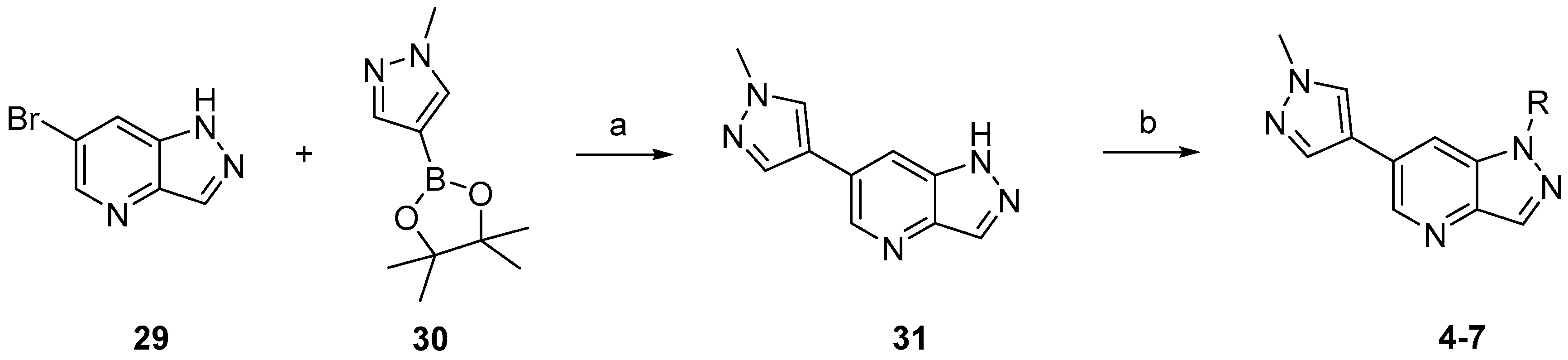
3.3. Chemistry
Compounds
4–
7 were synthesized according to the procedures outlined in
Scheme 1. Suzuki coupling of commercially available
29 with 1-methylpyrazole-4-boronic acid pinacol ester (
30) provided
31. Compounds
4–
7 were prepared by deprotonation of compound
31 followed by addition of the corresponding electrophile reagents.
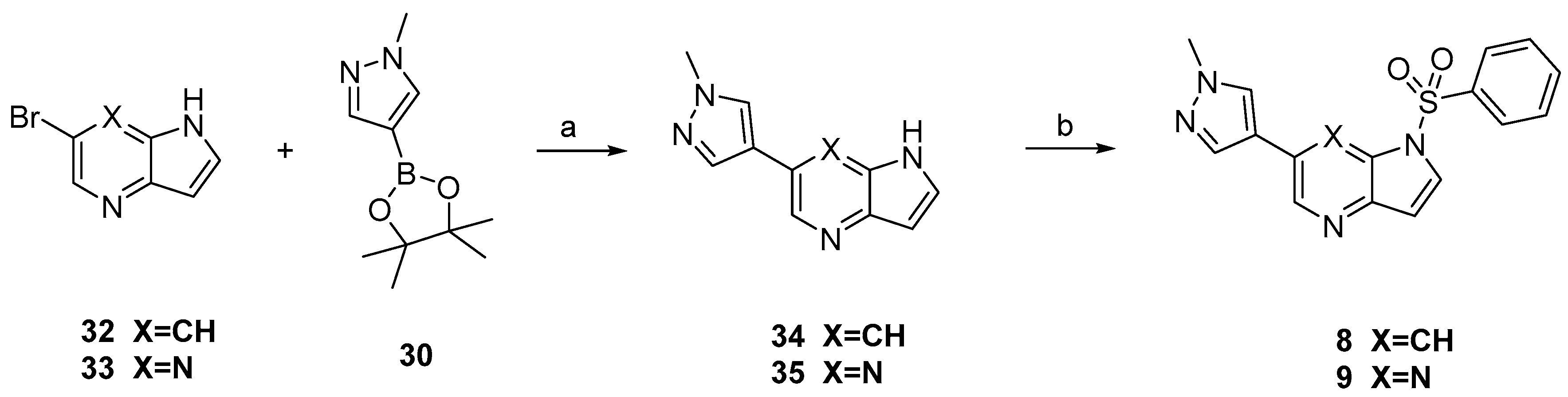
Compounds
8 and
9 were prepared according to the procedure in
Scheme 2. Treatment of compounds
32 and
33 with
30 via Suzuki coupling afforded compounds
34 and
35 which were sulfonylated to afford
8 and
9, respectively.
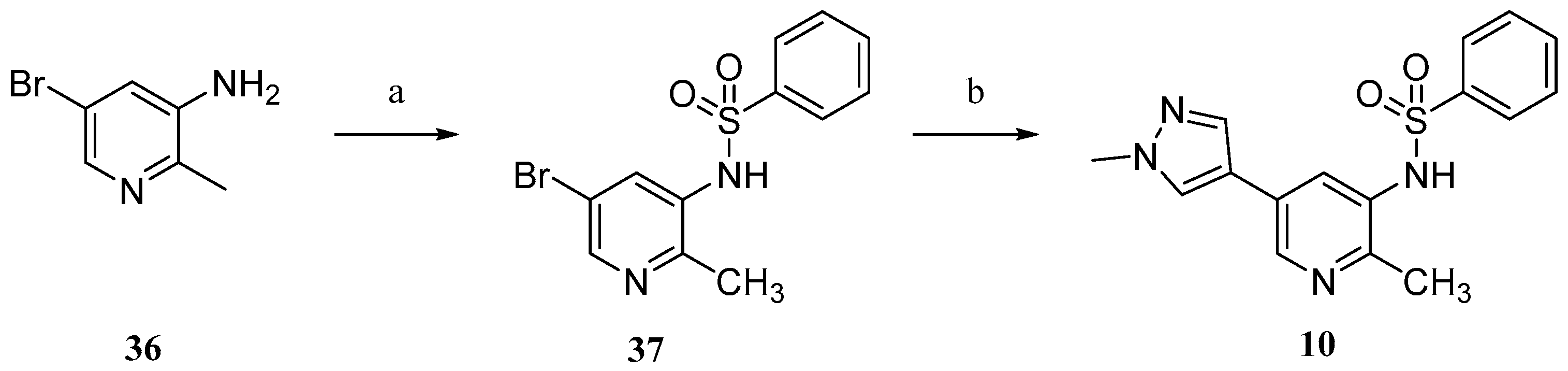
Compound
10 was prepared according to
Scheme 3. Commercially available 5-bromo-2-methylpyridin-3-amine (
36) were sulfonylated to afford
37. Conventional Suzuki coupling of
37 and 1-methylpyrazole-4-boronic acid pinacol ester (
30) afforded compound
10.
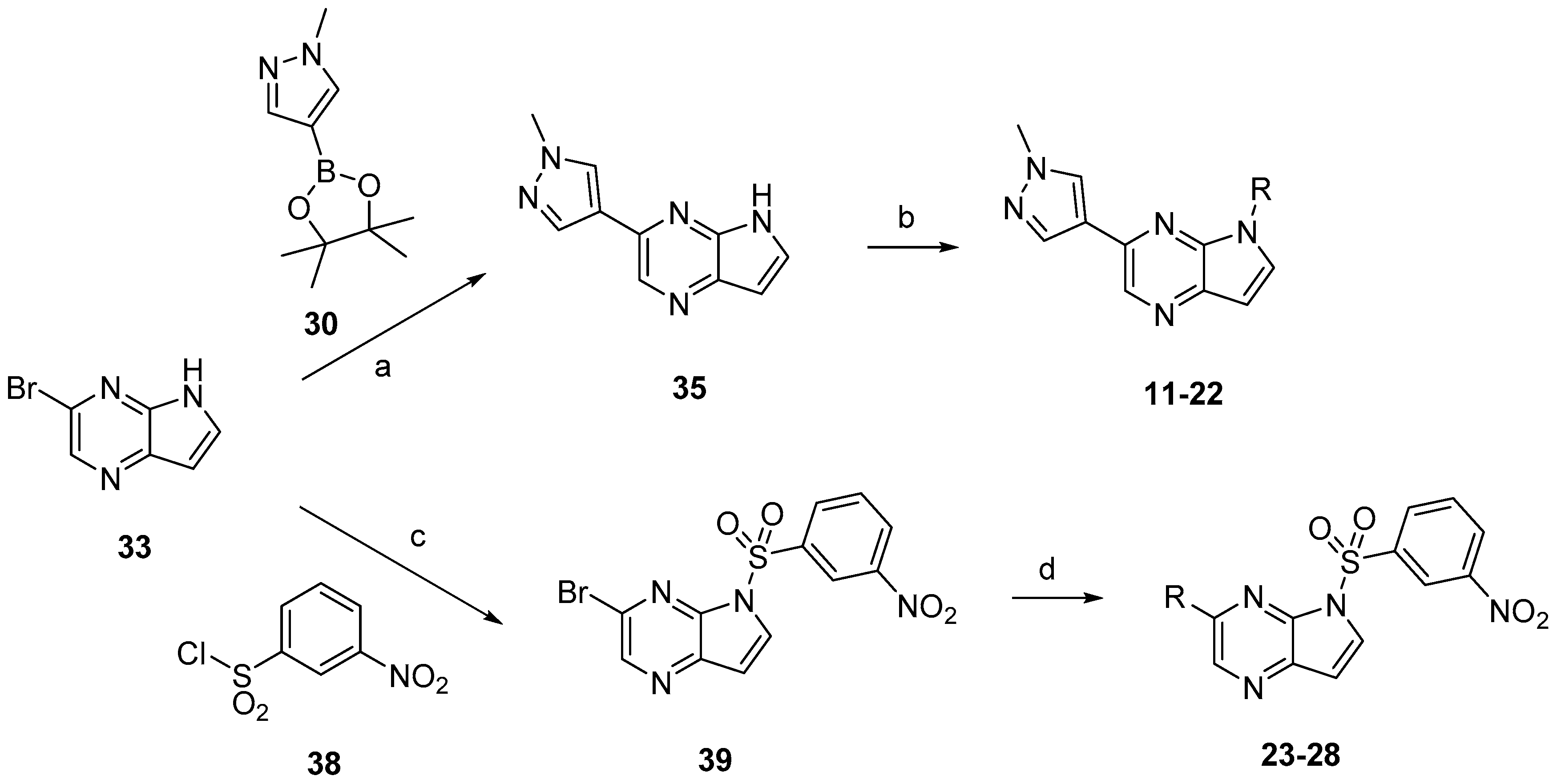
Compounds
11–
28 were prepared according to the procedures outlined in
Scheme 4. Compound
33 was first treated via Suzuki coupling then sulfonylated with corresponding sulfonyl chlorides to afford compounds
11–
22. Correspondingly, compound
33 firstly was sulfonylated with 3-nitrobenzenesulfonyl chloride (
38) then treated via Suzuki coupling with corresponding boric acid or boric acid ester to afford compounds
23–
28.
1H-NMR (400 MHz) and 13C-NMR (125 MHz or 151 MHz) spectra were recorded by using a Varian Mercury-400, a Mercury-500 and a Mercury-600 High Performance Digital FT-NMR spectrometer (Varian, Palo Alto, CA, USA) with tetramethylsilane (TMS) as an internal standard. Abbreviations for peak patterns in NMR spectra: br = broad, s = singlet, d = doublet, and m = multiplet. Low-resolution mass spectra were obtained with a Finnigan LCQ Deca XP mass spectrometer using a CAPCELL PAK C18 (50mm × 2.0 mm, 5 ZM) (Finnigan, Palo Alto, CA, USA) or an Agilent ZORBAX Eclipse XDB C18 (50 mm × 2.1 m, 5 ZM) (Agilent, Santa Clara, CA, USA) in positive or negative electrospray mode. Purity of all compounds was determined by analytical Gilson high-performance liquid chromatography (HPLC) using an YMC ODS3 column (50 mm × 4.6 mm, 5 ZM). Conditions were as follows: CH3CN/H2O eluent at 2.5 mL·min−1 flow (containing 0.1% trifluoroacetic acid (TFA)) at 35 °C, 8 min, gradient 5% CH3CN to 95% CH3CN, monitored by UV absorption at 214 nm and 254 nm. TLC analysis was carried out with glass precoated silica gel GF254 plates (Jiangyou, Qingdao, Shandong, China). TLC spots were visualized under UV light. Flash column chromatography was performed with a Teledyne ISCO CombiFlash Rf system (Teledyne, Huntsville, AL, USA). All solvents and reagents were used directly as obtained commercially unless otherwise noted. Anhydrous dimethylformamide was purchased from Acros (Shanghai, China) and was used without further drying. All air and moisture sensitive reactions were carried out under an atmosphere of dry Argon with heat-dried glassware and standard syringe techniques.
6-(1-Methyl-1H-pyrazol-4-yl)-1H-pyrazolo[4,3-b]pyridine (31). A solution of 6-bromo-1H-pyrazolo[4,3-b]pyridine (29) (330mg, 1.52 mmol), 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (30) (378 mg, 1.82 mmol), PdCl2(dppf)-CH2Cl2 (61.9 mg, 80 µmol) and K2CO3 (628 mg, 4.54 mmol) in 1,4-dioxane:water (15 mL, 2:1, v) in a microwave tube was flushed with N2 for 5 min then sealed. The tube was heated at 80 °C for 2 h. Then the reaction mixture was evaporated to dryness. The residue was purified by flash chromatography to give 31 (260 mg, 86% yield); LC–MS m/z (ESI) found (M + H)+ 200.0 (M + H)+; 1H-NMR (400 MHz, DMSO-d6) δ13.29 (s, 1H), 8.80 (s, 1H), 8.36 (s, 1H), 8.24 (s, 1H), 8.07 (s, 2H), 3.90 (s, 3 H).
6-(1-Methyl-1H-pyrazol-4-yl)-1-(phenylsulfonyl)-1H-pyrazolo[4,3-b]pyridine (4). Sodium hydride (29 mg, 1.2 mmol) was suspended in 3 mL of anhydrous DMF. 6-(1-methyl-1H-pyrazol-4-yl)-1H-pyrazolo[4,3-b]pyridine (31) (160 mg, 0.8 mmol) was dissolved in 5 mL of anhydrous DMF was slowly added dropwise, and the mixture was stirred for 30 min benzenesulfonyl chloride (214 mg, 0.94 mmol) dissolved in 5 mL of anhydrous DMF was slowly added in drops, and the mixture was stirred for 4 h at room temperature. The reaction solution was poured to 0.1 N hydrochloric acid, which was then brought to basic using aqueous sodium bicarbonate solution, and extracted with ethyl acetate. The organic layer was collected, and distilled under reduced pressure. The remaining substance was purified by flash column chromatography to give 4 (224 mg, 82% yield); LC-MS m/z (ESI) found (M + H)+ 340.0 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 8.84 (d, J = 1.7 Hz, 1 H), 8.49 (s, 1 H), 8.38 (s, 1 H), 8.02 (d, J = 7.4 Hz, 2 H), 7.94 (s, 1H), 7.86 (s, 1H), 7.62 (t, J = 7.4 Hz, 1 H), 7.51 (t, J = 7.4 Hz, 2 H), 4.03 (s, 3 H). 13C-NMR (126 MHz, CDCl3) δ 146.75, 142.02, 141.60, 137.29, 137.20, 134.61, 134.41, 129.42 (C × 2), 128.85, 128.03, 127.72 (C × 2), 119.32, 116.03, 39.38. Retention time 2.95 min, >98% purity.
Compounds 5–7 were prepared with a similar procedure as that used for 4.
1-(Benzylsulfonyl)-6-(1-methyl-1H-pyrazol-4-yl)-1H-pyrazolo[4,3-b]pyridine (5). LC–MS m/z (ESI) found (M + H)+ 354.1 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 8.72 (d, J = 1.9 Hz, 1H), 8.50 (d, J = 0.8 Hz, 1H), 7.74 (d, J = 0.8 Hz, 1H), 7.69 (dd, J = 1.9, 0.8 Hz, 1H), 7.67 (s, 1H), 7.11–7.03 (m, 3H), 6.97 (dd, J = 7.9, 1.6 Hz, 2H), 4.70 (s, 2H), 4.00 (s, 3H). 13C-NMR (126 MHz, CDCl3) δ 146.47, 141.71, 140.45, 137.16, 136.00, 130.55 (C × 2), 129.49, 128.56 (C × 2), 128.37, 127.73, 126.14, 119.12, 115.27, 60.31, 39.32. Retention time 2.97 min, >98% purity.
1-Benzyl-6-(1-methyl-1H-pyrazol-4-yl)-1H-pyrazolo[4,3-b]pyridine (6). LC–MS m/z (ESI) found (M + H)+ 290.1 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 8.73 (d, J = 1.8 Hz, 1H), 8.28 (d, J = 0.9 Hz, 1H), 7.81 (s, 1H), 7.72 (s, 1H), 7.64–7.59 (m, 1H), 7.38–7.30 (m, 3H), 7.25–7.21 (m, 2H), 5.64 (s, 2H), 4.00 (s, 3H). 13C-NMR (126 MHz, CDCl3) δ 140.67, 140.55, 137.55, 137.52, 137.10, 135.76, 131.18, 128.82 (C × 2), 128.54, 127.92, 127.73 (C × 2), 121.94, 101.51, 47.90, 39.22. Retention time 3.05 min, 98.25% purity.
6-(1-Methyl-1H-pyrazol-4-yl)-1-phenethyl-1H-pyrazolo[4,3-b]pyridine (7). LC–MS m/z (ESI) found (M + H)+ 304.1 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 8.74 (d, J = 1.9 Hz, 1H), 8.06 (d, J = 1.0 Hz, 1H), 7.96 (s, 1H), 7.88 (s, 1H), 7.75 (s, 1H), 7.32–7.24 (m, 3H), 7.14–7.10 (m, 2H), 4.68 (t, J = 7.3 Hz, 2H), 4.01 (s, 3H), 3.35 (t, J = 7.3 Hz, 2H). 13C-NMR (126 MHz, CDCl3) δ 153.72, 147.61, 145.87, 142.01, 140.34, 139.20, 137.77, 137.60, 136.48, 129.91, 129.02, 120.95, 110.86, 107.81, 105.68, 64.3, 39.36, 34.2. Retention time 3.08 min, >98% purity.
6-(1-Methyl-1H-pyrazol-4-yl)-1H-pyrrolo[3,2-b]pyridine (34). A solution of 6-bromo-1H-pyrrolo[3,2-b]pyridine (32) (100 mg, 0.51 mmol), 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (30) (116 mg, 0.56 mmol), PdCl2(dppf)-CH2Cl2 (37 mg, 50 µmol) and K2CO3 (140 mg, 1.01 mmol) in 1,4-dioxane:water(6 mL, 2:1, v) in a microwave tube was flushed with N2 for 5 mins then sealed. The tube was heated at 80 °C for 3 h. Then the reaction mixture was evaporated to dryness. The residue was purified by flash chromatography to give 34 (90 mg, 89% yield); LC–MS m/z (ESI) found (M + H)+ 199.1 (M + H)+; 1H-NMR (400 MHz, DMSO-d6) δ 11.31 (brs, 1H), 8.59 (s, 1H), 8.21 (s, 1H), 7.93 (s, 1H), 7.87 (s, 1H), 7.60 (t, J = 2.8 Hz, 1H), 6.53 (t, J = 2.8 Hz, 1H), 3.88 (s, 3H).
6-(1-Methyl-1H-pyrazol-4-yl)-1-(phenylsulfonyl)-1H-pyrrolo[3,2-b]pyridine (8). Sodium hydride (7 mg, 0.28 mmol) was suspended in 3 mL of anhydrous DMF. 6-(1-methyl-1H-pyrazol-4-yl)-1H-pyrrolo[3,2-b]pyridine (34) (50 mg, 0.25 mmol) was dissolved in 3 mL of anhydrous DMF was slowly added dropwise, and the mixture was stirred for 30 min benzenesulfonyl chloride (38 μL, 0.28 mmol) dissolved in 3 mL of anhydrous DMF was slowly added in drops, and the mixture was stirred for 2 h at room temperature. The reaction solution was poured to 0.1 N hydrochloric acid, which was then brought to basic using aqueous sodium bicarbonate solution, and extracted with ethyl acetate. The organic layer was collected, and distilled under reduced pressure. The remaining substance was purified by flash column chromatography to give 8 (70 mg, 82% yield); LC–MS m/z (ESI) found (M + H)+ 339.1 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 8.70 (d, J = 1.7 Hz, 1H), 8.32 (d, J = 1.2 Hz, 1H), 7.91 (s, 1H), 7.89 (t, J = 1.7 Hz, 1H), 7.87–7.85 (m, 1H), 7.77 (d, J = 3.8 Hz, 2H), 7.63–7.56 (m, 1H), 7.49 (t, J = 7.7 Hz, 2H), 6.88 (dd, J = 3.8, 0.7 Hz, 1H), 4.02 (s, 3H). 13C-NMR (126 MHz, CDCl3) δ 147.08, 144.47, 138.00, 136.93, 134.28, 129.53 (C × 2), 129.25, 128.86, 127.36, 126.70 (C × 2), 124.81, 120.18, 117.08, 110.45, 39.25. Retention time 2.92 min, >98% purity.
Compounds 9 were prepared with a similar procedure as that used for 8.
3-(1-Methyl-1H-pyrazol-4-yl)-5-(phenylsulfonyl)-5H-pyrrolo[2,3-b]pyrazine (9). LC–MS m/z (ESI) found (M + H)+ 340.0 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 8.71 (s, 1H), 8.28–8.19 (m, 2H), 8.09 (s, 1H), 8.03 (s, 1H), 7.94 (d, J = 4.1 Hz, 1H), 7.62 (d, J = 7.4 Hz, 1H), 7.54 (t, J = 7.7 Hz, 2H), 6.80 (d, J = 4.1 Hz, 1H), 4.04 (s, 3H). 13C-NMR (126 MHz, CDCl3) δ 142.22, 140.54, 139.05, 138.04, 137.96, 137.77, 134.47, 129.14 (C × 2), 129.10, 128.96, 128.24 (C × 2), 120.98, 106.58, 39.37. Retention time 2.99 min, >99% purity.
N-(5-Bromo-2-methylpyridin-3-yl)benzenesulfonamide (37). To a stirred solution of the 5-bromo-2-methylpyridin-3-amine (36) (200 mg, 1.07 mmol) in anhydrous dichloromethane (15 mL) was added benzenesulfonyl chloride (152 μL, 1.12 mmol). After 1 h, The mixture was then partially concentrated in vacuo, diluted with EtOAc (40 mL) and saturated NaHCO3 solution (20 mL) and partitioned. The aqueous layer was extracted with EtOAc (2 × 20 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated to afford 37 (300 mg, 85% yield); LC–MS m/z (ESI) found (M + H)+ 328.1 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 8.37 (d, J = 2.1 Hz, 1H), 7.92 (d, J = 2.1 Hz, 1H), 7.82–7.76 (m, 2H), 7.67–7.61 (m, 1H), 7.56–7.49 (m, 2H), 2.17 (s, 3H).
N-(2-Methyl-5-(1-methyl-1H-pyrazol-4-yl)pyridin-3-yl)benzenesulfonamide (10). A solution of N-(5-bromo-2-methylpyridin-3-yl)benzenesulfonamide (37) (100 mg, 0.31 mmol), 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (30) (70 mg, 0.34 mmol), PdCl2(dppf)-CH2Cl2 (20 mg, 24 µmol) and K2CO3 (84 mg, 0.61 mmol) in 1,4-dioxane:water(15 mL, 2:1, v) in a microwave tube was flushed with N2 for 5 mins then sealed. The tube was heated at 80 °C for 3 h. Then the reaction mixture was evaporated to dryness. The residue was purified by flash chromatography to give 10 (80 mg, 80% yield); LC–MS m/z (ESI) found (M + H)+ 329.1 (M + H)+; 1H-NMR (400 MHz, DMSO-d6) δ 8.45 (d, J = 2.0 Hz, 1H), 7.78–7.75 (m, 2H), 7.73 (d, J = 1.4 Hz, 1H), 7.71 (s, 1H), 7.67 (s, 1H), 7.59 (t, J = 7.5 Hz, 1H), 7.47 (t, J = 7.5 Hz, 2H), 5.30 (s, 1H), 3.97 (s, 3H), 2.16 (s, 3H). 13C-NMR (126 MHz, CDCl3) δ 149.37, 143.60, 139.24, 136.71, 133.43, 131.00, 129.32 (C × 2), 128.46, 127.44, 127.31, 127.03 (C × 2), 118.85, 39.23, 20.03. Retention time 2.54 min, >98% purity.
Compounds 11–28 were prepared with a similar procedure as that used for 8.
3-(1-Methyl-1H-pyrazol-4-yl)-5H-pyrrolo[2,3-b]pyrazine (35). LC–MS m/z (ESI) found (M + H)+ 199.1 (M + H)+; 1H-NMR (400 MHz, DMSO-d6) δ 9.30 (s, 1H), 8.72 (s, 1H), 8.11 (s, 1H), 7.97 (s, 1H), 7.28 (s, 1H), 6.74 (dd, J = 3.7, 1.9 Hz, 1H), 4.02 (s, 3H).
5-(Cyclopentylsulfonyl)-3-(1-methyl-1H-pyrazol-4-yl)-5H-pyrrolo[2,3-b]pyrazine (11). LC–MS m/z (ESI) found (M + H)+ 311.2 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 8.80 (s, 1H), 8.09 (d, J = 0.8 Hz, 1H), 8.07 (d, J = 0.8 Hz, 1H), 7.83 (d, J = 4.1 Hz, 1H), 6.83 (d, J = 4.1 Hz, 1H), 4.03 (s, 3H), 3.16–3.01 (m, 1H), 2.20 (m, 2H), 2.01 (m, 2H), 1.96–1.62 (m, 4H). 13C-NMR (126 MHz, CDCl3) δ 142.25, 140.70, 139.08, 138.03, 137.65, 129.80, 129.30, 120.83, 105.69, 64.00, 40.57, 27.62 (C × 2), 25.86 (C × 2). Retention time 2.57 min, >99% purity.
5-(Cyclohexylsulfonyl)-3-(1-methyl-1H-pyrazol-4-yl)-5H-pyrrolo[2,3-b]pyrazine (12). LC–MS m/z (ESI) found (M + H)+ 345.1 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 8.81 (s, 1H), 8.07 (d, J = 0.7 Hz, 1H), 8.05 (d, J = 0.7 Hz, 1H), 7.84 (d, J = 4.0 Hz, 1H), 6.85 (d, J = 4.0 Hz, 1H), 4.01 (s, 3H), 3.82 (m, 1H), 2.01–1.85 (m, 4H), 1.69 (dd, J = 10.9, 4.8 Hz, 2H), 1.32–1.17 (m, 4H). Retention time 2.59 min, 98.54% purity.
2-((3-(1-Methyl-1H-pyrazol-4-yl)-5H-pyrrolo[2,3-b]pyrazin-5-yl)sulfonyl)benzonitrile (13). LC–MS m/z (ESI) found (M + H)+ 364.0 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 8.71 (s, 1H), 8.66–8.62 (m, 1H), 8.16 (d, J = 4.2 Hz, 1H), 8.01–7.96 (m, 2H), 7.89–7.83 (m, 2H), 7.77 (dd, J = 7.6, 1.3 Hz, 1H), 6.89 (d, J = 4.2 Hz, 1H), 4.02 (s, 3H). 13C-NMR (126 MHz, CDCl3) δ 142.29, 140.40, 139.90, 139.20, 138.37, 137.78, 135.52, 134.39, 132.82, 131.76, 130.15, 129.00, 120.65, 115.00, 111.37, 106.99, 39.36. Retention time 3.04 min, >99% purity.
5-((4-Chlorophenyl)sulfonyl)-3-(1-methyl-1H-pyrazol-4-yl)-5H-pyrrolo[2,3-b]pyrazine (14). HPLC 96.66%; LC–MS m/z (ESI) found (M + H)+ 373.0 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 8.73 (s, 1H), 8.19 (d, J = 8.5 Hz, 2H), 8.09 (s, 1H), 8.03 (s, 1H), 7.91 (d, J = 4.1 Hz, 1H), 7.51 (d, J = 8.5 Hz, 2H), 6.82 (d, J = 4.1 Hz, 1H), 4.05 (s, 3H). 13C-NMR (126 MHz, CDCl3) δ 142.31, 141.36, 140.46, 139.06, 138.17, 137.77, 136.36, 129.69 (C × 2), 129.53 (C × 2), 128.92, 128.89, 120.87, 106.91, 39.39. Retention time 2.98 min, 96.66% purity.
5-((2,4-Difluorophenyl)sulfonyl)-3-(1-methyl-1H-pyrazol-4-yl)-5H-pyrrolo[2,3-b]pyrazine (15). LC–MS m/z (ESI) found (M + H)+ 375.1 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 8.71 (s, 1H), 8.44 (td, J = 8.5, 6.0 Hz, 1H), 8.01 (dd, J = 4.1, 1.3 Hz, 1H), 7.99 (d, J = 0.7 Hz, 1H), 7.91 (s, 1H), 7.16–7.10 (m, 1H), 6.90 (ddd, J = 10.4, 8.5, 2.4 Hz, 1H), 6.85 (d, J = 4.1 Hz, 1H), 4.02 (s, 3H). 13C-NMR (126 MHz, CDCl3) δ 168.08, 166.01, 161.34, 159.25, 142.19, 140.38, 139.07, 138.16, 137.76, 133.92, 129.52, 128.76, 120.74, 112.10, 106.53, 39.34. Retention time 2.97 min, 94.53% purity.
5-((3,4-Dichlorophenyl)sulfonyl)-3-(1-methyl-1H-pyrazol-4-yl)-5H-pyrrolo[2,3-b]pyrazine (16). LC–MS m/z (ESI) found (M + H)+ 407.0 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 8.75 (s, 1H), 8.50 (d, J = 2.2 Hz, 1H), 8.10 (d, J = 0.7 Hz, 1H), 8.06 (s, 1H), 8.03 (dd, J = 8.5, 2.2 Hz, 1H), 7.90 (d, J = 4.1 Hz, 1H), 7.62 (d, J = 8.5 Hz, 1H), 6.84 (d, J = 4.1 Hz, 1H), 4.06 (s, 3H). 13C-NMR (126 MHz, CDCl3) δ 142.44, 140.33, 139.75, 139.07, 138.37, 137.69, 137.34, 133.79, 131.27, 130.76, 129.06, 128.62, 127.09, 120.71, 107.13, 39.38. Retention time 3.02 min, >99% purity.
3-(1-Methyl-1H-pyrazol-4-yl)-5-((3-nitrophenyl)sulfonyl)-5H-pyrrolo[2,3-b]pyrazine (17). LC–MS m/z (ESI) found (M + H)+ 384.2 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 9.40 (t, J = 2.1 Hz, 1H), 8.77 (s, 1H), 8.49 (dd, J = 10.8, 8.0 Hz, 2H), 8.24 (s, 1H), 8.06 (s, 1H), 7.92 (d, J = 4.2 Hz, 1H), 7.78 (t, J = 8.0 Hz, 1H), 6.86 (dd, J = 4.2, 0.7 Hz, 1H), 4.08 (s, 3H). 13C-NMR (126 MHz, CDCl3) δ 148.09, 142.68, 140.32, 139.87, 139.00, 138.45, 137.37, 133.39, 130.67, 129.68, 128.93, 128.39, 124.61, 120.43, 107.41, 39.37. Retention time 3.01 min, >99% purity.
3-(1-Methyl-1H-pyrazol-4-yl)-5-((4-nitrophenyl)sulfonyl)-5H-pyrrolo[2,3-b]pyrazine (18). LC–MS m/z (ESI) found (M + H)+ 384.0 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 8.75 (s, 1H), 8.48–8.44 (m, 2H), 8.37 (d, J = 8.8 Hz, 2H), 8.10 (s, 1H), 8.03 (s, 1H), 7.91 (d, J = 4.1 Hz, 1H), 6.87 (d, J = 4.1 Hz, 1H), 4.06 (s, 3H). Retention time 3.02 min, 98.86% purity.
3-((3-(1-Methyl-1H-pyrazol-4-yl)-5H-pyrrolo[2,3-b]pyrazin-5-yl)sulfonyl)aniline (19). LC–MS m/z (ESI) found (M + H)+ 354.1 (M + H)+; 1H-NMR (600 MHz, CDCl3) δ 9.05 (d, J = 1.9 Hz, 1H), 8.67 (s, 1H), 8.17–8.14 (m, 2H), 8.06 (dd, J = 8.4, 0.8 Hz, 1H), 8.04 (d, J = 0.8 Hz, 1H), 7.98 (d, J = 4.1 Hz, 1H), 7.42 (d, J = 8.4 Hz, 1H), 6.80 (d, J = 4.1 Hz, 1H), 4.05 (s, 3H). Retention time 2.79 min, 94.56% purity.
4-((3-(1-Methyl-1H-pyrazol-4-yl)-5H-pyrrolo[2,3-b]pyrazin-5-yl)sulfonyl)aniline (20). LC–MS m/z (ESI) found (M + H)+ 355.07 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 8.69 (s, 1H), 8.09 (d, J = 0.8 Hz, 1H), 8.03 (s, 1H), 8.01 (d, J = 2.1 Hz, 1H), 7.99 (d, J = 2.1 Hz, 1H), 7.93 (d, J = 4.1 Hz, 1H), 6.76 (d, J = 4.1 Hz, 1H), 6.66 (d, J = 2.0 Hz, 1H), 6.64 (d, J = 2.0 Hz, 1H), 4.04 (s, 3H). Retention time 2.75 min, 98.68% purity.
N-(3-((3-(1-Methyl-1H-pyrazol-4-yl)-5H-pyrrolo[2,3-b]pyrazin-5-yl)sulfonyl)phenyl)acetamide (21). HPLC 94.55%; LC–MS m/z (ESI) found (M + H)+ 396.1 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 8.72 (s, 1H), 8.61 (s, 1H), 8.20 (s, 1H), 8.06 (s, 1H), 7.91 (t, J = 6.0, 6.3 Hz, 2H), 7.66 (d, J = 8.2 Hz, 1H), 7.47 (d, J = 8.2 Hz, 1H), 7.44 (d, J = 12.3 Hz, 1H), 6.80 (d, J = 4.1 Hz, 1H), 4.01 (s, 3H), 2.04 (s, 3H). Retention time 3.12 min, 94.55% purity.
N-(3-((3-(1-Methyl-1H-pyrazol-4-yl)-5H-pyrrolo[2,3-b]pyrazin-5-yl)sulfonyl)phenyl)propionamide (22). LC–MS m/z (ESI) found (M + H)+ 410.1 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 8.71 (s, 1H), 8.61 (s, 1H), 8.22 (s, 1H), 8.07 (s, 1H), 7.92 (t, J = 6.1 Hz, 2H), 7.67 (d, J = 8.1 Hz, 1H), 7.47 (d, J = 8.1 Hz, 1H), 7.42 (d, J = 12.5 Hz, 1H), 6.81 (d, J = 4.1 Hz, 1H), 4.04 (s, 3H), 2.41 (q, J = 7.6 Hz, 2H), 1.24 (t, J = 7.6 Hz, 3H). Retention time 3.13 min, 97.72% purity.
1-(4-(5-((3-Nitrophenyl)sulfonyl)-5H-pyrrolo[2,3-b]pyrazin-3-yl)-1H-pyrazol-1-yl)ethan-1-one (23). LC–MS m/z (ESI) found (M + H)+ 412.0 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 9.28 (s, 1H), 8.91 (s, 1H), 8.86 (s, 1H), 8.58 (d, J = 8.2 Hz, 1H), 8.51 (d, J = 8.2 Hz, 1H), 8.40 (s, 1H), 8.02 (d, J = 4.2 Hz, 1H), 7.81 (t, J = 8.2 Hz, 1H), 6.91 (d, J = 4.2 Hz, 1H), 2.83 (s, 3H). 13C-NMR (126 MHz, CDCl3) δ 169.38, 148.24, 142.03, 140.64, 140.34, 140.19, 139.73, 139.00, 133.57, 130.68, 129.81, 129.04, 126.08, 124.04, 123.66, 107.39, 21.67. Retention time 2.64 min, 90.95% purity.
1-(4-(5-((3-Nitrophenyl)sulfonyl)-5H-pyrrolo[2,3-b]pyrazin-3-yl)-1H-pyrazol-1-yl)propan-1-one (24). LC–MS m/z (ESI) found (M + H)+ 426.3 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 9.27 (t, J = 2.0, 2.0 Hz, 1H), 8.91 (d, J = 0.8 Hz, 1H), 8.86 (s, 1H), 8.61–8.56 (m, 1H), 8.53–8.48 (m, 2H), 8.38 (d, J = 0.8 Hz, 1H), 8.32 (s, 1H), 8.02 (d, J = 4.2 Hz, 1H), 6.91 (d, J = 4.2 Hz, 1H), 3.27 (q, J = 7.4 Hz, 2H), 1.38 (t, J = 7.4 Hz, 3H). 13C-NMR (126 MHz, CDCl3) δ 172.88, 148.25, 141.86, 140.77, 140.28, 139.75, 139.00, 138.69, 133.60, 130.66, 129.74, 129.04, 126.18, 124.36, 124.02, 123.35, 107.39, 29.70. Retention time 2.65 min, 95.32% purity.
1-(4-(5-((3-Nitrophenyl)sulfonyl)-5H-pyrrolo[2,3-b]pyrazin-3-yl)-1H-pyrazol-1-yl)propan-2-one (25). LC–MS m/z (ESI) found (M + H)+ 426.2 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 9.40 (t, J = 2.0 Hz, 1H), 8.79 (s, 1H), 8.48 (m, 2H), 8.30 (d, J = 0.8 Hz, 1H), 8.14 (d, J = 0.8 Hz, 1H), 7.95 (d, J = 4.1 Hz, 1H), 7.79 (t, J = 8.0 Hz, 1H), 6.87 (d, J = 4.1 Hz, 1H), 5.11 (s, 2H), 2.29 (s, 3H). 13C-NMR (151 MHz, CDCl3) δ 201.10, 147.99, 142.15, 140.19, 139.74, 139.23, 138.40, 138.03, 133.42, 130.72, 130.44, 128.98, 128.59, 124.75, 121.22, 107.31, 61.19, 27.02. Retention time 2.66 min, 94.67% purity.
1-(4-(5-((3-Nitrophenyl)sulfonyl)-5H-pyrrolo[2,3-b]pyrazin-3-yl)phenyl)ethan-1-one (26). LC–MS m/z (ESI) found (M + H)+ 422.3 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 9.32 (t, J = 2.0 Hz, 1H), 9.12 (s, 1H), 8.60–8.55 (m, 1H), 8.53–8.48 (m, 1H), 8.25 (d, J = 8.4 Hz, 2H), 8.18 (d, J = 8.4 Hz, 2H), 8.09 (d, J = 4.2 Hz, 1H), 7.79 (t, J = 8.1 Hz, 1H), 6.96 (d, J = 4.2 Hz, 1H), 3.73 (d, J = 0.8 Hz, 3H). Retention time 2.65 min, 91.84% purity.
(4-(5-((3-Nitrophenyl)sulfonyl)-5H-pyrrolo[2,3-b]pyrazin-3-yl)phenyl)methanol (27). LC–MS m/z (ESI) found (M + H)+ 410.0 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 9.30 (t, J = 2.0 Hz, 1H), 9.02 (s, 1H), 8.57 (dt, J = 7.9, 1.5 Hz, 1H), 8.48 (ddd, J = 8.2, 2.0, 1.1 Hz, 1H), 8.13 (d, J = 1.5 Hz, 1H), 8.11 (s, 1H), 8.02 (d, J = 4.2 Hz, 1H), 7.77 (t, J = 8.1 Hz, 1H), 7.57 (d, J = 8.1 Hz, 2H), 6.91 (d, J = 4.2 Hz, 1H), 4.83 (s, 2H). 13C-NMR (126 MHz, CDCl3) δ 148.17, 147.07, 142.88, 140.39, 140.11, 139.84, 139.22, 135.31, 133.66, 130.68, 129.64, 128.97, 127.64 (C × 2), 127.20 (C × 2), 124.29, 107.25, 64.85. Retention time 2.71 min, 94.62% purity.
3-(7-Methoxy-1H-indol-2-yl)-5-((3-nitrophenyl)sulfonyl)-5H-pyrrolo[2,3-b]pyrazine (28). LC–MS m/z (ESI) found (M + H)+ 449.1 (M + H)+; 1H-NMR (400 MHz, CDCl3) δ 9.43 (s, 1H), 9.19 (s, 1H), 9.07 (s, 1H), 8.58 (s, 1H), 8.50 (s, 1H), 7.99 (s, 1H), 7.81 (s, 1H), 7.23–7.03 (m, 2H), 6.97–6.87 (m, 1H), 6.78 (d, J = 8.4 Hz, 1H), 4.12 (s, 3H). Retention time 2.94 min, 96.21% purity.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





























