
Antisense Oligonucleotide-Based Therapy for Neuromuscular Disease

 ,
,
Abstract
:1. Introduction
1.1. ASO Therapy in Duchenne Muscular Dystrophy
1.2. ASO Therapy in Spinal Muscular Atrophy
2. Cellular Uptake Mechanism of Antisense Oligonucleotides
2.1. ASO Internalization Process Mediated by Endocytosis
2.2. ASO Uptake by Cell-Penetrating Peptides
2.3. Limiting Factors for ASOs Uptake
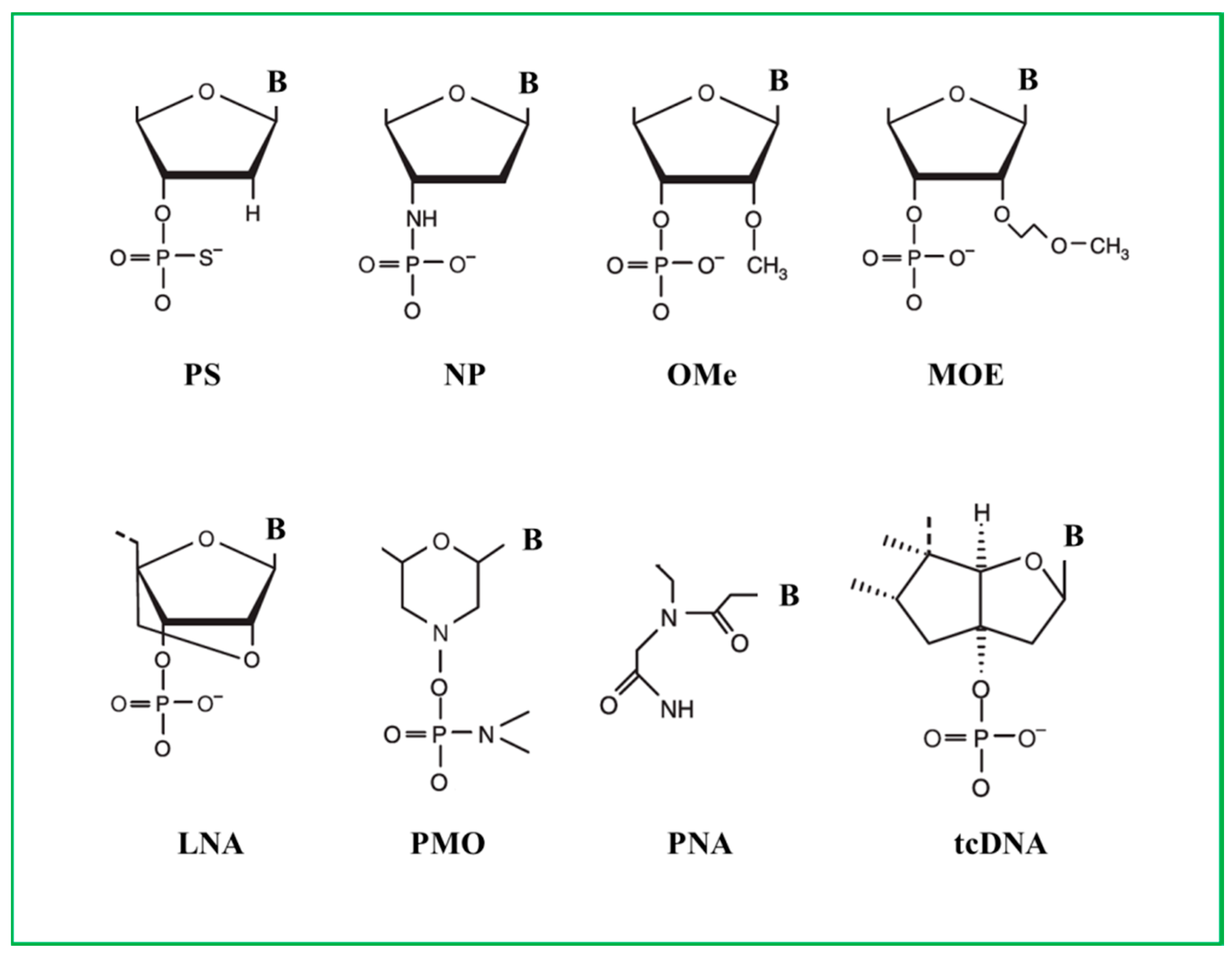
3. Different Chemistries of Antisense Oligonucleotides
3.1. Phosphorothioate ASOs
3.2. 2′-O-Methyl and 2’-O-Methoxyethyl ASOs
3.3. Locked Nuclei Acid ASOs
3.4. Phosphorodiamidate Morpholino ASOs
3.5. Peptide Nucleic Acids ASOs
3.6. Tricyclo-DNA ASOs
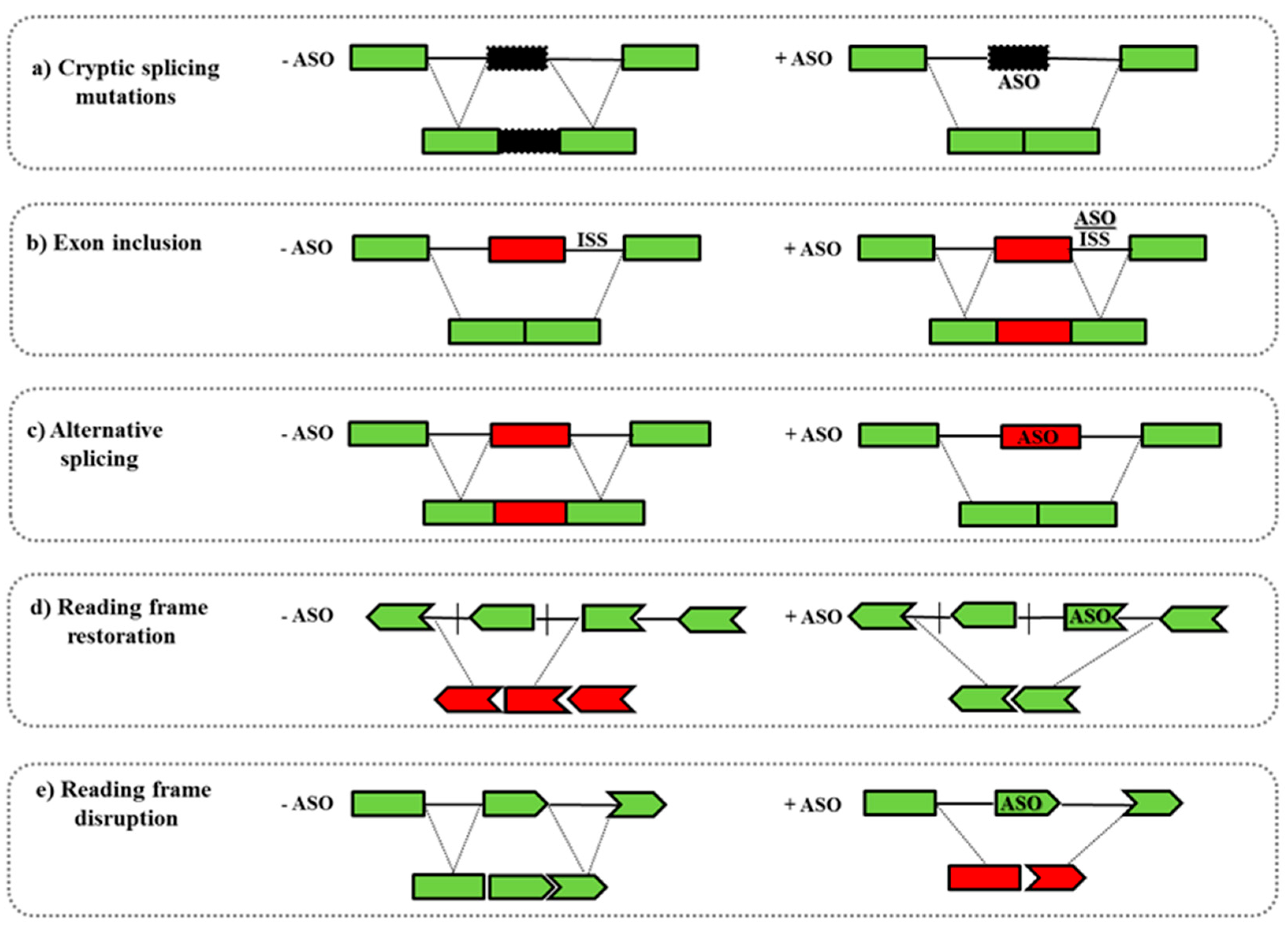
4. Mechanism of Action of ASOs
- -
- Another well-studied application is the splicing modulation of the nuclear pre-mRNA. ASOs can target specific regions as 5′/3′ splice junctions or exonic/intronic splicing enhancer/silencer sites (ESEs or ISEs, ESSs or ISSs) leading to the skipping/inclusion of an exon. This strategy can be used to: (i) restore the mRNA reading frame by exon skipping in diseases such as DMD in which frame-shift deletions or non-sense mutations cause the functional protein loss; (ii) promote the inclusion of exons, as occurs in SMA where ASOs induce the inclusion of the exon 7 in the SMN2 gene; (iii) introduce an out-of-frame deletion for reducing protein expression, as in Alzheimer disease or in Amyotrophic Lateral Sclerosis (Figure 2) [67].
- -
- ASOs can also target microRNA (miRNAs) that are involved in many disease mechanisms and could be used as therapeutic tools. miRNAs are able to silence the target mRNA as well as influencing multiple mRNA expression profiles. Targeting miRNA regulation by using ASOs could be very effective in clinical interventions, as has been demonstrated for hepatitis C infection (antimiR122), breast cancer (anti-miR221), and brain tumors (anti-miR155) [68,69]. Catapano and colleagues showed that the systemic treatment of SMA mice with a single dose of oligomer PMO25 is able to reverse the altered miR-132 levels in spinal cord, muscle, and serum [70].
- -
- -
- ASOs can also interfere in the binding between proteins and pathogenic RNA species. Myotonic dystrophy type 1 (DM1) is a neuromuscular disease caused by expanded CUG repeats in the 3′-untranslated region of the DM protein kinase (DMPK) transcript [73]. A morpholino ASO has been developed, CAG25, and it is able to form a stable RNA-morpholino heteroduplex with the pathogenic DMPK transcripts carrying the CUG repeats. In this way, CAG25 blocks the interaction of these abnormal RNA species with other proteins such as muscleblind-like 1 (MBNL1), which has a fundamental role in the control of the splicing machinery [74]. Mulders and colleagues developed a 2′-O-methyl-phosphorothioate modified (CAG)7 ASO that silences the toxic DMPK transcript and induce a normalizing effect on aberrant pre-mRNA splicing [75].
5. Pre-Clinical and Clinical Approaches of ASOs in DMD
5.1. Phosphorothioate ASOs
5.2. 2′OMe ASOs
5.3. Morpholino ASO
6. Pre-Clinical and Clinical Approaches of ASOs in SMA
7. ASO Therapy in Other Neuromuscular Disorders
7.1. Myotonic Dystrophy
7.2. Facioscapulohumeral Muscular Dystrophy
8. Delivery of ASOs
- -
- Solid Lipid Nanoparticles (SLNs) that, made of natural, semi-synthetic or synthetic lipids, are biocompatible and produced easily at large scale systems, can protect drugs from degradation and control the release of the molecules [141].
- -
- Polymer nanoparticles (including micelles, nanocapsules, nanospheres, colloids, dendrimers, core-shells) in which the drug can be loaded by different methods, i.e., entrapment, dispersion, dissolution, or adsorption [142]. Examples of polymer nanoparticles used to conjugate ASOs for exon skipping application in the DMD field are cationic core-shell NPs, named T1 and ZM2, made up of a polymethyl methacrylate (PMMA) core surrounded by a cationic shell for the ASO binding. Both T1 and ZM2 NPs showed the capability to delivery 2′OMePS M23D ASO in mdx mice. In particular, intraperitoneal and oral administrations of ZM2-ASOs complexes induced dystrophin restoration in preclinical studies [143,144,145,146,147].
- -
- Lipid-based Nanoparticles (LNPs), composed of cationic lipid and other compounds called “helper lipids” that contribute to their stability and delivery efficiency. LNPs for antisense delivery usually encapsulate the ASO inside the aqueous core and between bilayers [148].
- -
- Carbon-based nanomaterials, like graphene, nanodiamond, fullerenes, nanotubes, are used for various applications including medicine for drug delivery and imaging [149].
9. Conclusions
Acknowledgments
Conflicts of Interest
Funding
References
- Bull, S.C.; Doig, A.J. Properties of protein drug target classes. PLoS ONE 2015, 10, e0117955. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, K.; Gogtay, N.J. Therapeutic nucleic acids: Current clinical status. Br. J. Clin. Pharmacol. 2016, 82, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L. Gene therapy returns to centre stage. Nature 2015, 526, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Potaczek, D.; Garn, H.; Unger, S.D.; Renz, H. Antisense molecules: A new class of drugs. J. Allergy Clin. Immunol. 2016, 137, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- Grijalvo, S.; Aviñó, A.; Eritja, R. Oligonucleotide delivery: A patent review (2010–2013). Expert Opin. Ther. Pat. 2014, 24, 801–819. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Muntoni, F. Muscular dystrophy: New challenges and review of the current clinical trials. Curr. Opin. Pediatr. 2013, 25, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef] [PubMed]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Van der Ree, M.H.; de Vree, J.M.; Stelma, F.; Willemse, S.; van der Valk, M.; Rietdijk, S.; Molenkamp, R.; Schinkel, J.; van Nuenen, A.C.; Beuers, U.; et al. Safety, tolerability, and antiviral effect of RG-101 in patients with chronic hepatitis C: A phase 1B, double-blind, randomised controlled trial. Lancet 2017, 389, 709–717. [Google Scholar] [CrossRef]
- Straub, V.; Balabanov, P.; Bushby, K.; Ensini, M.; Goemans, N.; De Luca, A.; Pereda, A.; Hemmings, R.; Campion, G.; Kaye, E.; et al. Stakeholder cooperation to overcome challenges in orphan medicine development: The example of Duchenne muscular dystrophy. Lancet Neurol. 2016, 15, 882–890. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Van Deutekom, J.C.; Fokkema, I.F.; Van Ommen, G.J.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 12, 731–740. [Google Scholar] [CrossRef]
- Pramono, Z.A.; Takeshima, Y.; Alimsardjono, H.; Ishii, A.; Takeda, S.; Matsuo, M. Induction of exon skipping of the dystrophin transcript in lymphoblastoid cells by transfecting an antisense oligodeoxynucleotide complementary to an exon recognition sequence. Biochem. Biophys. Res. Commun. 1996, 226, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Dunckley, M.G.; Manoharan, M.; Villiet, P.; Eperon, I.C.; Dickson, G. Modification of splicing in the dystrophin gene in cultured Mdx muscle cells by antisense oligoribonucleotides. Hum. Mol. Genet. 1998, 7, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Dis. Mod. Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Van Ommen, G.J.; van Deutekom, J.; Aartsma-Rus, A. The therapeutic potential of antisense-mediated exon skipping. Curr. Opin. Mol. Ther. 2008, 10, 140–149. [Google Scholar] [PubMed]
- Feldkötter, M.; Schwarzer, V.; Wirth, R.; Wienker, T.F.; Wirth, B. Quantitative analyses of SMN1 and SMN2 based on real-time light-Cycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 2002, 70, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Pearn, J. Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J. Med. Genet. 1978, 15, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef]
- McAndrew, P.E.; Parsons, D.W.; Simard, L.R.; Rochette, C.; Ray, P.N.; Mendell, J.R.; Prior, T.W.; Burghes, A.H. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet. 1997, 60, 1411–1422. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.E.; Thomas, N.H.; Lewis, C.M.; Abbs, S.J.; Rodrigues, N.R.; Davies, K.E.; Mathew, C.G. Correlation of SMNt and SMNc gene copy number with age of onset and survival in spinal muscular atrophy. Eur. J. Hum. Genet. 1998, 6, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Cartegni, L.; Krainer, A.R. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 2002, 30, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Kashima, T.; Manley, J.L. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet. 2003, 34, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef] [PubMed]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Passini, M.A.; Bu, J.; Richards, A.M.; Kinnecom, C.; Sardi, S.P.; Stanek, L.M.; Hua, Y.; Rigo, F.; Matson, J.; Hung, G.; et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 2011, 3, 72ra18. [Google Scholar] [CrossRef] [PubMed]
- Porensky, P.N.; Mitrpant, C.; McGovern, V.L.; Bevan, A.K.; Foust, K.D.; Kaspar, B.K.; Wilton, S.D.; Burghes, A.H. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum. Mol. Genet. 2012, 21, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Janghra, N.; Mitrpant, C.; Dickinson, R.L.; Anthony, K.; Price, L.; Eperon, I.C.; Wilton, S.D.; Morgan, J.; Muntoni, F. A Novel morpholino oligomer targeting ISS-N1 improves rescue of severe SMA transgenic mice. Hum. Gene Ther. 2013, 24, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Mitrpant, C.; Porensky, P.; Zhou, H.; Price, L.; Muntoni, F.; Fletcher, S.; Wilton, S.D.; Burghes, A.H. Improved antisense oligonucleotide design to suppress aberrant SMN2 gene transcript processing: Towards a treatment for spinal muscular atrophy. PLoS ONE 2013, 8, e62114. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, E.; Marais, T.; Chatauret, N.; Benkhelifa-Ziyyat, S.; Duque, S.; Ravassard, P.; Carcenac, R.; Astord, S.; Pereira de Moura, A.; Voit, T.; et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum. Mol. Genet. 2011, 20, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Foust, K.D.; Wang, X.; McGovern, V.L.; Braun, L.; Bevan, A.K.; Haidet, A.M.; Le, T.T.; Morales, P.R.; Rich, M.M.; Burghes, A.H.; et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010, 28, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Passini, M.A.; Bu, J.; Roskelley, E.M.; Richards, A.M.; Sardi, S.P.; O’Riordan, C.R.; Klinger, K.W.; Shihabuddin, L.S.; Cheng, S.H. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J. Clin. Investig. 2010, 120, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Valori, C.F.; Ning, K.; Wyles, M.; Mead, R.J.; Grierson, A.J.; Shaw, P.J.; Azzouz, M. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci. Transl. Med. 2010, 2, 35ra42. [Google Scholar] [CrossRef] [PubMed]
- Avila, A.M.; Burnett, B.G.; Taye, A.A.; Gabanella, F.; Knight, M.A.; Hartenstein, P.; Cizman, Z.; Di Prospero, N.A.; Pellizzoni, L.; Fischbeck, K.H.; et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J. Clin. Investig. 2007, 117, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, J.; Zabidi-Hussin, Z.A.; Sasongko, T.H. Histone deacetylase inhibitors as potential treatment for spinal muscular atrophy. Genet. Mol. Biol. 2007, 36, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Naryshkin, N.A.; Weetall, M.; Dakka, A.; Narasimhan, J.; Zhao, X.; Feng, Z.; Ling, K.K.Y.; Karp, G.M.; Qi, H.; Woll, M.G.; et al. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014, 345, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Palacino, J.; Swalley, S.E.; Song, C.; Cheung, A.K.; Shu, L.; Zhang, X.; Van Hoosear, M.; Shin, Y.; Chin, D.N.; Keller, C.G.; et al. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol. 2015, 11, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Gaisina, I.N.; El-Khodor, B.F.; Ramboz, S.; Makhortova, N.R.; Rubin, L.L.; Kozikowski, A.P. Identification of a Maleimide-Based Glycogen Synthase Kinase-3 (GSK-3) Inhibitor, BIP-135, that Prolongs the Median Survival Time of Delta7 SMA KO Mouse Model of Spinal Muscular Atrophy. ACS Chem. Neurosci. 2012, 3, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Corti, S.; Nizzardo, M.; Nardini, M.; Donadoni, C.; Salani, S.; Ronchi, D.; Simone, C.; Falcone, M.; Papadimitriou, D.; Locatelli, F.; et al. Embryonic stem cell-derived neural stem cells improve spinal muscular atrophy phenotype in mice. Brain 2010, 133, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Corti, S.; Nizzardo, M.; Simone, C.; Falcone, M.; Nardini, M.; Ronchi, D.; Donadoni, C.; Salani, S.; Riboldi, G.; Magri, F.; et al. Genetic correction of human induced pluripotent stem cells from patients with spinal muscular atrophy. Sci. Transl. Med. 2012, 4, 165ra162. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Farr, S.A.; Butt, W.; Kumar, V.B.; Franko, M.W.; Morley, J.E. Delivery across the blood-brain barrier of antisense directed against amyloid beta: Reversal of learning and memory deficits in mice overexpressing amyloid precursor protein. J. Pharmacol. Exp. Ther. 2001, 297, 1113–1121. [Google Scholar] [PubMed]
- Owen, D.; Harrison, S.C. Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harb. Perspect. Biol. 2014, 6, a016725. [Google Scholar]
- Lajoie, P.; Nabi, I.R. Lipid rafts, caveolae, and their endocytosis. Int. Rev. Cell Mol. Biol. 2010, 282, 135–163. [Google Scholar] [PubMed]
- Juliano, R.L.; Carver, K. Cellular uptake and intracellular trafficking of oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, J.D.; Flynn, N.H. Cell-penetrating peptides transport therapeutics into cells. Pharmacol. Ther. 2015, 154, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Park, J.; Ryu, J.; Lee, H.J.; Bahn, J.H.; Han, K.; Choi, E.Y.; Lee, K.S.; Kwon, H.Y.; Choi, S.Y. Mutational analysis of a human immunodeficiency virus type 1 Tat protein transduction domain which is required for delivery of an exogenous protein into mammalian cells. J. Gen. Virol. 2002, 83, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Joliot, A.; Pernelle, C.; Deagostini-Bazin, H.; Prochiantz, A. Antennapedia homeobox peptide regulates neural morphogenesis. Proc. Natl. Acad. Sci. USA 1991, 88, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Betts, C.; Saleh, A.F.; Arzumanov, A.A.; Hammond, S.M.; Godfrey, C.; Coursindel, T.; Gait, M.J.; Wood, M.J. Pip-6-PMO, A new generation of peptide-oligonucleotide conjugates with improved cardiac exon skipping activity for DMD treatment. Mol. Ther. Nucleic Acids 2012, 1, e38. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Kayali, R.; Bertoni, C.; Fike, F.; Hu, H.; Iversen, P.L.; Gatti, R.A. Arginine-rich cell-penetrating peptide dramatically enhances AMO-mediated ATM aberrant splicing correction and enables delivery to brain and cerebellum. Hum. Mol. Genet. 2011, 20, 3151–3160. [Google Scholar] [CrossRef] [PubMed]
- Boisguérin, P.; Deshayes, S.; Gait, M.J.; O’Donovan, L.; Godfrey, C.; Betts, C.A.; Wood, M.J.; Lebleu, B. Delivery of therapeutic oligonucleotides with cell penetrating peptides. Adv. Drug Deliv. Rev. 2015, 87, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, G.D.; Arzumanov, A.; Abes, R.; Yin, H.; Wood, M.J.; Lebleu, B.; Gait, M.J. Improved cell-penetrating peptide-PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle. Nucleic Acids Res. 2008, 36, 6418–6428. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Saleh, A.F.; Betts, C.; Camelliti, P.; Seow, Y.; Ashraf, S.; Arzumanov, A.; Hammond, S.; Merritt, T.; Gait, M.J.; et al. Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice. Mol. Ther. 2011, 19, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.A.; Subasinghe, C.; Shinozuka, K.; Cohen, J.S. Physicochemical properties of phosphorothioate oligodeoxynucleotides. Nucleic Acids Res. 1988, 16, 3209–3221. [Google Scholar] [CrossRef] [PubMed]
- Gryaznov, S.M.; Lloyd, D.H.; Chen, J.K.; Schultz, R.G.; DeDionisio, L.A.; Ratmeyer, L.; Wilson, W.D. Oligonucleotide N3’-->P5’ phosphoramidates. Proc. Natl. Acad. Sci. USA 1995, 92, 5798–5802. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S. Importance of nucleotide sequence and chemical modifications of antisense oligonucleotides. Biochim. Biophys. Acta 1999, 1489, 53–67. [Google Scholar] [CrossRef]
- Swayze, E.E.; Siwkowski, A.M.; Wancewicz, E.V.; Migawa, M.T.; Wyrzykiewicz, T.K.; Hung, G.; Monia, B.P.; Bennett, C.F. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res. 2007, 35, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Kaman, W.E.; Bremmer-Bout, M.; Janson, A.A.; den Dunnen, J.T.; van Ommen, G.J.; van Deutekom, J.C. Comparative analysis of antisense oligonucleotide analogs for targeted DMD exon 46 skipping in muscle cells. Gene Ther. 2004, 11, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Swayze, E.E. RNA targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef] [PubMed]
- Braasch, D.A.; Corey, D.R. Novel antisense and peptide nucleic acid strategies for controlling gene expression. Biochemistry 2002, 41, 4503–4510. [Google Scholar] [CrossRef] [PubMed]
- Sazani, P.; Gemignani, F.; Kang, S.H.; Maier, M.A.; Manoharan, M.; Persmark, M.; Bortner, D.; Kole, R. Systemically delivered antisense oligomers upregulate gene expression in mouse tissues. Nat. Biotechnol. 2002, 20, 1228–1233. [Google Scholar] [CrossRef] [PubMed]
- Renneberg, D.; Bouliong, E.; Reber, U.; Schümperli, D.; Leumann, C.J. Antisense properties of tricyclo-DNA. Nucleic Acids Res. 2002, 30, 2751–2757. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A.; Griffith, G.; Babbs, A.; El Andaloussi, S.; Ezzat, K.; Avril, A.; Dugovic, B.; Chaussenot, R.; Ferry, A.; Voit, T.; et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricycle-DNa oligomers. Nat. Med. 2015, 21, 270–275. [Google Scholar] [PubMed]
- Deconinck, A.E.; Rafael, J.A.; Skinner, J.A.; Brown, S.C.; Potter, A.C.; Metzinger, L.; Watt, D.J.; Dickson, J.G.; Tinsley, J.M.; Davies, K.E. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 1997, 90, 717–727. [Google Scholar] [CrossRef]
- Grady, R.M.; Teng, H.; Nichol, M.C.; Cunningham, J.C.; Wilkinson, R.S.; Sanes, J.R. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: A model for Duchenne muscular dystrophy. Cell 1997, 90, 729–738. [Google Scholar] [CrossRef]
- Evers, M.M.; Toonen, L.J.; van Roon-Mom, W.M. Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv. Drug Deliv. Rev. 2015, 87, 90–103. [Google Scholar] [CrossRef] [PubMed]
- McClorey, G.; Wood, M.J. An overview of the clinical application of antisense oligonucleotides for RNA-targeting therapies. Curr. Opin. Pharmacol. 2015, 24, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Lundin, K.E.; Gissberg, O.; Smith, C.I. Oligonucleotide Therapies: The Past and the Present. Hum. Gene Ther. 2015, 26, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Catapano, F.; Zaharieva, I.; Scoto, M.; Marrosu, E.; Morgan, J.; Muntoni, F.; Zhou, H. Altered Levels of MicroRNA-9, -206, and -132 in Spinal Muscular Atrophy and Their Response to Antisense Oligonucleotide Therapy. Mol. Ther. Nucleic Acids 2016, 5, e331. [Google Scholar] [CrossRef] [PubMed]
- Marsollier, A.C.; Ciszewski, L.; Mariot, V.; Popplewell, L.; Voit, T.; Dickson, G.; Dumonceaux, J. Antisense targeting of 3′ end elements involved in DUX4 mRNA processing is an efficient therapeutic strategy for facioscapulohumeral dystrophy: A new gene-silencing approach. Hum. Mol. Genet. 2016, 25, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; King, O.D.; Zhang, Y.; Clayton, N.P.; Spencer, C.; Wentworth, B.M.; Wagner, K.R. Morpholino-mediated Knockdown of DUX4 toward Facioscapulohumeral Muscular Dystrophy Therapeutics. Mol. Ther. 2016, 24, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- González-Barriga, A.; Mulders, S.A.; van de Giessen, J.; Hooijer, J.D.; Bijl, S.; van Kessel, I.D.; van Beers, J.; van Deutekom, J.C.; Fransen, J.A.; Wieringa, B.; et al. Design and analysis of effects of triplet repeat oligonucleotides in cell models for Myotonic Dystrophy. Mol. Ther. Nucleic Acids 2013, 2, e81. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.M.; Sobczak, K.; Lueck, J.D.; Osborne, R.J.; Lin, X.; Dirksen, R.T.; Thornton, C.A. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 2009, 325, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Mulders, S.A.; van den Broek, W.J.; Wheeler, T.M.; Croes, H.J.; van Kuik-Romeijn, P.; de Kimpe, S.J.; Furling, D.; Platenburg, G.J.; Gourdon, G.; Thornton, C.A.; et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 13915–13920. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S.; Henry, S.P.; Grillone, L.R. Fomivirsen: Clinical pharmacology and potential drug interactions. Clin. Pharmacokinet. 2002, 41, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, Y.; Yagi, M.; Wada, H.; Ishibashi, K.; Nishiyama, A.; Kakumoto, M.; Sakaeda, T.; Saura, R.; Okumura, K.; Matsuo, M. Intravenous infusion of an antisense oligonucleotide results in exon skipping in muscle dystrophin mRNA of Duchenne muscular dystrophy. Pediatr. Res. 2006, 59, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Van Deutekom, J.C.; Janson, A.A.; Ginjaar, I.B.; Frankhuizen, W.S.; Aartsma-Rus, A.; Bremmer-Bout, M.; den Dunnen, J.T.; Koop, K.; van der Kooi, A.J.; Goemans, N.M.; et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 2007, 357, 2677–2686. [Google Scholar] [CrossRef] [PubMed]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.A.; et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Goemans, N.M.; Tulinius, M.; van den Hauwe, M.; Kroksmark, A.K.; Buyse, G.; Wilson, R.J.; van Deutekom, J.C.; de Kimpe, S.J.; Lourbakos, A.; Campion, G. Long-Term Efficacy, Safety, and Pharmacokinetics of Drisapersen in Duchenne Muscular Dystrophy: Results from an Open-Label Extension Study. PLoS ONE 2016, 11, e0161955. [Google Scholar] [CrossRef] [PubMed]
- Voit, T.; Topaloglu, H.; Straub, V.; Muntoni, F.; Deconinck, N.; Campion, G.; De Kimpe, S.J.; Eagle, M.; Guglieri, M.; Hood, S.; et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): An exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 2014, 13, 987–996. [Google Scholar] [CrossRef]
- GSK and Prosensa Announce Primary Endpoint Not Met in Phase III Study of Drisapersen in Patients With Duchenne Muscular Dystrophy. Available online: https://globenewswire.com/news-release/2013/09/20/574726/10049265/en/GSK-and-Prosensa-Announce-Primary-Endpoint-Not-Met-in-Phase-III-Study-of-Drisapersen-in-Patients-With-Duchenne-Muscular Dystrophy.html (accessed on 22 March 2017).
- Biomarin. Available online: http://investors.biomarin.com/2014-11-24-BioMarin-and-Prosensa-Holding-N-V-Reach-Agreement-on-Intended-Public-Offer-for-100-of-Prosensas-Outstanding-Stock-Will-Add-Duchenne-Muscular-Dystrophy-Products-to-Rare-Disease-Portfolio (accessed on 29 March 2017).
- BioMarin Announces that FDA Has Advised It Will Not Take Action on the Kyndrisa™ (drisapersen) New Drug Application by the PDUFA Date. Available online: http://investors.biomarin.com/2015-12-18-BioMarin-Announces-That-FDA-Has-Advised-it-Will-Not-Take-Action-on-the-Kyndrisa-drisapersen-New-Drug-Application-by-the-PDUFA-Date (accessed on 23 March 2017).
- BioMarin Announces Withdrawal of Market Authorization Application for Kyndrisa™ (drisapersen) in Europe. Available online: http://investors.biomarin.com/2016-05-31-BioMarin-Announces-Withdrawal-of-Market-Authorization-Application-for-Kyndrisa-drisapersen-in-Europe (accessed on 23 March 2017).
- Sazani, P.; Ness, K.P.; Weller, D.L.; Poage, D.W.; Palyada, K.; Shrewsbury, S.B. Repeat-dose toxicology evaluation in cynomolgus monkeys of AVI-4658, a phosphorodiamidate morpholino oligomer (PMO) drug for the treatment of duchenne muscular dystrophy. Int. J. Toxicol. 2011, 30, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Kinali, M.; Arechavala-Gomeza, V.; Feng, L.; Cirak, S.; Hunt, D.; Adkin, C.; Guglieri, M.; Ashton, E.; Abbs, S.; Nihoyannopoulos, P.; et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009, 8, 918–928. [Google Scholar] [CrossRef]
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.J.; et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605. [Google Scholar] [CrossRef]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E. Eteplirsen Study Group and Telethon Foundation DMD Italian Network. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef] [PubMed]
- FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. Available online: www.fda.org https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm521263.htm (accessed on 23 March 2017).
- Sarepta Therapeutics Announces EMA Validation of Eteplirsen Authorization Application for Treatment of Duchenne Muscular Dystrophy Amenable to Exon Skipping 51. Available online: http://investorrelations.sarepta.com/phoenix.zhtml?c=64231&p=irol-newsArticle&ID=2230729 (accessed on 23 March 2017).
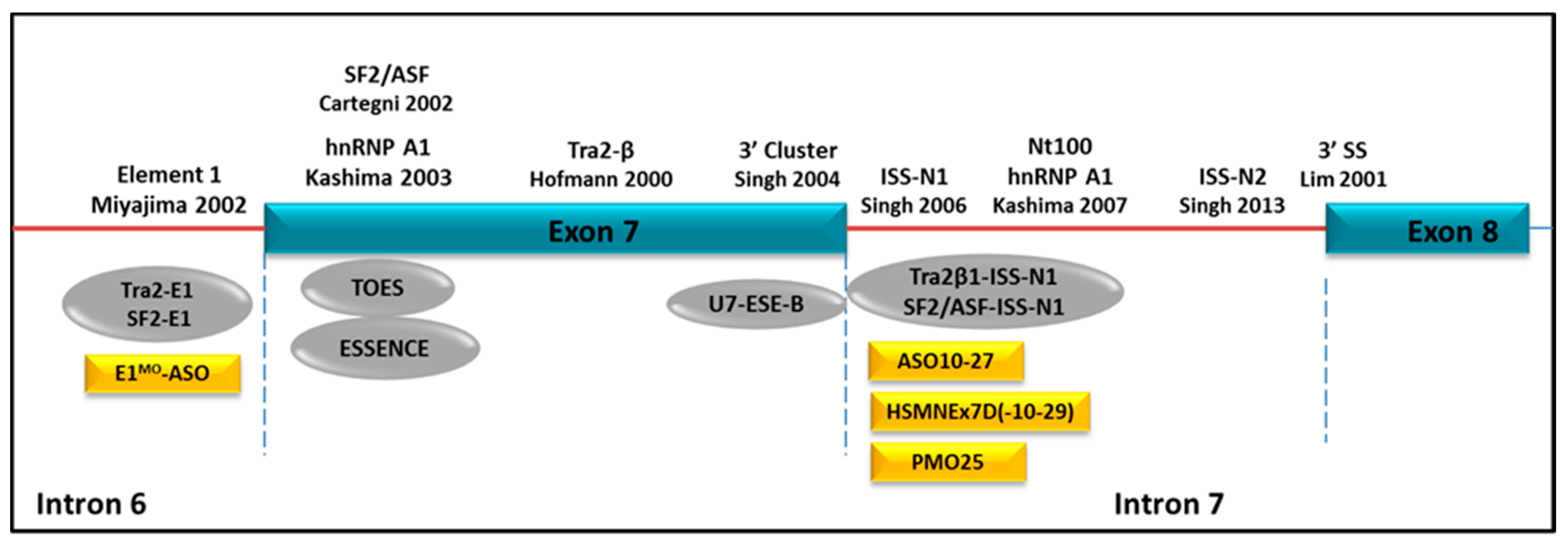
- Hofmann, Y.; Lorson, C.L.; Stamm, S.; Androphy, E.J.; Wirth, B. Htra2-beta 1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2). Proc. Natl. Acad. Sci. USA 2000, 97, 9618–9623. [Google Scholar] [CrossRef] [PubMed]
- Kashima, T.; Rao, N.; Manley, J.L. An intronic element contributes to splicing repression in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2007, 104, 3426–3431. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.R.; Hertel, K.J. Modulation of survival motor neuron pre-mRNA splicing by inhibition of alternative 3′ splice site pairing. J. Biol. Chem. 2001, 276, 45476–45483. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, H.; Miyaso, H.; Okumura, M.; Kurisu, J.; Imaizumi, K. Identification of a cis-acting element for the regulation of SMN exon 7 splicing. J. Biol. Chem. 2002, 277, 23271–23277. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.K.; Singh, N.N.; Androphy, E.J.; Singh, R.N. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol. Cell. Biol. 2006, 26, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.N.; Lawler, M.N.; Ottesen, E.W.; Upreti, D.; Kaczynski, J.R.; Singh, R.N. An intronic structure enabled by a long-distance interaction serves as a novel target for splicing correction in spinal muscular atrophy. Nucleic Acids Res. 2013, 41, 8144–8165. [Google Scholar] [CrossRef] [PubMed]
- Osman, E.Y.; Miller, M.R.; Robbins, K.L.; Lombardi, A.M.; Atkinson, A.K.; Brehm, A.J.; Lorson, C.L. Morpholino antisense oligonucleotides targeting intronic repressor Element1 improve phenotype in SMA mouse models. Hum. Mol. Genet. 2014, 23, 4832–4845. [Google Scholar] [CrossRef] [PubMed]
- Owen, N.; Zhou, H.; Malygin, A.A.; Sangha, J.; Smith, L.D.; Muntoni, F.; Eperon, I.C. Design principles for bifunctional targeted oligonucleotide enhancers of splicing. Nucleic Acids Res. 2011, 39, 7194–7208. [Google Scholar] [CrossRef] [PubMed]
- Skordis, L.A.; Dunckley, M.G.; Yue, B.; Eperon, I.C.; Muntoni, F. Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc. Natl. Acad. Sci. USA 2003, 100, 4114–4119. [Google Scholar] [CrossRef] [PubMed]
- Cartegni, L.; Krainer, A.R. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat. Struct. Biol. 2003, 10, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Baughan, T.D.; Dickson, A.; Osman, E.Y.; Lorson, C.L. Delivery of bifunctional RNAs that target an intronic repressor and increase SMN levels in an animal model of spinal muscular atrophy. Hum. Mol. Genet. 2009, 18, 1600–1611. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Vickers, T.A.; Okunola, H.L.; Bennett, C.F.; Krainer, A.R. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 2008, 82, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.H.; Schray, R.C.; Patterson, C.A.; Ayitey, S.O.; Tallent, M.K.; Lutz, G.J. Oligonucleotide-mediated survival of motor neuron protein expression in CNS improves phenotype in a mouse model of spinal muscular atrophy. J. Neurosci. 2009, 29, 7633–7638. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Meng, J.; Marrosu, E.; Janghra, N.; Morgan, J.; Muntoni, F. Repeated low doses of morpholino antisense oligomer: An intermediate mouse model of spinal muscular atrophy to explore the window of therapeutic response. Hum. Mol. Genet. 2015, 24, 6265–6277. [Google Scholar] [CrossRef] [PubMed]
- Nizzardo, M.; Simone, C.; Salani, S.; Ruepp, M.D.; Rizzo, F.; Ruggieri, M.; Zanetta, C.; Brajkovic, S.; Moulton, H.M.; Müehlemann, O.; et al. Effect of combined systemic and local morpholino treatment on the spinal muscular atrophy Delta7 mouse model phenotype. Clin. Ther. 2014, 36, 340–356. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves First Drug for Spinal Muscular Atrophy. Available online: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm534611.htm (accessed on 23 March 2017).
- U.S. FDA Approves Biogen’s SPINRAZA™ (nusinersen), The First Treatment for Spinal Muscular Atrophy. Available online: http://media.biogen.com/press-release/neurodegenerative-diseases/us-fda-approves-biogens-spinraza-nusinersen-first-treatment (accessed on 23 March 2017).
- Shababi, M.; Lorson, C.L.; Rudnik-Schoneborn, S.S. Spinal muscular atrophy: A motor neuron disorder or a multi-organ disease? J. Anat. 2014, 224, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G.; Gillingwater, T.H. Spinal muscular atrophy: Going beyond the motor neuron. Trends Mol. Med. 2013, 19, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Somers, E.; Stencel, Z.; Wishart, T.M.; Gillingwater, T.H.; Parson, S.H. Density, calibre and ramification of muscle capillaries are altered in a mouse model of severe spinal muscular atrophy. Neuromuscul. Disord. 2012, 22, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Somers, E.; Lees, R.D.; Hoban, K.; Sleigh, J.N.; Zhou, H.; Muntoni, F.; Talbot, K.; Gillingwater, T.H.; Parson, S.H. Vascular defects and spinal cord hypoxia in spinal muscular atrophy. Ann. Neurol. 2015, 79, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Sintusek, P.; Catapano, F.; Angkathunkayul, N.; Marrosu, E.; Morgan, J.; Muntoni, F.; Zhou, H. Histopathological defects in intestine in severe spinal muscular atrophy mice are improved by systemic antisense oligonucleotide treatment. PLoS ONE 2016, 11, e0155032. [Google Scholar] [CrossRef] [PubMed]
- Szunyogova, E.; Zhou, H.; Maxwell, G.K.; Powis, R.A.; Francesco, M.; Gillingwater, T.H.; Parson, S.H. Survival Motor Neuron (SMN) protein is required for normal mouse liver development. Sci. Rep. 2016, 6, 34635. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, E.W.; Howell, M.D.; Singh, N.N.; Seo, J.; Whitley, E.M.; Singh, R.N. Severe impairment of male reproductive organ development in a low SMN expressiong mouse model of spinal muscualr atrophy. Sci. Rep. 2016, 6, 20193. [Google Scholar] [CrossRef] [PubMed]
- Gogliotti, R.G.; Quinlan, K.A.; Barlow, C.B.; Heier, C.R.; Jeckman, C.J.; Didonato, C.J. Motor neuron rescue in spinal muscular atrophy mice demonstrates that sensory-motor defects are a consequence, not, a cause of motor neuron dysfunction. J. Neurosci. 2012, 11, 3818–3829. [Google Scholar] [CrossRef] [PubMed]
- Kariya, S.; Obis, T.; Garone, C.; Akay, T.; Sera, F.; Iwata, S.; Homma, S.; Monani, U.R. Requirement of enhanced Survival Motoneuron protein imposed during neuromuscular junction maturation. J. Clin. Investig. 2014, 124, 785–800. [Google Scholar] [CrossRef] [PubMed]
- Iyer, C.C.; McGovern, V.L.; Murray, J.D.; Gombash, S.E.; Zaworski, P.G.; Foust, K.D.; Janssen, P.M.; Burghes, A.H. Low levels of Survival Motor Neuron protein are sufficient for normal muscle function in the SMNΔ7 mouse model of SMA. Hum. Mol. Genet. 2015, 24, 6160–6173. [Google Scholar] [CrossRef] [PubMed]
- Monani, U.R.; Sendtner, M.; Coovert, D.D.; Parsons, D.W.; Andreassi, C.; Le, T.T.; Jablonka, S.; Schrank, B.; Rossoll, W.; Prior, T.W.; et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 2000, 9, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Schrank, B.; Götz, R.; Gunnersen, J.M.; Ure, J.M.; Toyka, K.V.; Smith, A.G.; Sendtner, M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl. Acad. Sci. USA 1997, 94, 9920–9925. [Google Scholar] [CrossRef] [PubMed]
- Bogdanik, L.P.; Osborne, M.A.; Davis, C.; Martin, W.P.; Austin, A.; Rigo, F.; Bennett, C.F.; Lutz, C.M. Systemic, post-symptomatic antisense oligonucleotide rescues motor unit maturation delay in a new mouse model for type II/III spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2015, 112, E5863–E5872. [Google Scholar] [CrossRef] [PubMed]
- Jirka, S.; Aartsma-Rus, A. An update on RNA-targeting therapies for neuromuscular disorders. Curr. Opin. Neurol. 2015, 5, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.F.; Dastidar, S.; Furling, D.; Chuah, M.K. Therapeutic Approaches for Dominant Muscle Diseases: Highlight on Myotonic Dystrophy. Curr. Gene Ther. 2015, 15, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.M.; Leger, A.J.; Pandey, S.K.; MacLeod, A.R.; Nakamori, M.; Cheng, S.H.; Wentworth, B.M.; Bennett, C.F.; Thornton, C.A. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 2012, 488, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Letter from Ionis Pharmaceuticals & Biogen to the MDF Community. Available online: http://us8.campaign-archive2.com/?u=8f5969cac3271759ce78c8354&id=1109538bcf&e=[UNIQID] (accessed on 23 March 2017).
- Pandey, S.N.; Lee, Y.C.; Yokota, T.; Chen, Y.W. Morpholino treatment improves muscle function and pathology of Pitx1 transgenic mice. Mol. Ther. 2014, 22, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Statland, J.; Tawil, R. Facioscapulohumeral muscular dystrophy. Neurol Clin. 2014, 32, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Van der Maarel, S.M.; Tawil, R.; Tapscott, S.J. Facioscapulohumeral muscular dystrophy and DUX4: Breaking the silence. Trends Mol. Med. 2011, 5, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.C.; Lopes, C.M.; Sousa Lobo, J.M.; Amaral, M.H. Nucleic Acids Delivery Systems: A Challenge for Pharmaceutical Technologists. Curr. Drug Metab. 2015, 16, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Ming, X.; Laing, B. Bioconjugates for targeted delivery of therapeutic oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Boisguerin, P.; O’Donovan, L.; Gait, M.J.; Lebleu, B. In Vitro Assays to Assess Exon Skipping in Duchenne Muscular Dystrophy. Meth. Mol. Biol. 2015, 1324, 317–329. [Google Scholar]
- Lehto, T.; Ezzat, K.; Wood, M.J.; El Andaloussi, S. Peptides for nucleic acid delivery. Adv. Drug Deliv. Rev. 2016, 106, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Al-Dosari, M.S.; Gao, X. Nonviral gene delivery: Principle, limitations, and recent progress. AAPS J. 2009, 11, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Lu, P.; Benrashid, E.; Malik, S.; Ashar, J.; Doran, T.J.; Lu, Q.L. Dose-dependent restoration of dystrophin expression in cardiac muscle of dystrophic mice by systemically delivered morpholino. Gene Ther. 2010, 17, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Amantana, A.; Moulton, H.M.; Cate, M.L.; Reddy, M.T.; Whitehead, T.; Hassinger, J.N.; Youngblood, D.S.; Iversen, P.L. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide-morpholino oligomer conjugate. Bioconjug. Chem. 2007, 18, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Hazell, G.; Shabanpoor, F.; Saleh, A.F.; Bowerman, M.; Sleigh, J.N.; Meijboom, K.E.; Zhou, H.; Muntoni, F.; Talbot, K.; et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2016, 27, 10962–10967. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Nakamura, A.; Nagata, T.; Saito, T.; Kobayashi, M.; Aoki, Y.; Echigoya, Y.; Partridge, T.; Hoffman, E.P.; Takeda, S. Extensive and prolonged restoration of dystrophin expression with vivo-morpholino-mediated multiple exon skipping in dystrophic dogs. Nucleic Acid. Ther. 2012, 22, 306–315. [Google Scholar] [PubMed]
- Wang, M.; Wu, B.; Tucker, J.D.; Lu, P.; Lu, Q. Cationic polyelectrolyte-mediated delivery of antisense morpholino oligonucleotides for exon-skipping in vitro and in mdx mice. Int. J. Nanomed. 2015, 10, 5635–5646. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.; Conejos-Sánchez, I.; Griffin, B.T.; O’Driscoll, C.M.; Alonso, M.J. Lipid-based nanocarriers for oral peptide delivery. Adv. Drug Deliv. Rev. 2016, 337, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Bennet, D.; Sanghyo, K. Polymer Nanoparticles for Smart Drug Delivery. In Application of Nanotechnology in Drug Delivery; Sezer, A.D., Ed.; IN TECH d.o.o: Rijeka, Croatia, 2014; Chapter 8; pp. 257–310. ISBN 978-953-51-1628-8. [Google Scholar]
- Rimessi, P.; Sabatelli, P.; Fabris, M.; Braghetta, P.; Bassi, E.; Spitali, P.; Vattemi, G.; Tomelleri, G.; Mari, L.; Perrone, D.; et al. Cationic PMMA nanoparticles bind and deliver antisense oligoribonucleotides allowing restoration of dystrophin expression in the mdx mouse. Mol. Ther. 2009, 17, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Ferlini, A.; Sabatelli, P.; Fabris, M.; Bassi, E.; Falzarano, S.; Vattemi, G.; Perrone, D.; Gualandi, F.; Maraldi, N.M.; Merlini, L.; et al. Dystrophin restoration in skeletal, heart and skin arrector pili smooth muscle of mdx mice by ZM2 NP-AON complexes. Gene Ther. 2010, 17, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Bassi, E.; Falzarano, S.; Fabris, M.; Gualandi, F.; Merlini, L.; Vattemi, G.; Perrone, D.; Marchesi, E.; Sabatelli, P.; Sparnacci, K.; et al. Persistent dystrophin protein restoration 90 days after a course of intraperitoneally administered naked 2’OMePS AON and ZM2 NP-AON complexes in mdx mice. J. Biomed. Biotechnol. 2012, 2012, 8970–8976. [Google Scholar] [CrossRef] [PubMed]
- Falzarano, M.S.; Passarelli, C.; Bassi, E.; Fabris, M.; Perrone, D.; Sabatelli, P.; Maraldi, N.M.; Donà, S.; Selvatici, R.; Bonaldo, P.; et al. Biodistribution and molecular studies on orally administered nanoparticle-AON complexes encapsulated with alginate aiming at inducing dystrophin rescue in mdx mice. BioMed Res. Int. 2013, 2013, 527418. [Google Scholar] [CrossRef] [PubMed]
- Falzarano, M.S.; Passarelli, C.; Ferlini, A. Nanoparticle delivery of antisense oligonucleotides and their application in the exon skipping strategy for Duchenne muscular dystrophy. Nucleic Acid Ther. 2014, 24, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Lee, R.J. The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv. Drug Deliv. Rev. 2016, 99, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, K.; Mukherjee, S.P.; Gallud, A.; Burkert, S.C.; Bistarelli, S.; Bellucci, S.; Bottini, M.; Star, A.; Fadeel, B. Biological interactions of carbon-based nanomaterials: From coronation to degradation. Nanomedicine 2016, 12, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 6213. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | ASO | Number of Patients | Reference |
|---|---|---|---|
| DMD | |||
| PS | 1 | [77] | |
| 2′OMe (Drisarpersen) | 4 | [78] | |
| 2′OMe (Drisarpersen) | 12 | [79] | |
| 2′OMe (Drisarpersen) | 53 | [81] | |
| PMO (Eteplirsen) | 7 | [87] | |
| PMO (Eteplirsen) | 19 | [88] | |
| PMO (Eteplirsen) | 12 (*) | [89] | |
| PMO (Eteplirsen) | 12 (*) | [90] | |
| SMA | |||
| 2′MOE (Nusinersen) | 28 | [8] | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sardone, V.; Zhou, H.; Muntoni, F.; Ferlini, A.; Falzarano, M.S. Antisense Oligonucleotide-Based Therapy for Neuromuscular Disease. Molecules 2017, 22, 563. https://doi.org/10.3390/molecules22040563
Sardone V, Zhou H, Muntoni F, Ferlini A, Falzarano MS. Antisense Oligonucleotide-Based Therapy for Neuromuscular Disease. Molecules. 2017; 22(4):563. https://doi.org/10.3390/molecules22040563
Chicago/Turabian StyleSardone, Valentina, Haiyan Zhou, Francesco Muntoni, Alessandra Ferlini, and Maria Sofia Falzarano. 2017. "Antisense Oligonucleotide-Based Therapy for Neuromuscular Disease" Molecules 22, no. 4: 563. https://doi.org/10.3390/molecules22040563
APA StyleSardone, V., Zhou, H., Muntoni, F., Ferlini, A., & Falzarano, M. S. (2017). Antisense Oligonucleotide-Based Therapy for Neuromuscular Disease. Molecules, 22(4), 563. https://doi.org/10.3390/molecules22040563