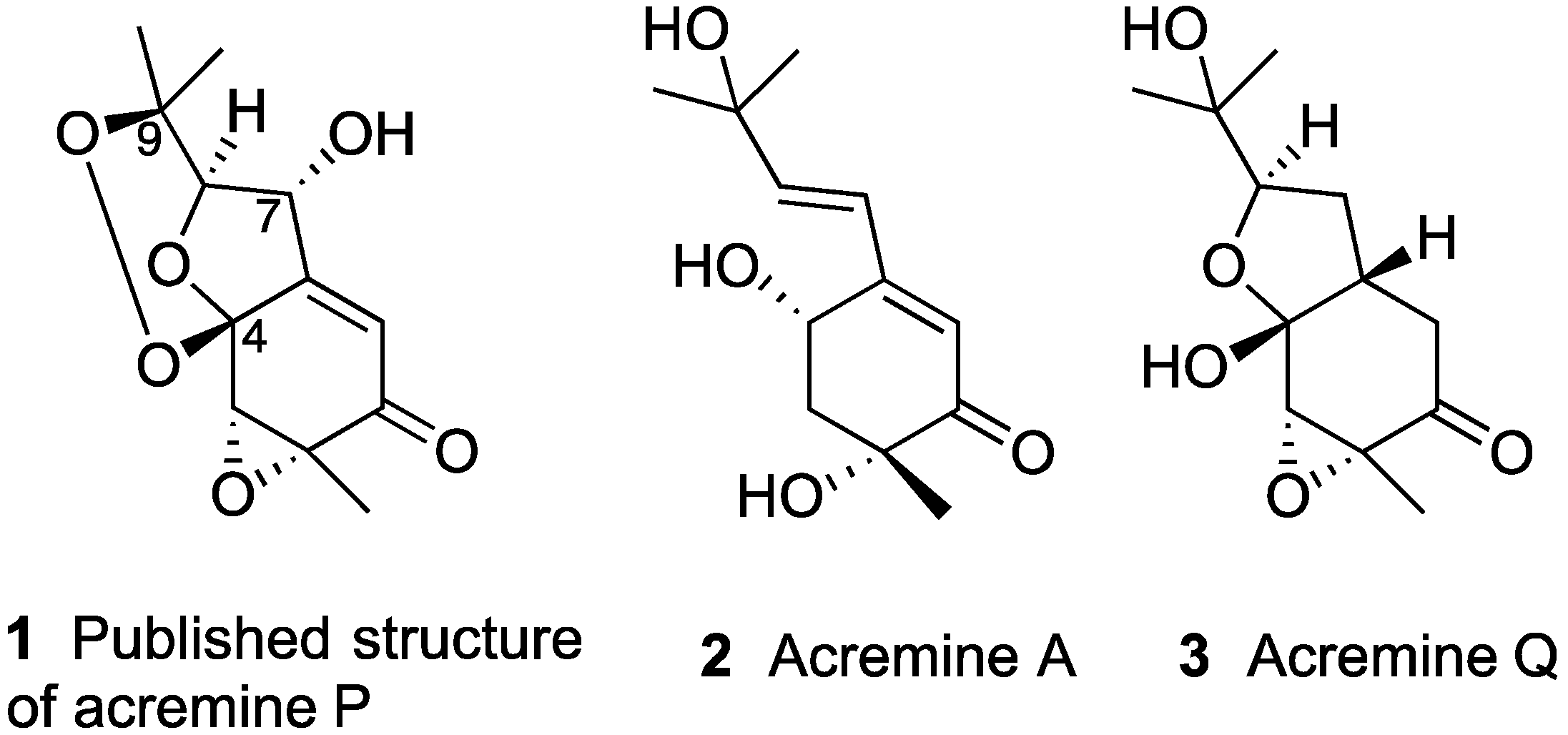
:1. Introduction
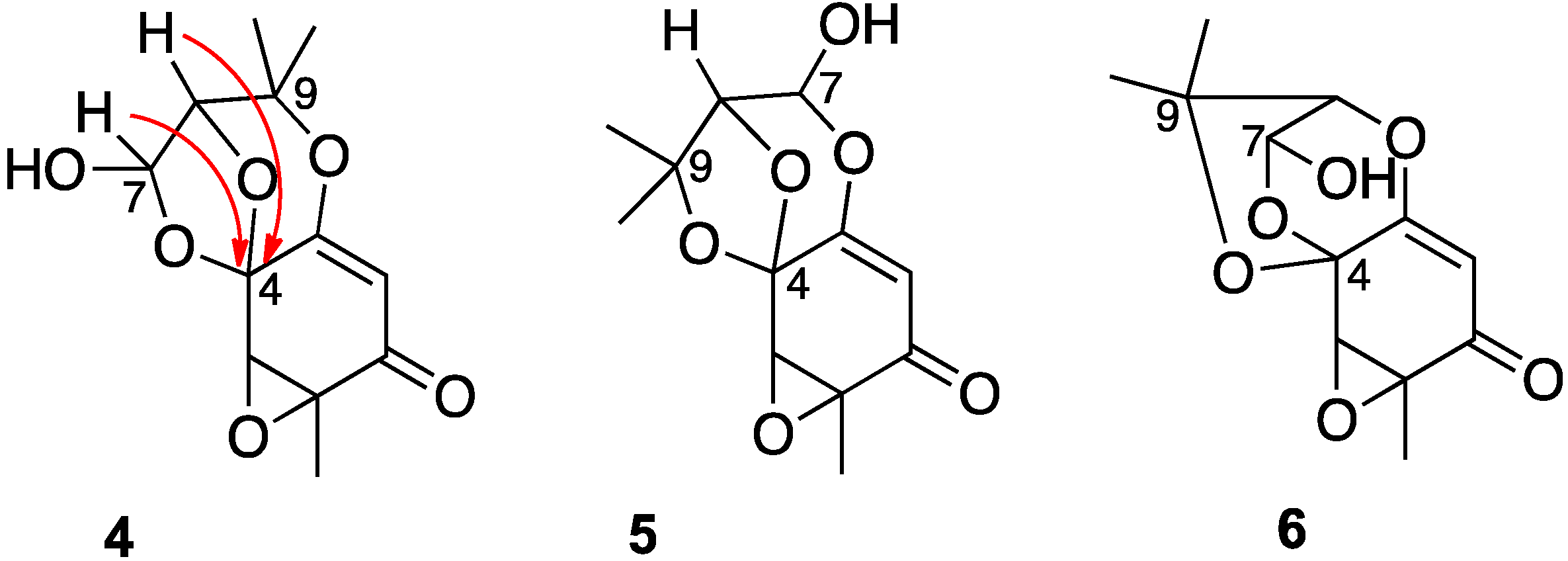
2. Results and Discussions
3. Materials and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Patterson, E.L.; Vanmeter, J.C.; Bohonos, N. Isolation of cephalosporin C. J. Med. Chem. 1964, 7, 689. [Google Scholar] [CrossRef] [PubMed]
- Assante, G.; Dallavalle, S.; Malpezzi, L.; Burruano, S.; Torta, L. Acremines A-F, novel secondary metabolites produced by a strain of an endophytic Acremonium, isolated from sporangiophores of Plasmopara viticola in grapevine leaves. Tetrahedron 2005, 61, 7686–7692. [Google Scholar] [CrossRef]
- Arnone, A.; Assante, G.; Bava, A.; Dallavalle, S.; Nasini, G. Acremines H-N, novel prenylated polyketide metabolites produced by a strain of Acremonium byssoides. Tetrahedron 2009, 65, 786–791. [Google Scholar] [CrossRef]
- Januar, L.A.; Molinski, T.F. Acremolin from Acremonium strictum is 2,3-etheno-2′-isopropyl-1-methylguanine, not an 1H-azirine. Synthesis and structural revision. Org. Lett. 2013, 15, 2370–2373. [Google Scholar] [CrossRef] [PubMed]
- Boot, C.M.; Tenney, K.; Valeriote, F.A.; Crews, P. Highly N-methylated linear peptides produced by an atypical sponge-derived Acremonium sp. J. Nat. Prod. 2006, 69, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Song, Y.; Chen, Y.; Huang, H.; Zhang, W.; Ju, J. Cyclic heptapeptides, cordyheptapeptides C-E from the marine-derived fungus Acremonium persicinum SCSIO 115 and their cytotoxic activities. J. Nat. Prod. 2012, 75, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Frazer, J.A.; Lambert, L.K.; Pierens, G.K.; Bernhardt, P.V.; Garson, M.J. Secondary metabolites of the sponge-derived fungus Acremonium persicinum. J. Nat. Prod. 2013, 76, 1432–1440. [Google Scholar]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic and synthetic organic chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef] [PubMed]
- Tantillo, D.J. Walking in the woods with quantum chemistry-applications of quantum chemical calculations in natural products research. Nat. Prod. Rep. 2013, 30, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, P.H.; Jansma, M.J.; Hoye, T.R. A guide to small molecule structure assignment through computation of (1H and 13C) NMR chemical shifts. Nat. Prot. 2014, 9, 643–659. [Google Scholar] [CrossRef] [PubMed]
- Saielli, G.; Nicolaou, K.C.; Ortiz, A.; Zhang, H.; Bagno, A. Addressing the stereochemistry of complex organic molecules by density functional theory-NMR: Vannusal B in retrospective. J. Am. Chem. Soc. 2011, 133, 6072–6077. [Google Scholar] [CrossRef] [PubMed]
- Lodewyk, M.W.; Tantillo, D.J. Prediction of the structure of nobilisitine using computed NMR chemical shifts. J. Nat. Prod. 2011, 74, 1339–1342. [Google Scholar] [CrossRef] [PubMed]
- Spartan Software. Available online: https://www.wavefun.com./products/spartan.html (accessed on 21 February 2017).
- Sarotti, A.M.; Pellegrinet, S.C. A multi-standard approach for GIAO 13C-NMR calculations. J. Org. Chem. 2009, 74, 7254–7260. [Google Scholar] [CrossRef] [PubMed]
- Kutateladze, A.G.; Mukhina, O.A. Parametrization of 13C-1H nuclear spin-spin coupling constants. J. Org. Chem. 2015, 80, 10838–10848. [Google Scholar] [CrossRef] [PubMed]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An improved probability for the stereochemical assignment of isomeric compounds using quantum chemical assignments of NMR shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- De Voss, J.J.; Cryle, M.J. Carbon-carbon bond cleavage by P450 systems. Met. Ions Life Sci. 2007, 3, 397–435. [Google Scholar]
Sample Availability: A sample of acremine P is available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carbon | Exptl. | Calc. (1) | Calc. (4a) | Calc. (4b) | Calc. (4c) | Calc. (4d) |
|---|---|---|---|---|---|---|
| 1 | 192.3, C | 197.6 | 193.3 | 193.5 | 193.0 | 192.3 |
| 2 | 102.4, CH | 122.8 | 108.7 | 104.0 | 108.7 | 102.4 |
| 3 | 162.5, C | 159.0 | 164.6 | 163.3 | 165.5 | 162.5 |
| 4 | 99.0, C | 108.0 | 101.3 | 116.5 | 101.0 | 99.0 |
| 5 | 59.1, CH | 55.5 | 60.3 | 58.7 | 60.4 | 59.1 |
| 6 | 57.4, C | 57.2 | 57.3 | 57.5 | 57.7 | 57.4 |
| 7 | 95.0, CH | 72.0 | 95.9 | 99.4 | 98.8 | 95.0 |
| 8 | 86.2, CH | 89.0 | 88.7 | 82.2 | 82.0 | 86.2 |
| 9 | 78.2, C | 81.5 | 80.4 | 80.8 | 83.4 | 78.2 |
| 10 | 25.8, CH3 | 21.9 | 23.9 | 27.6 | 26.2 | 25.8 |
| 11 | 23.4, CH3 | 21.9 | 23.2 | 23.1 | 22.9 | 23.4 |
| 12 | 14.4, CH3 | 13.4 | 15.9 | 15.8 | 15.9 | 14.5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garson, M.J.; Hehre, W.; Pierens, G.K.; Suciati. Revision of the Structure of Acremine P from a Marine-Derived Strain of Acremonium persicinum. Molecules 2017, 22, 521. https://doi.org/10.3390/molecules22040521
Garson MJ, Hehre W, Pierens GK, Suciati. Revision of the Structure of Acremine P from a Marine-Derived Strain of Acremonium persicinum. Molecules. 2017; 22(4):521. https://doi.org/10.3390/molecules22040521
Chicago/Turabian StyleGarson, Mary J., Warren Hehre, Gregory K. Pierens, and Suciati. 2017. "Revision of the Structure of Acremine P from a Marine-Derived Strain of Acremonium persicinum" Molecules 22, no. 4: 521. https://doi.org/10.3390/molecules22040521
APA StyleGarson, M. J., Hehre, W., Pierens, G. K., & Suciati. (2017). Revision of the Structure of Acremine P from a Marine-Derived Strain of Acremonium persicinum. Molecules, 22(4), 521. https://doi.org/10.3390/molecules22040521