
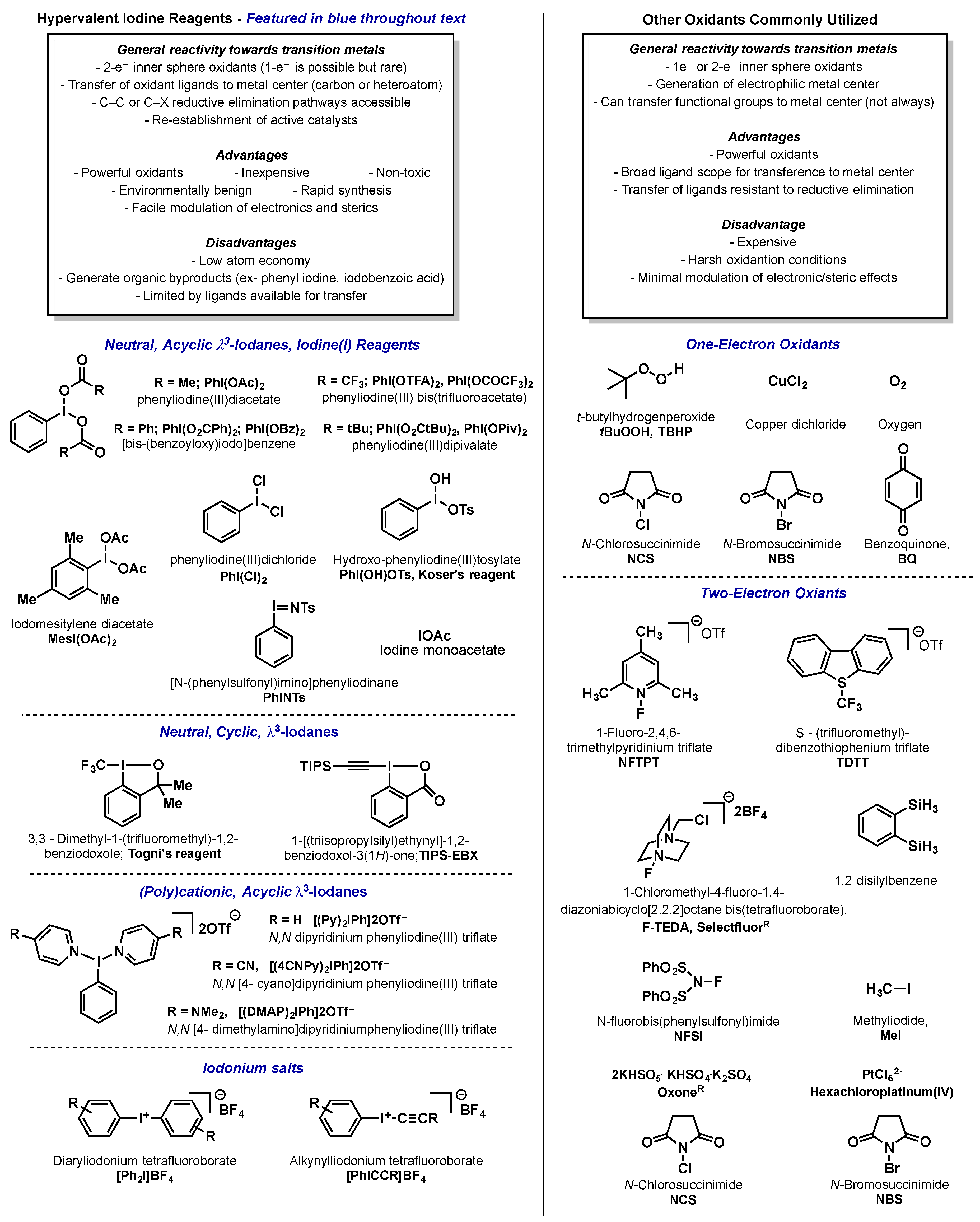
Hypervalent Iodine Reagents in High Valent Transition Metal Chemistry

{kind=link}
{kind=link}
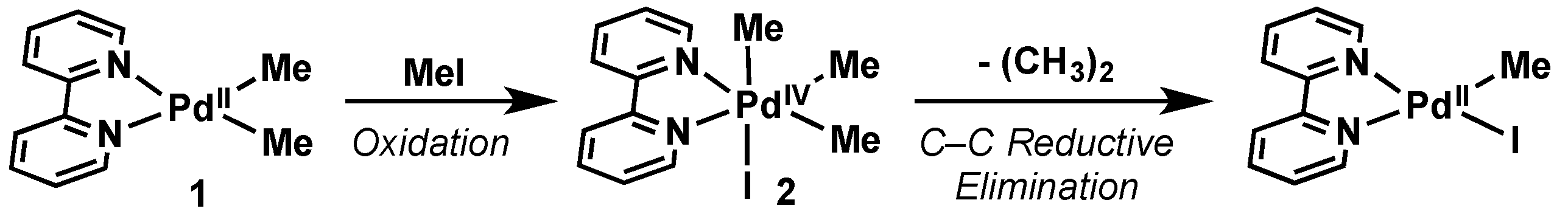{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
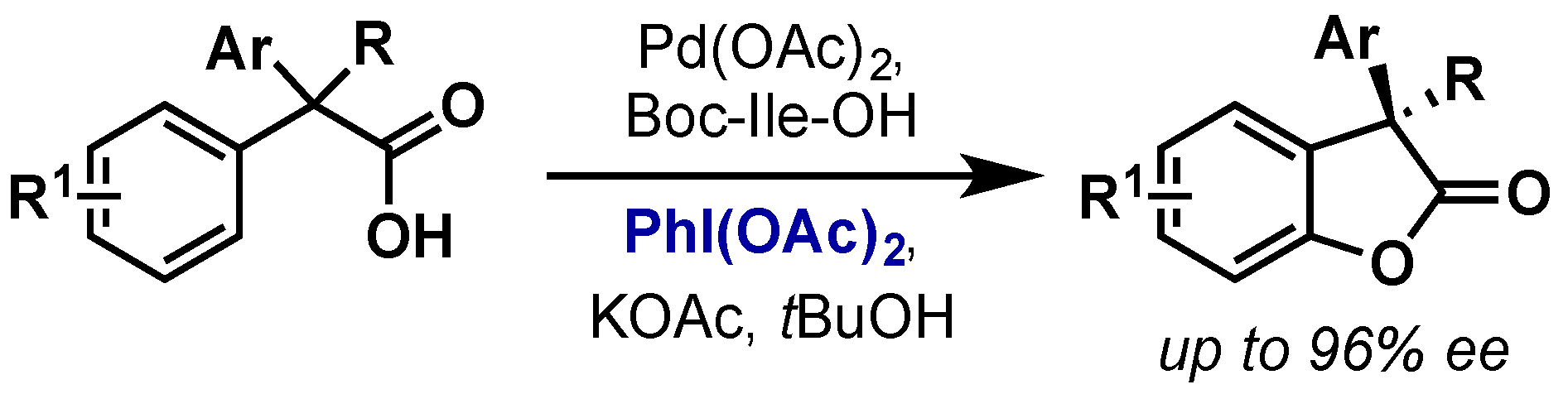{kind=link}
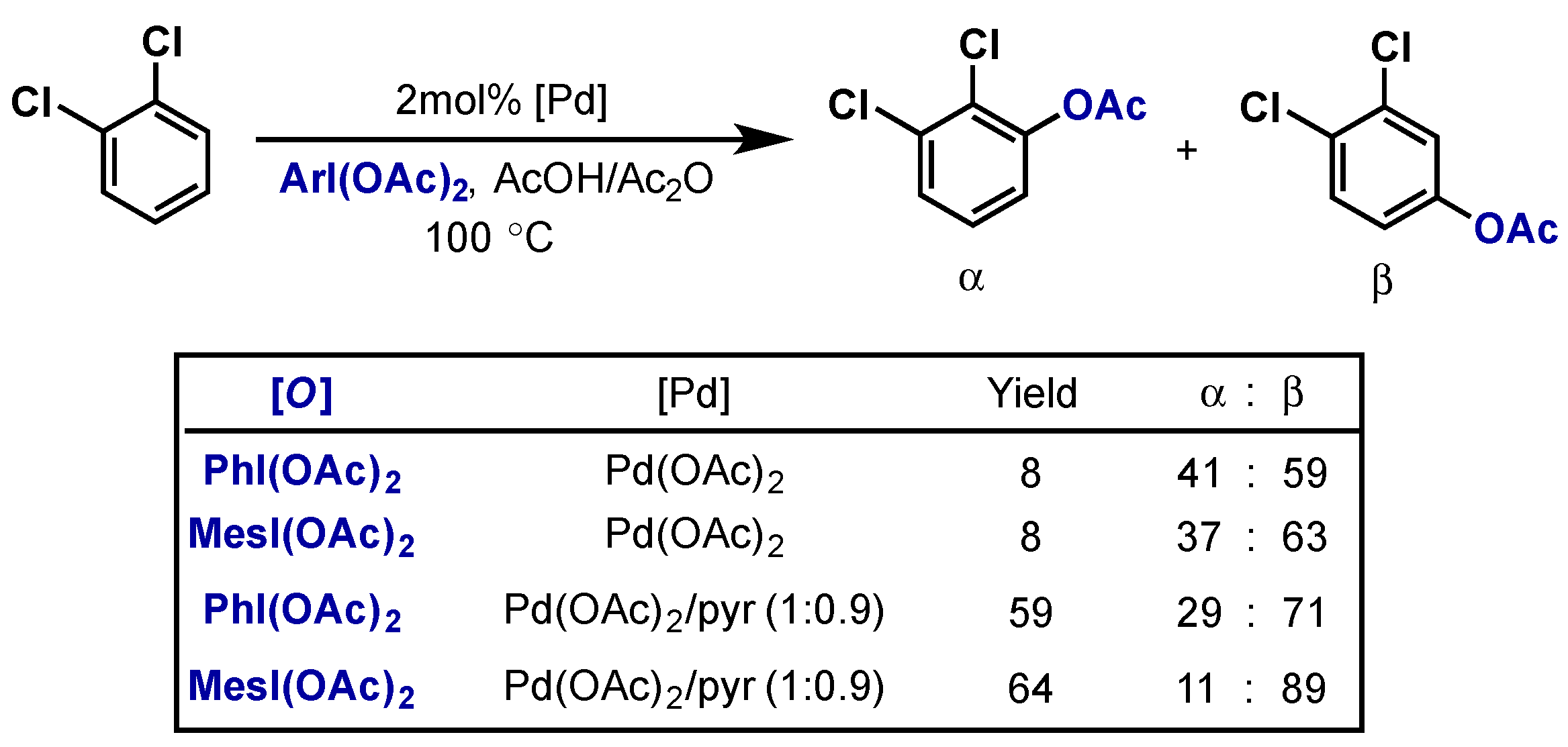{kind=link}
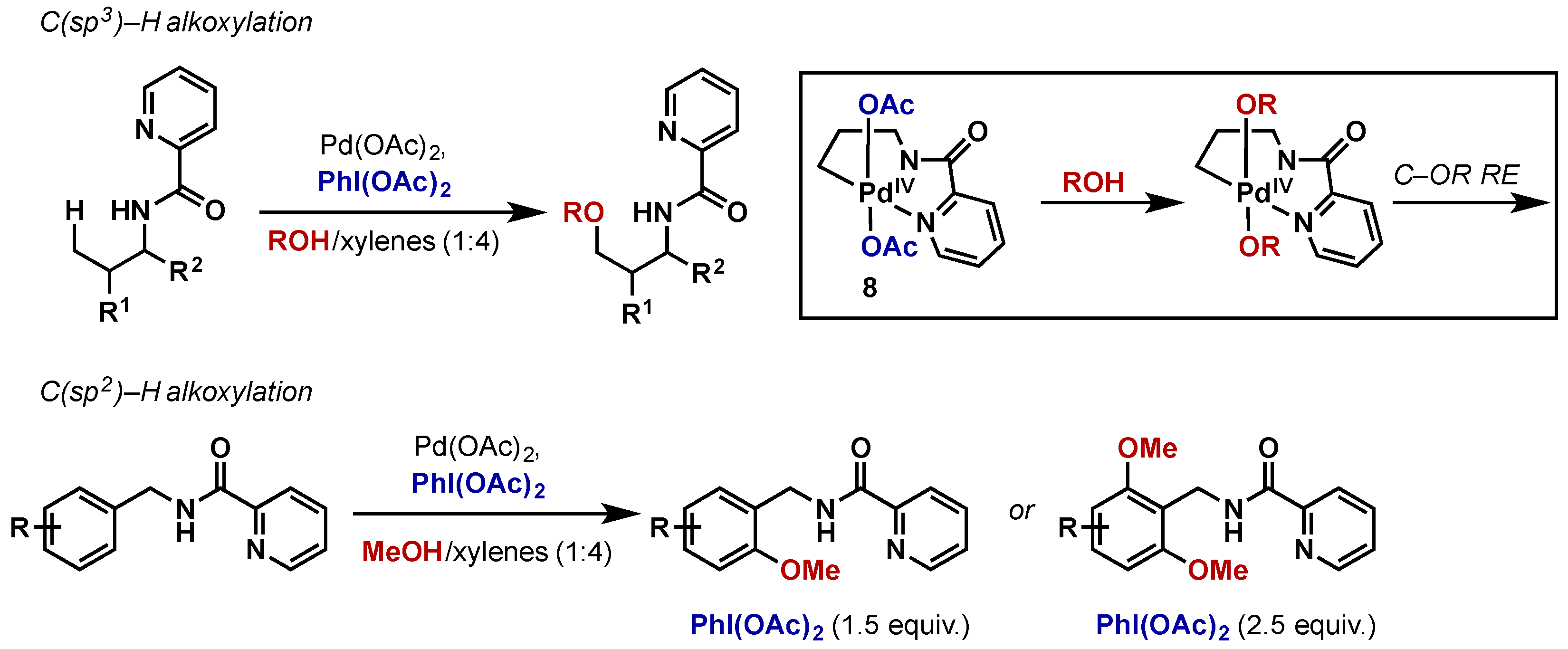{kind=link}
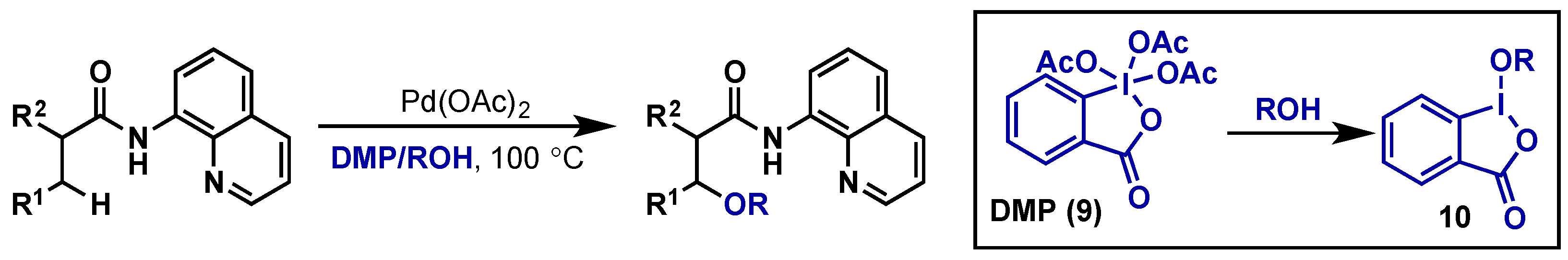{kind=link}
{kind=link}
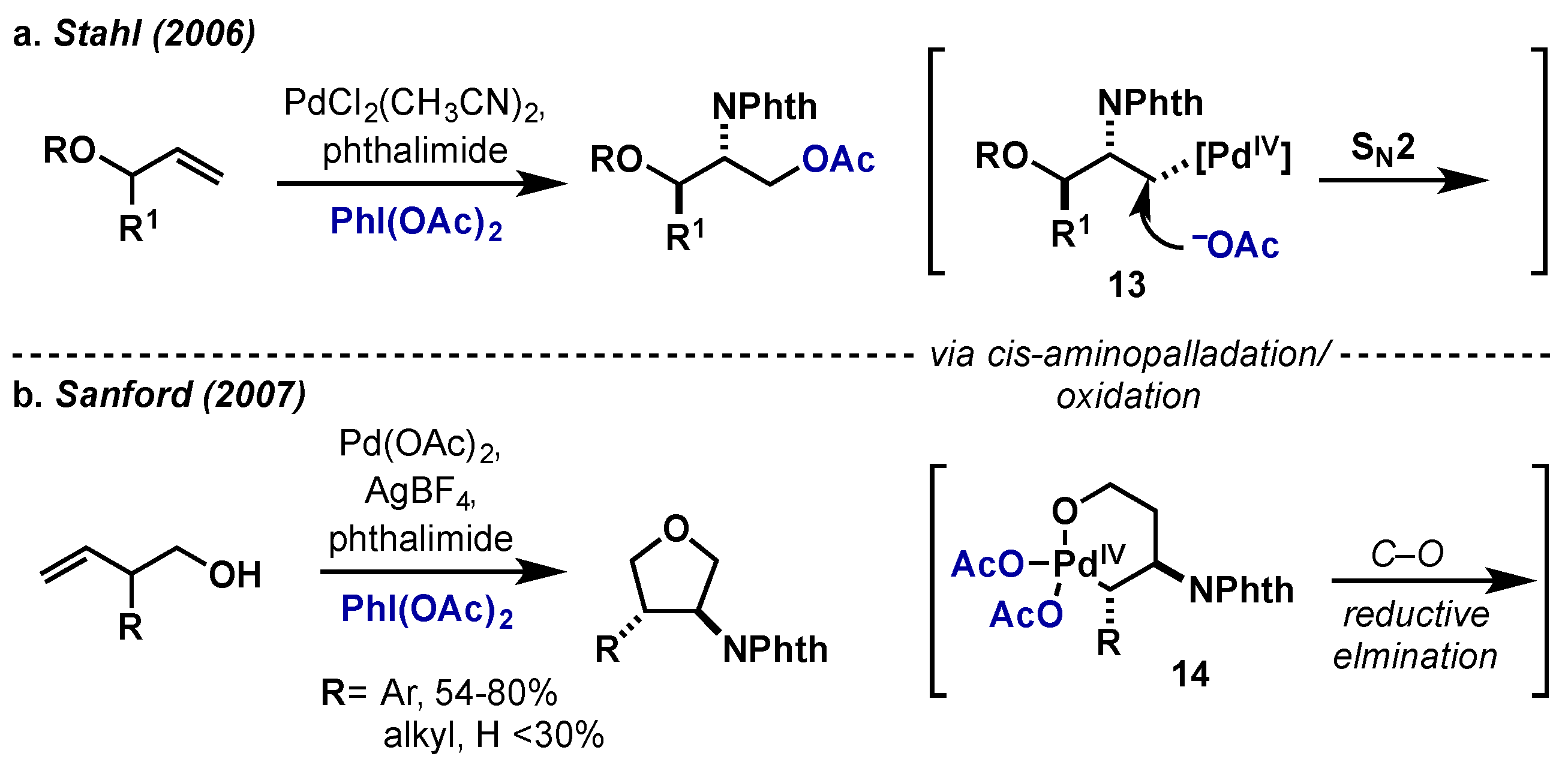{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
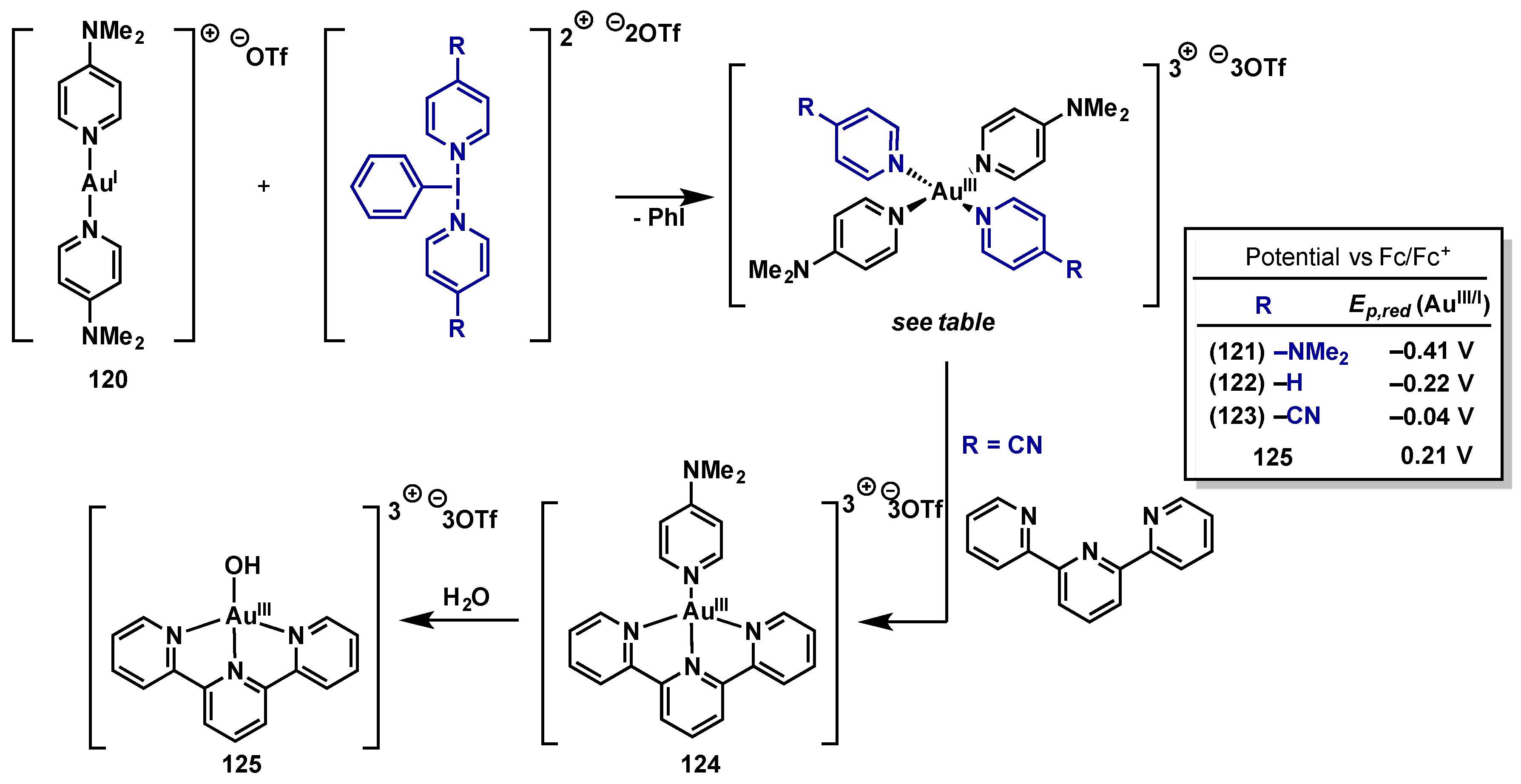{kind=link}
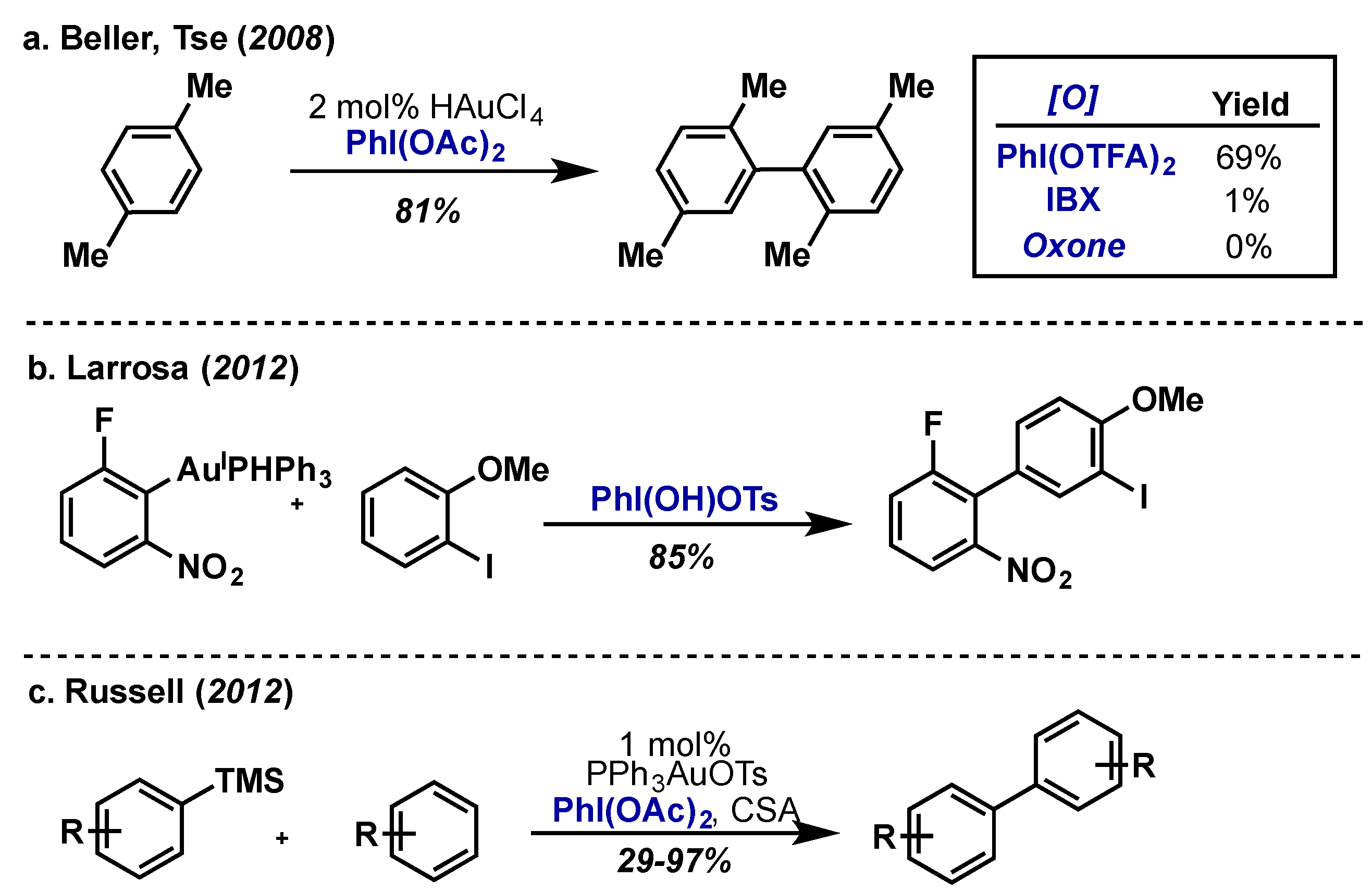{kind=link}
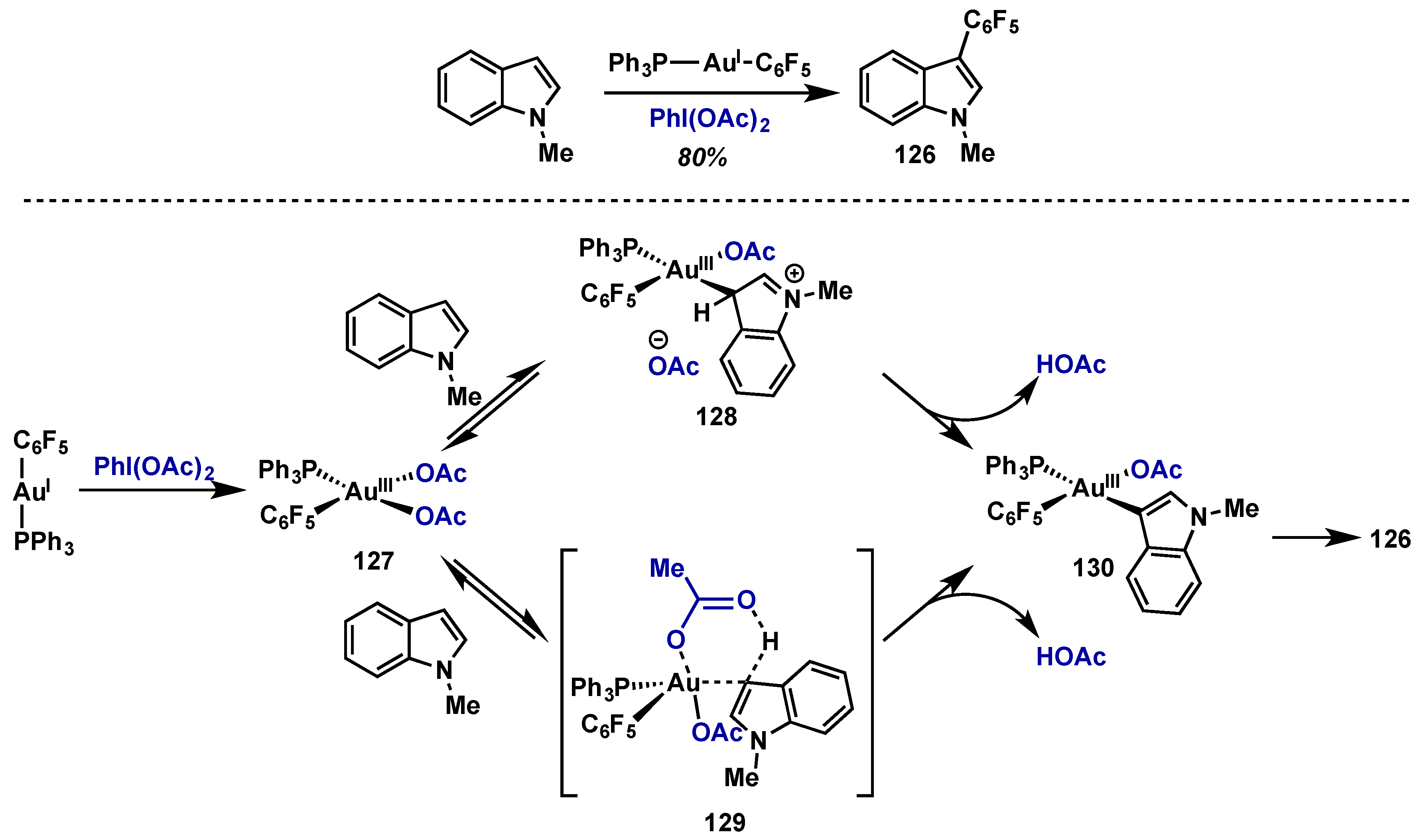{kind=link}
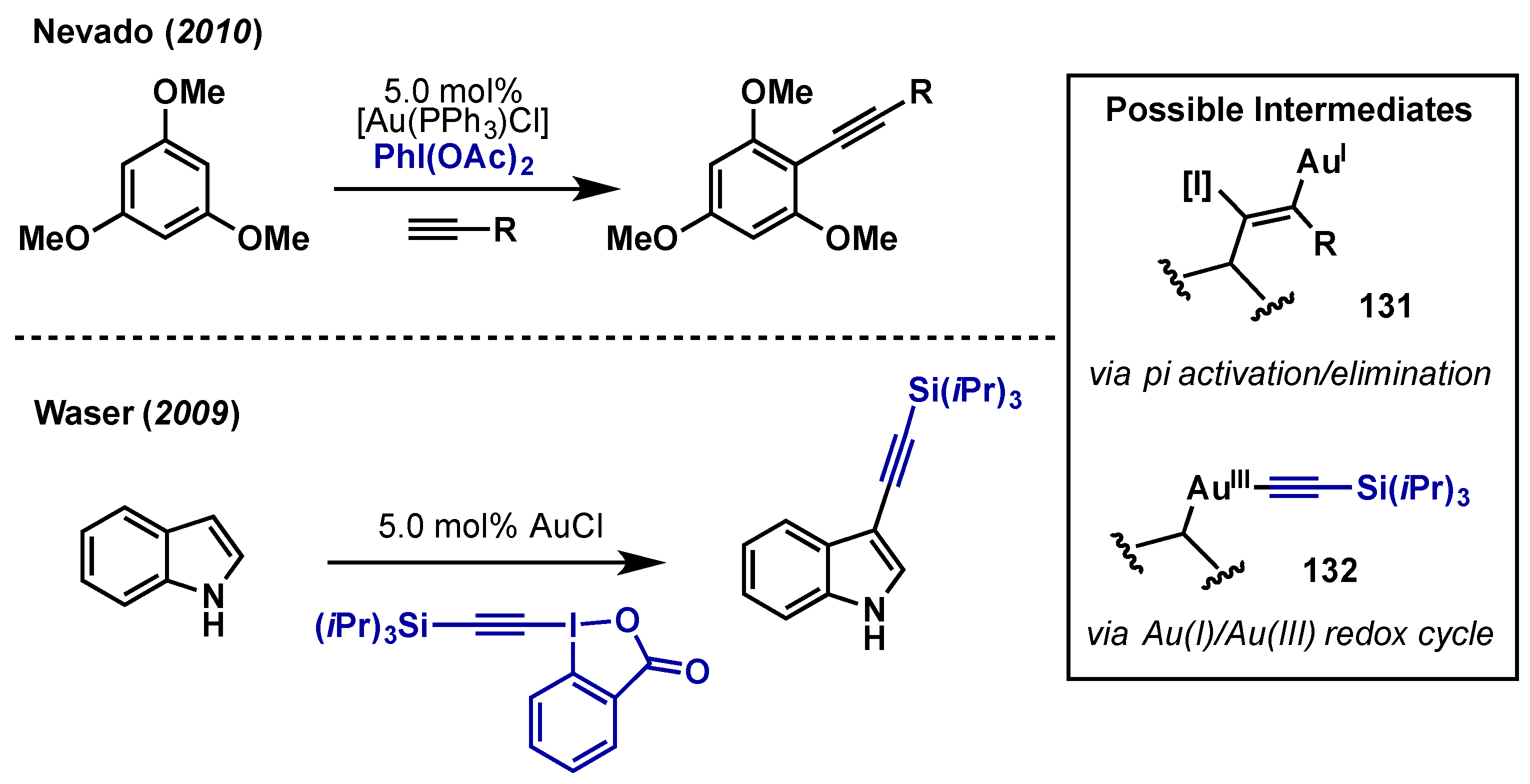{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Palladium
2.1. Introduction
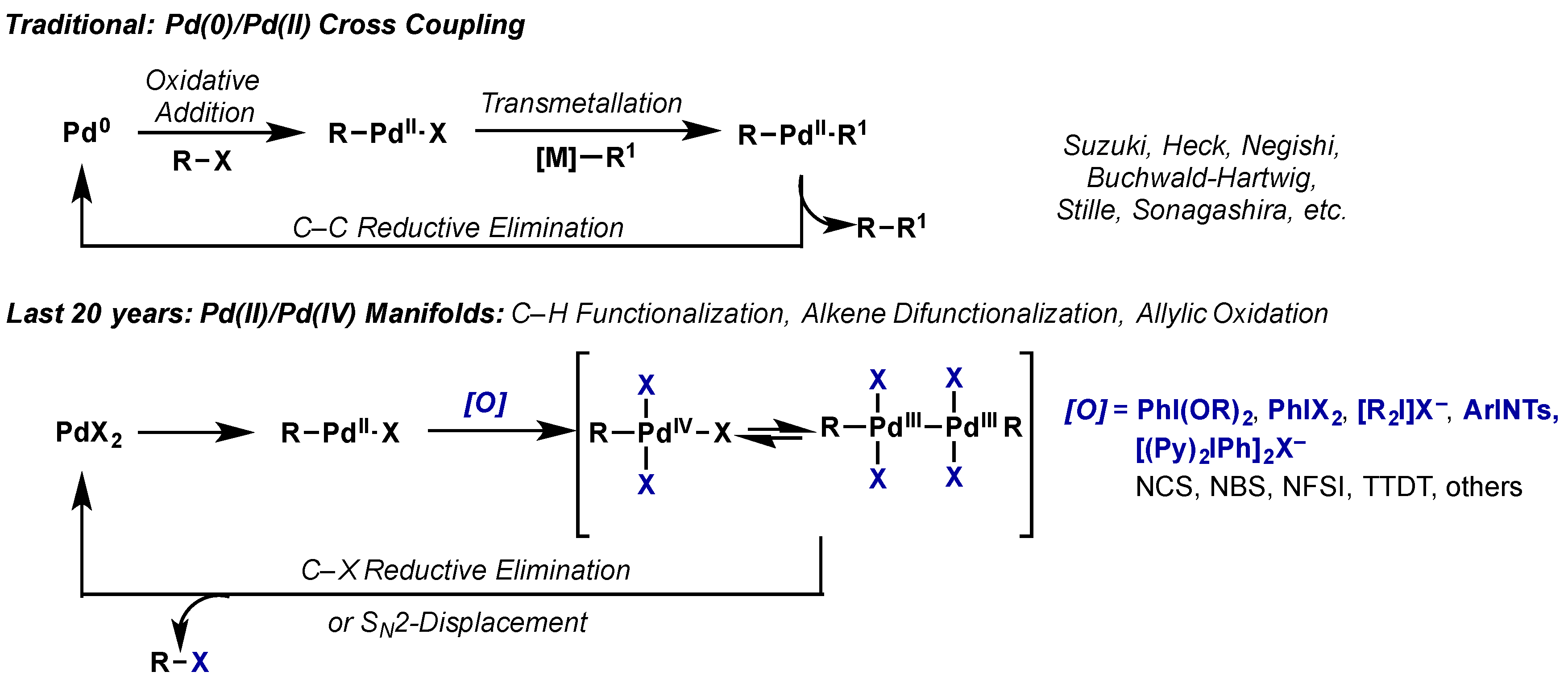
2.2. Palladium(IV)
2.2.1. Introduction
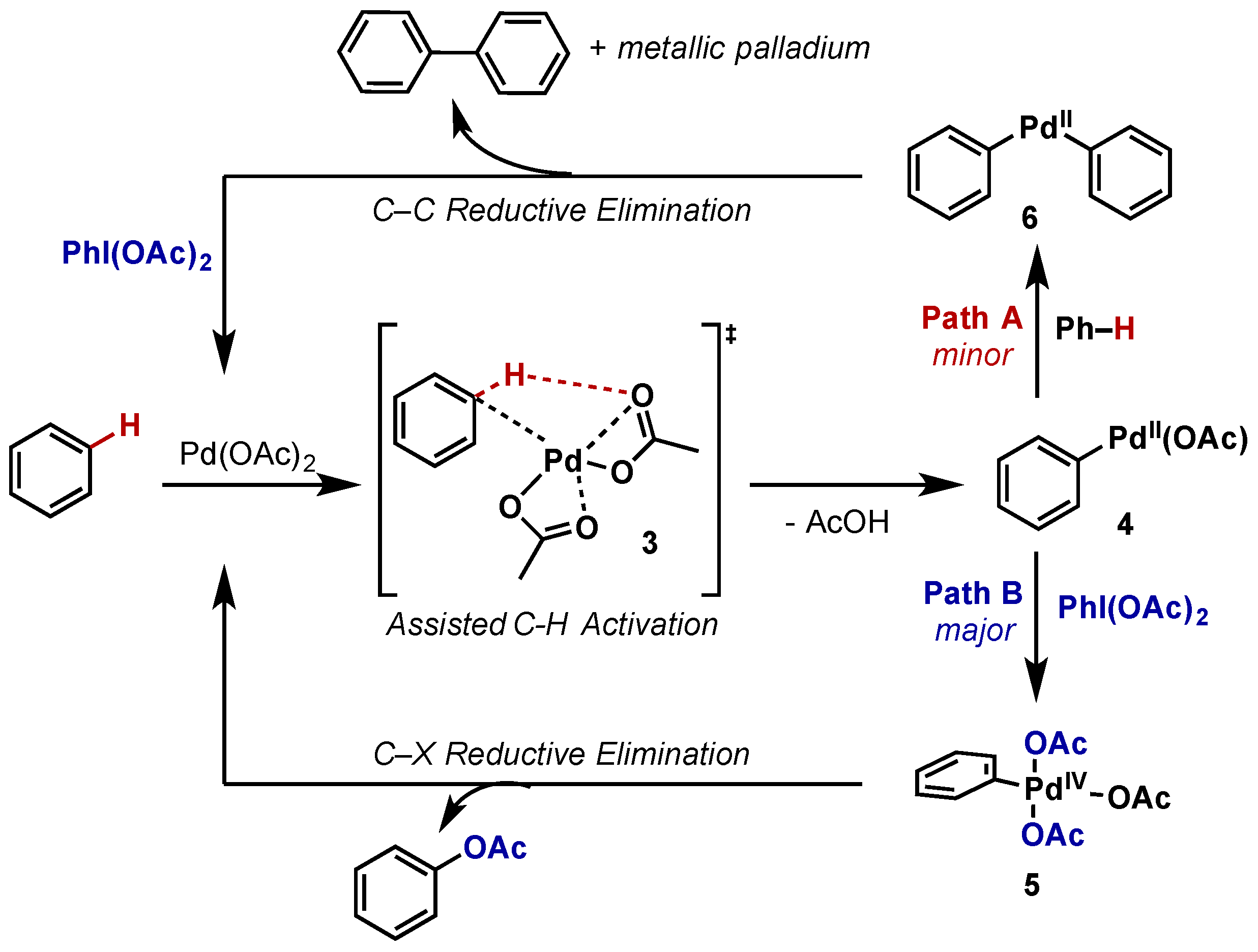
2.2.2. Carbon–Oxygen Bond Formation
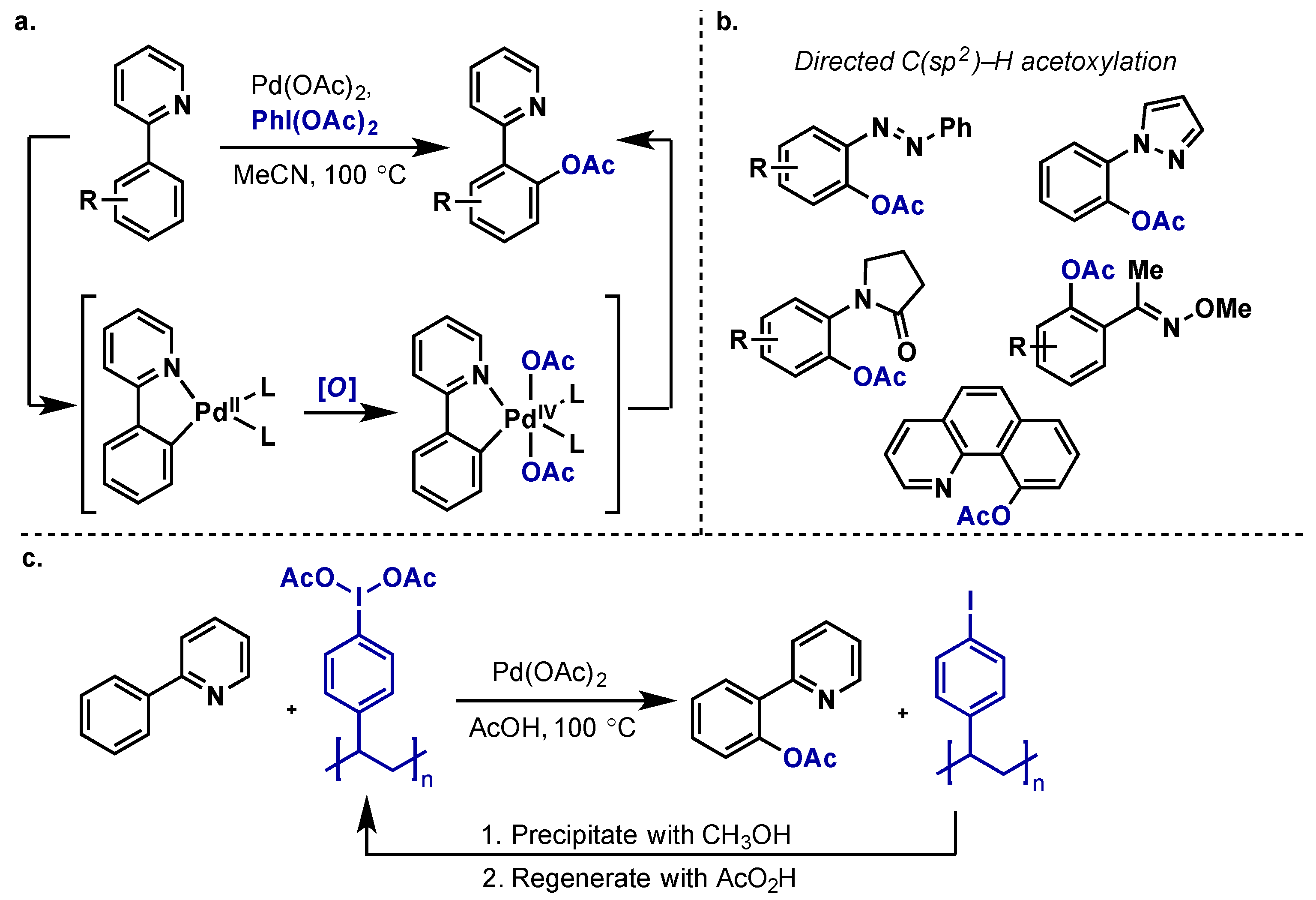
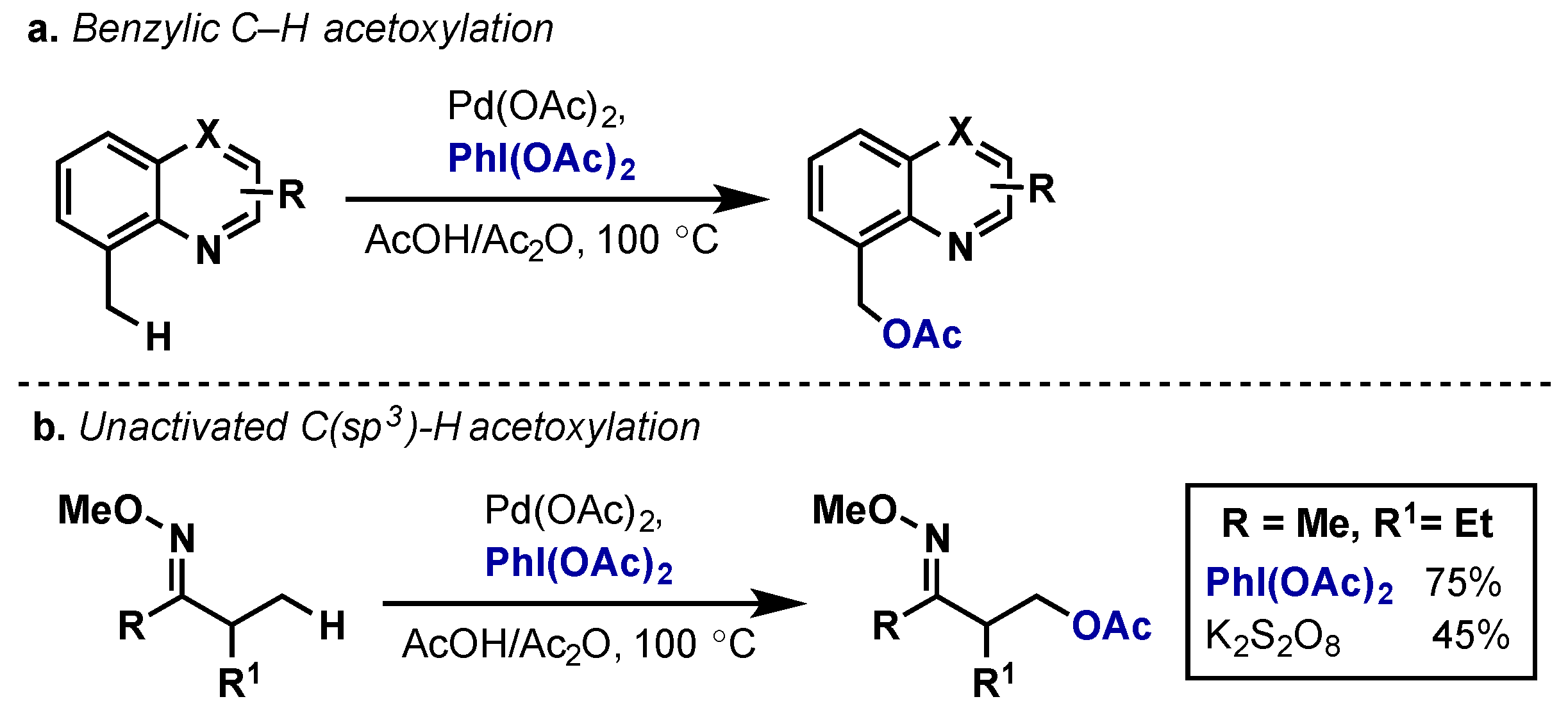
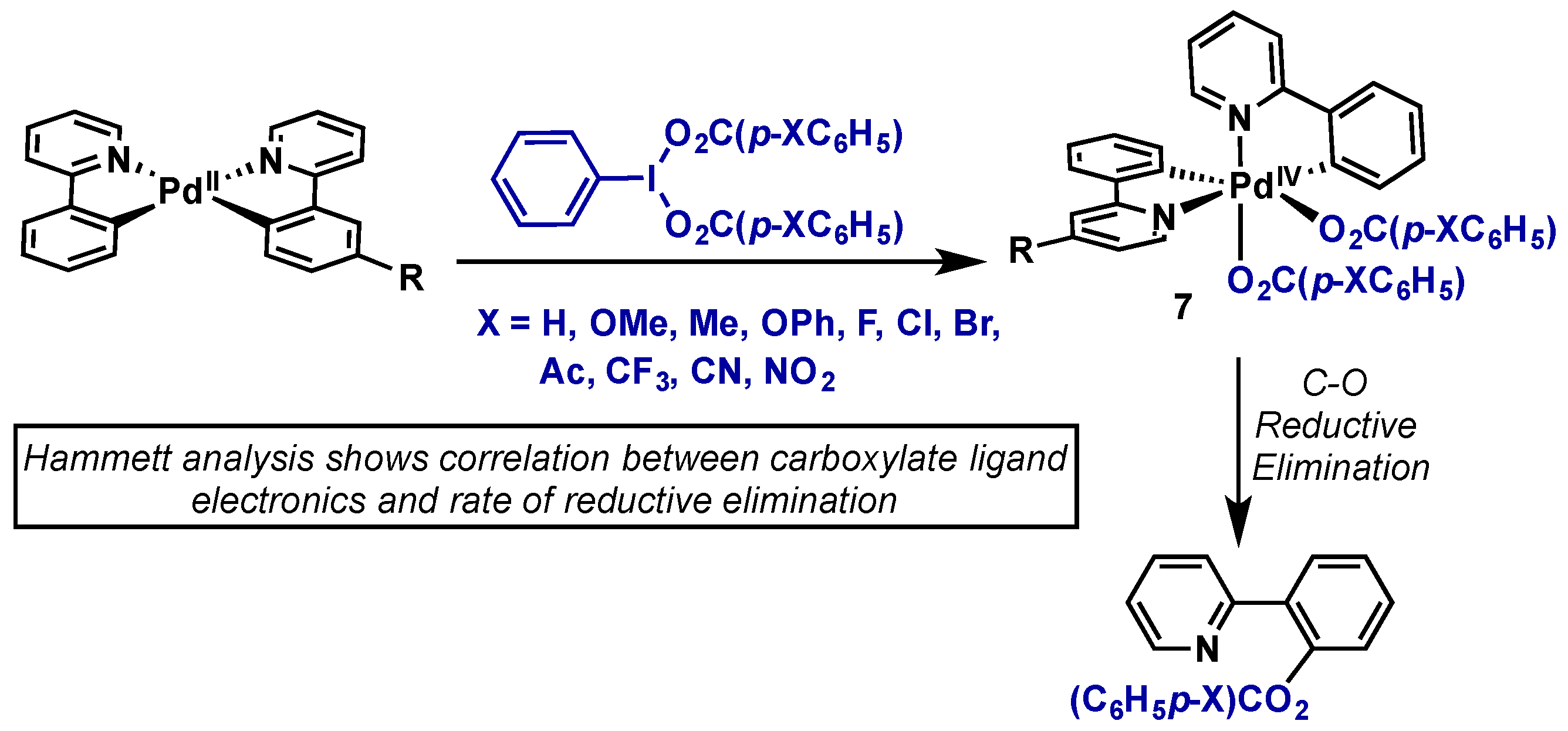
2.2.2.1. C–H Functionalization
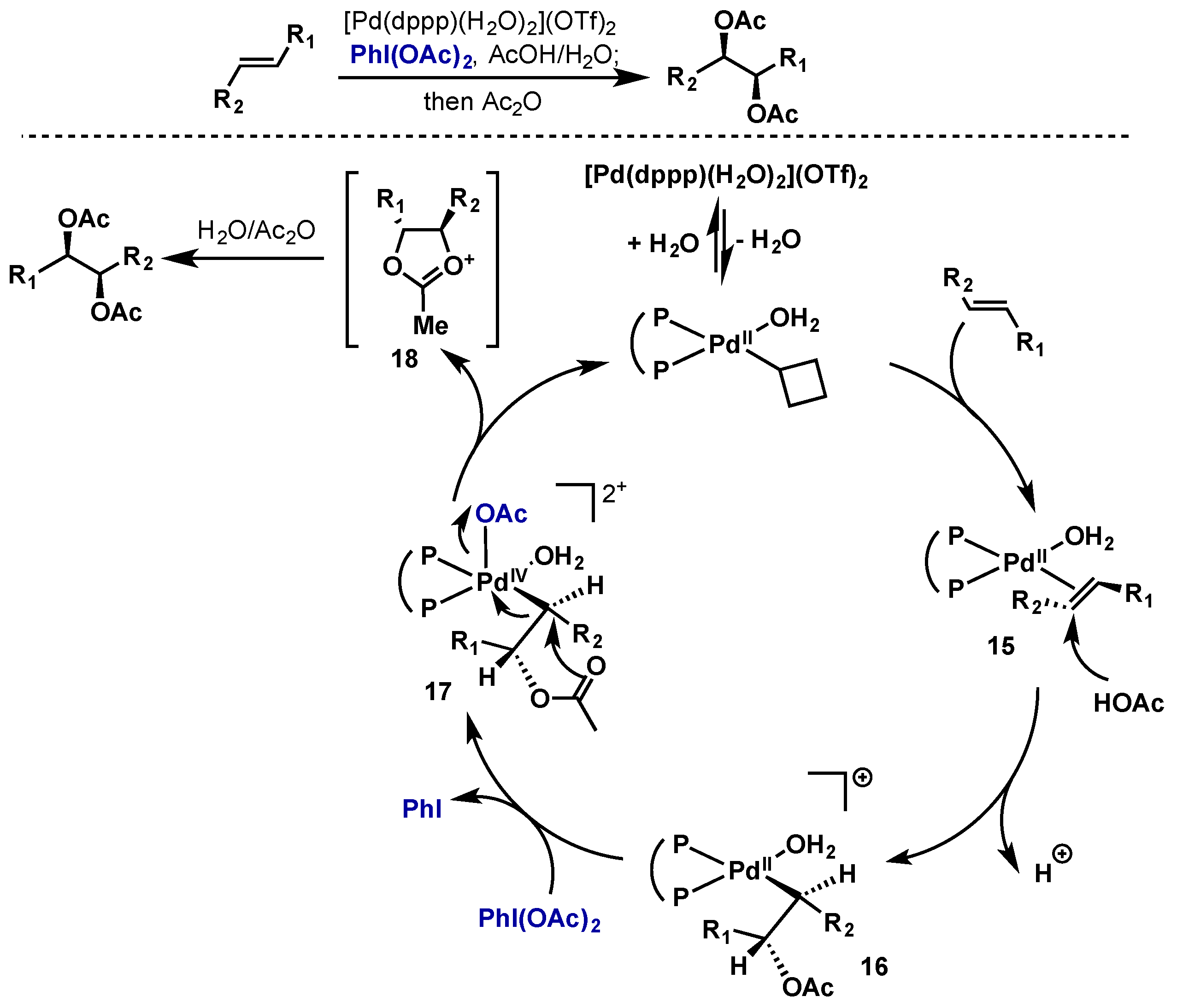
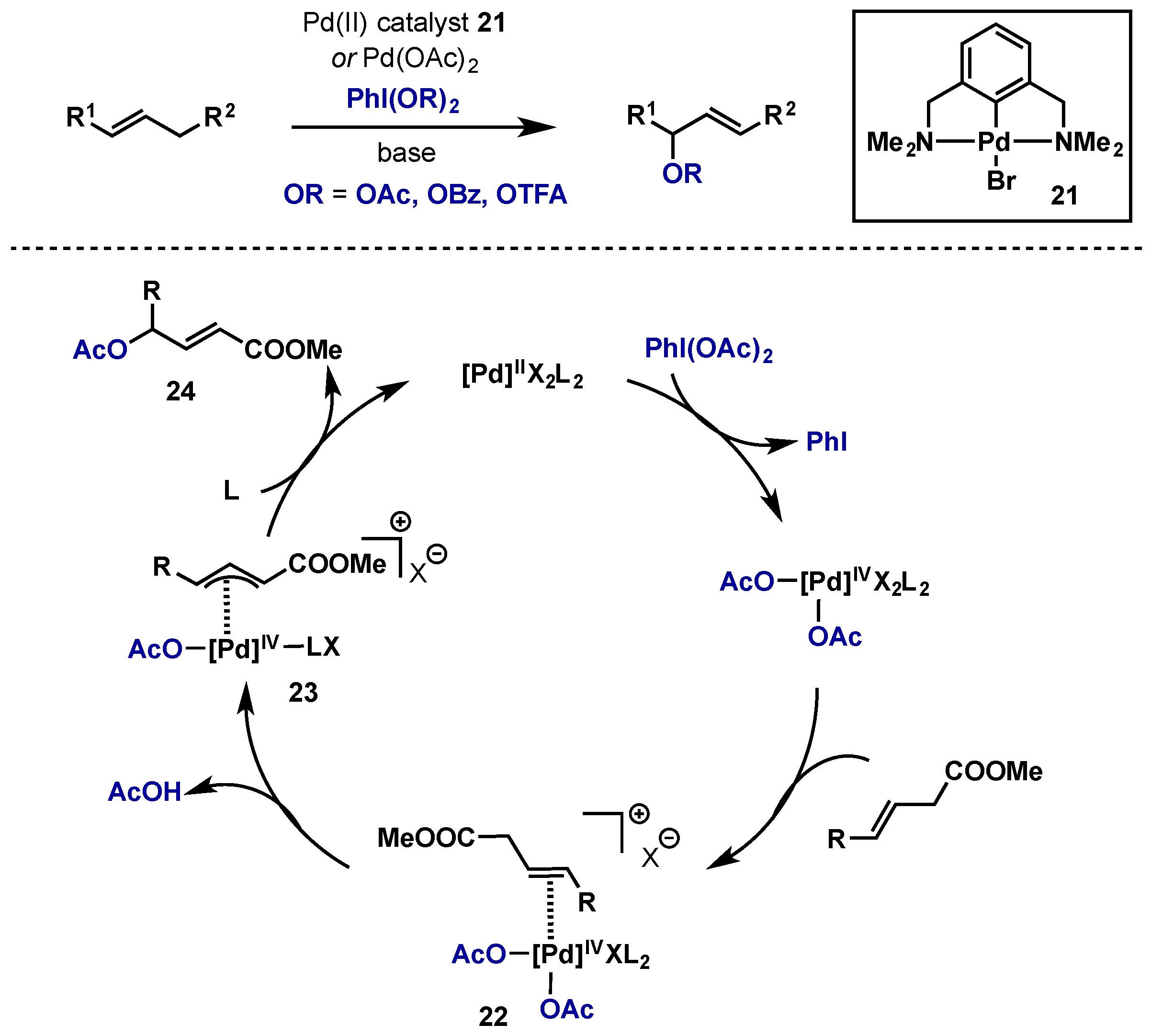
2.2.2.2. Alkene Difunctionalization, Allylic Oxidation
2.2.3. Carbon-Halogen, Carbon-Boron Bond Formation
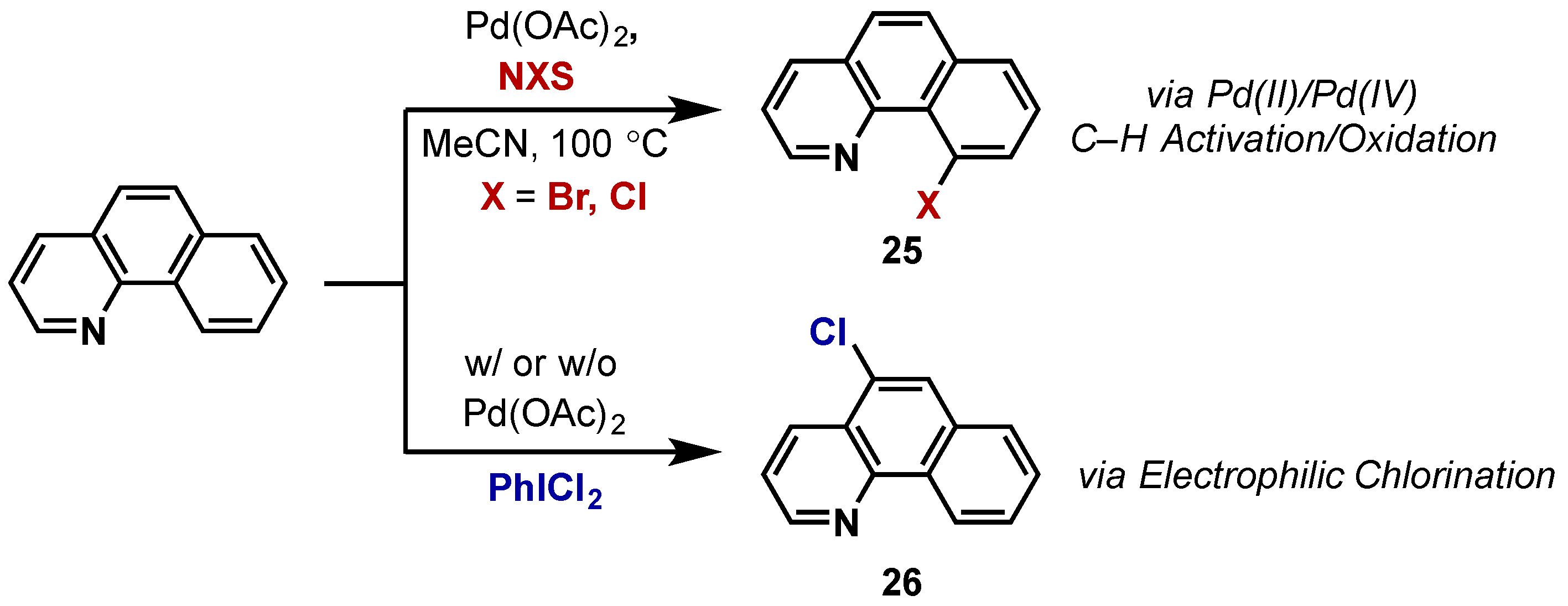
2.2.3.1. Carbon-Halogen Bond Formation
2.2.3.2. Carbon-Boron Bond Formation
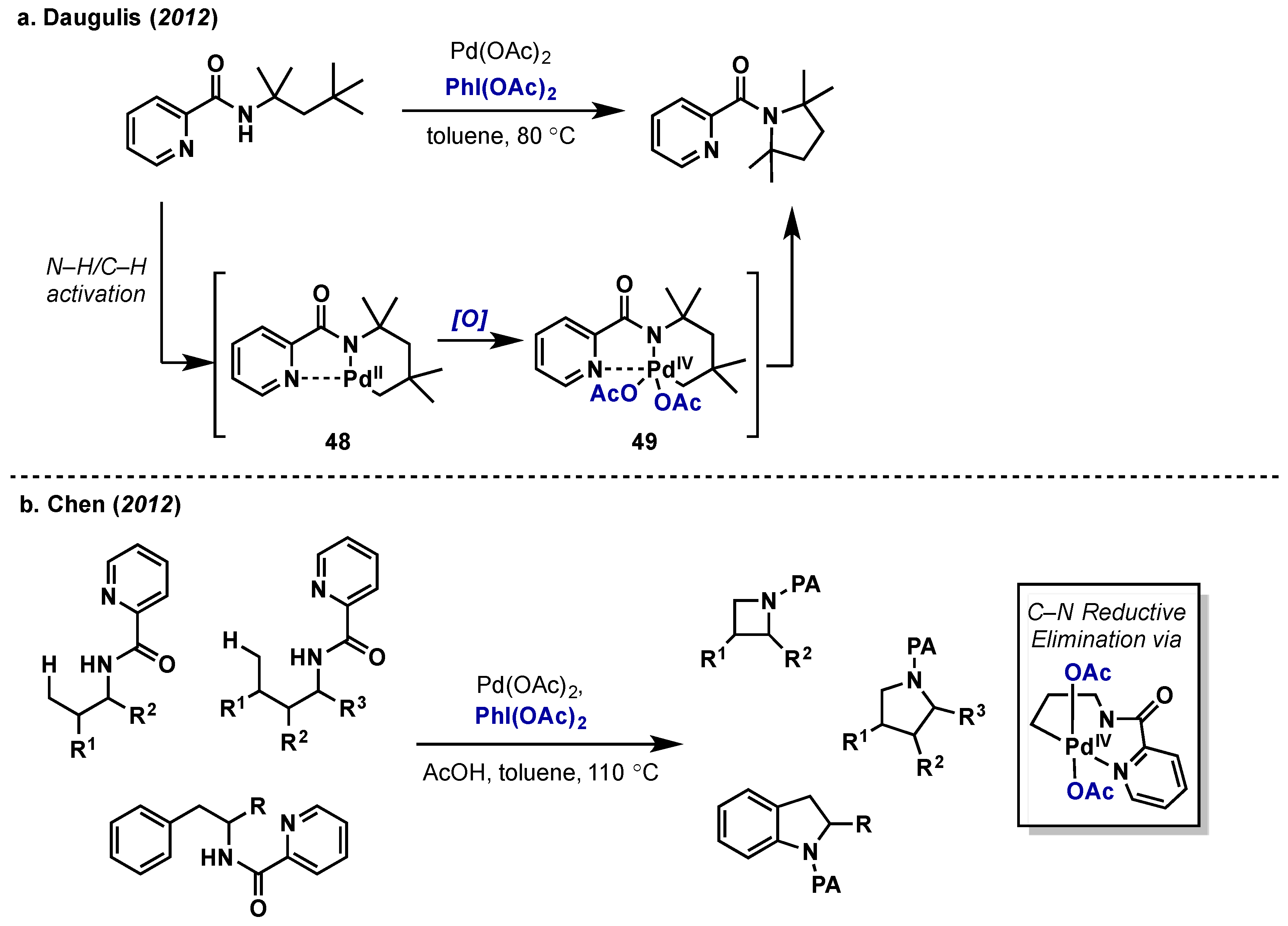
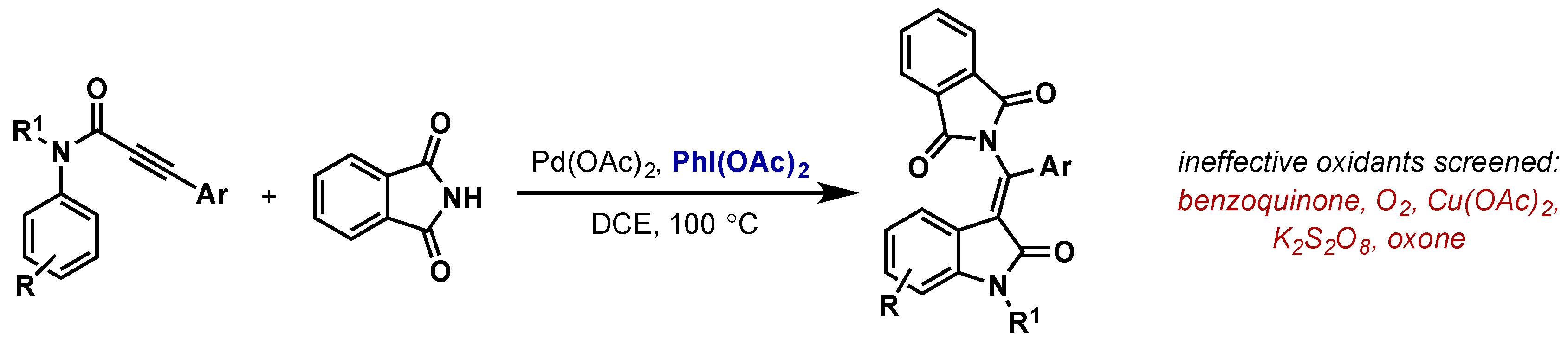
2.2.4. Carbon-Nitrogen Bond Formation
2.2.4.1. Alkene Diamination
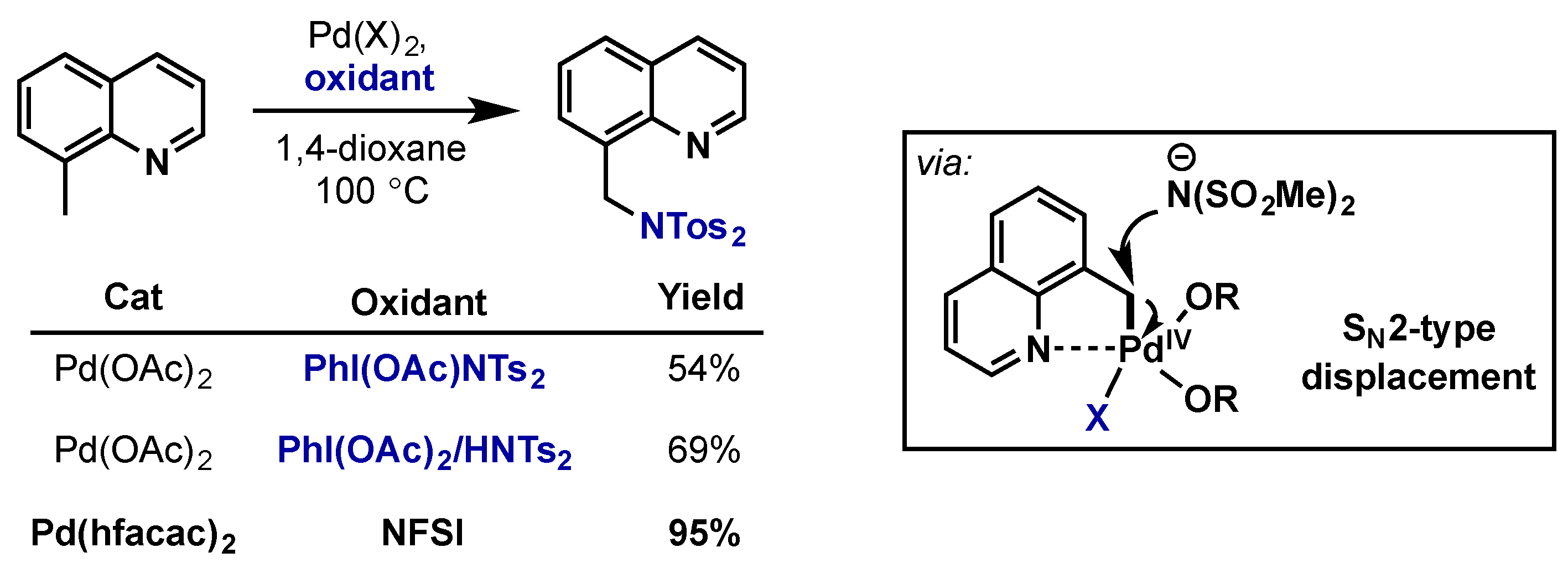
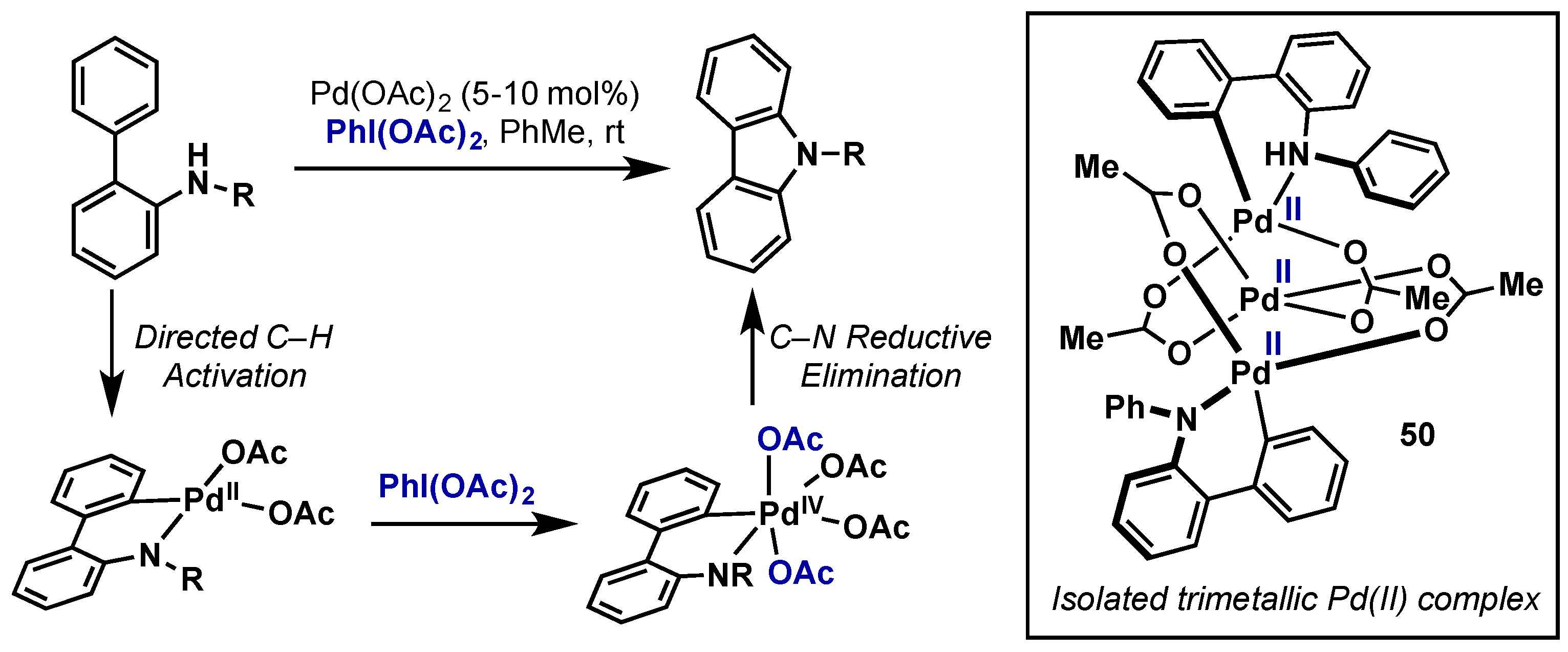
2.2.4.2. C–H Amination
2.2.5. C–C bond Formation
2.2.5.1. Diaryliodonium Salts
Stoichiometric Studies
Catalytic Applications
2.2.5.2. Benzoiodoxolones
2.2.5.3. Trifluoromethylation
2.2.6. Miscellaneous
2.3. Palladium(III)
2.3.1. Introduction
2.3.2. Complex Isolation
2.3.3. Conclusions
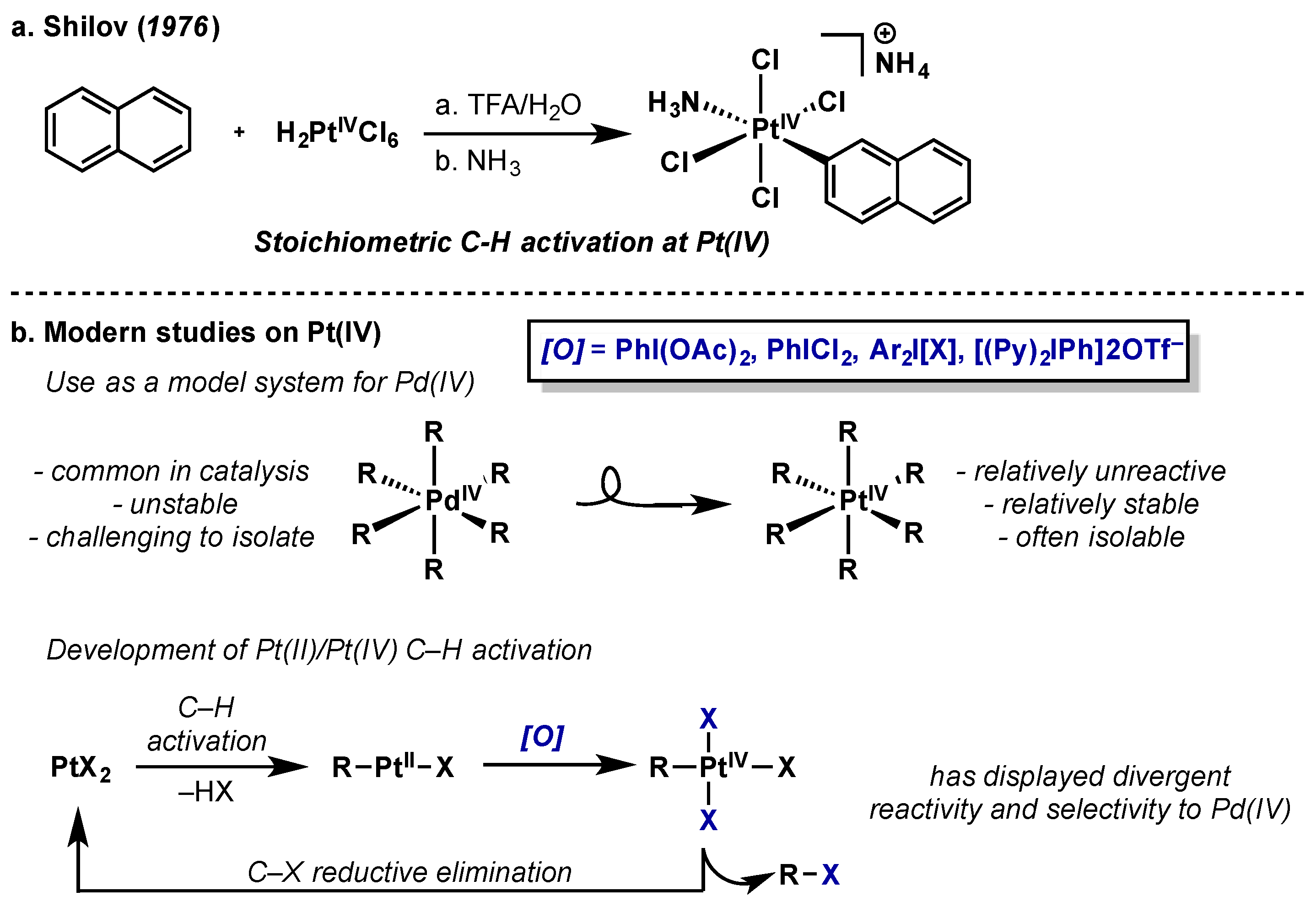
3. Platinum
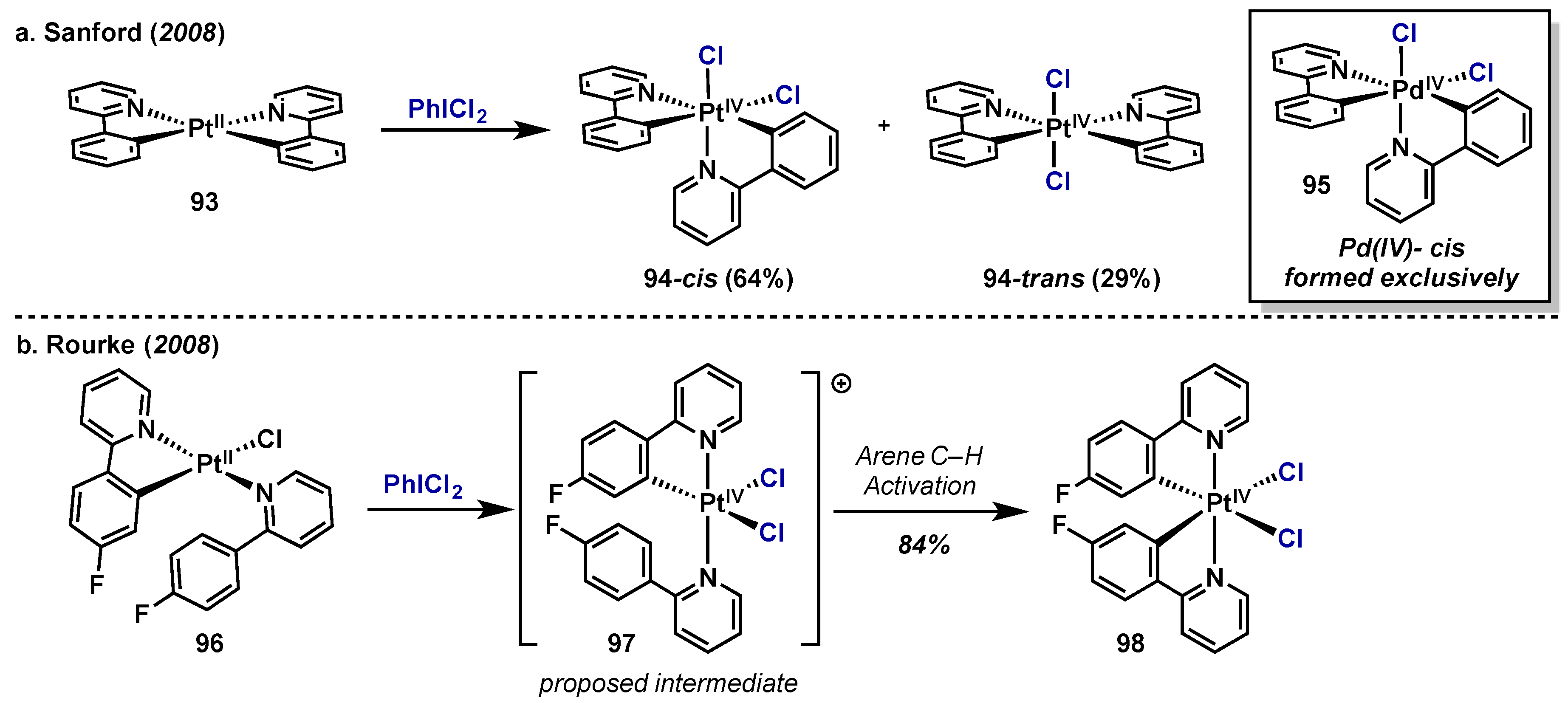
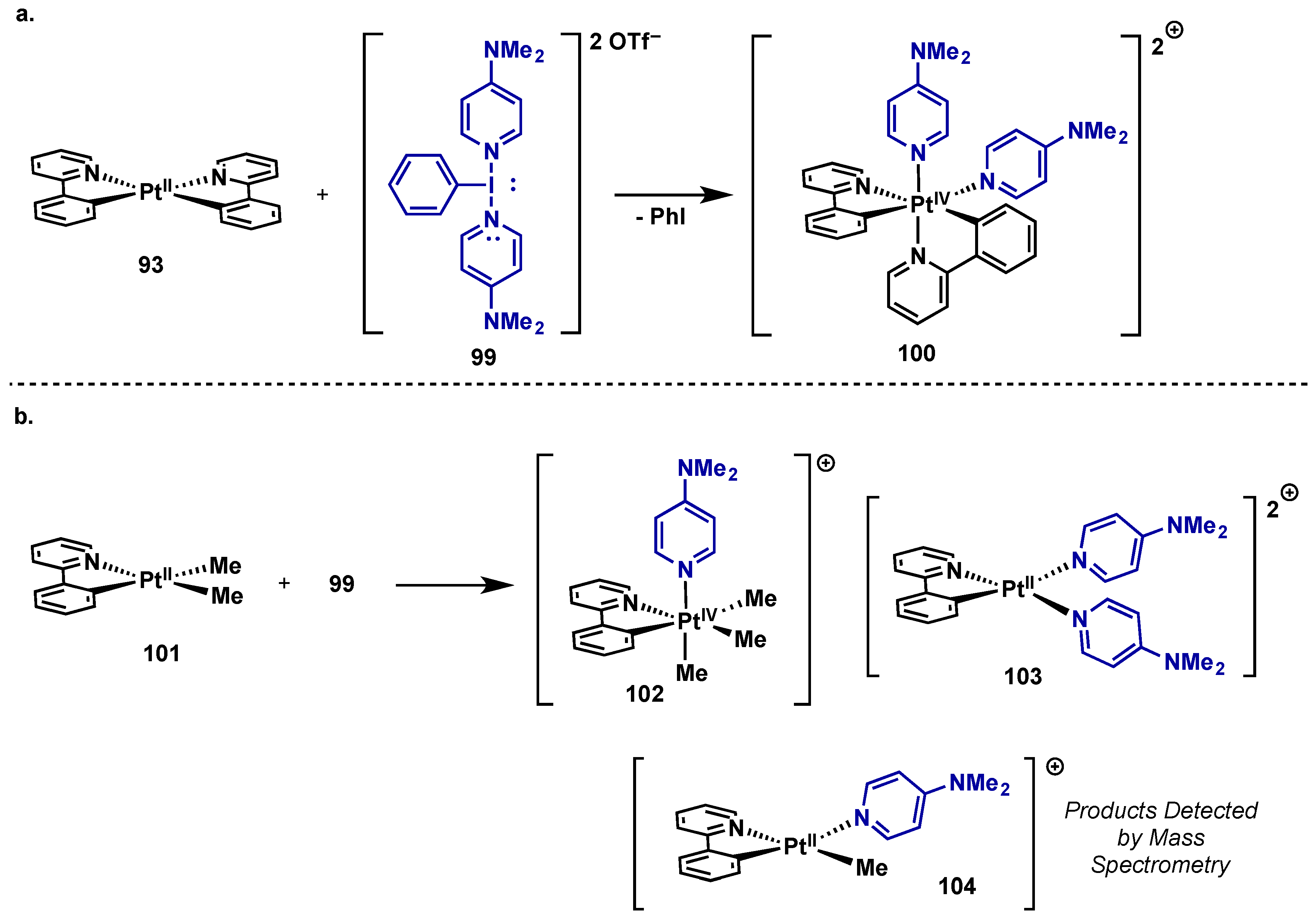
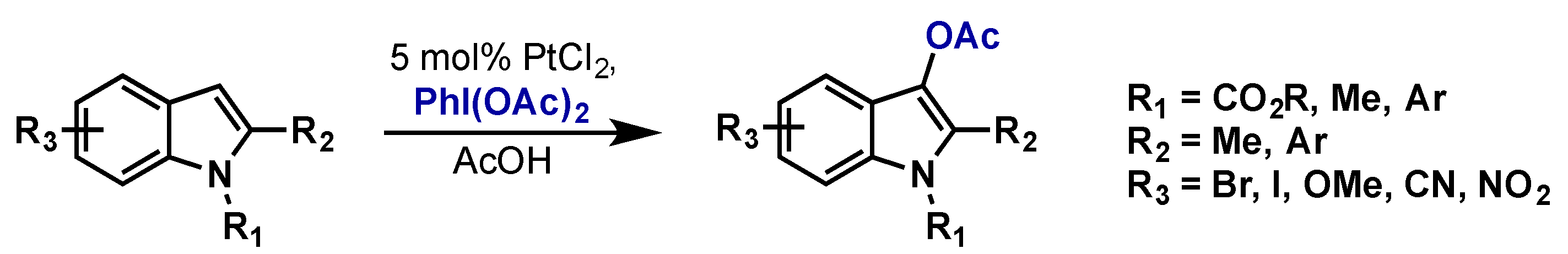
3.1. Complex Isolation
3.2. Catalytic Applications
3.3. Conclusions
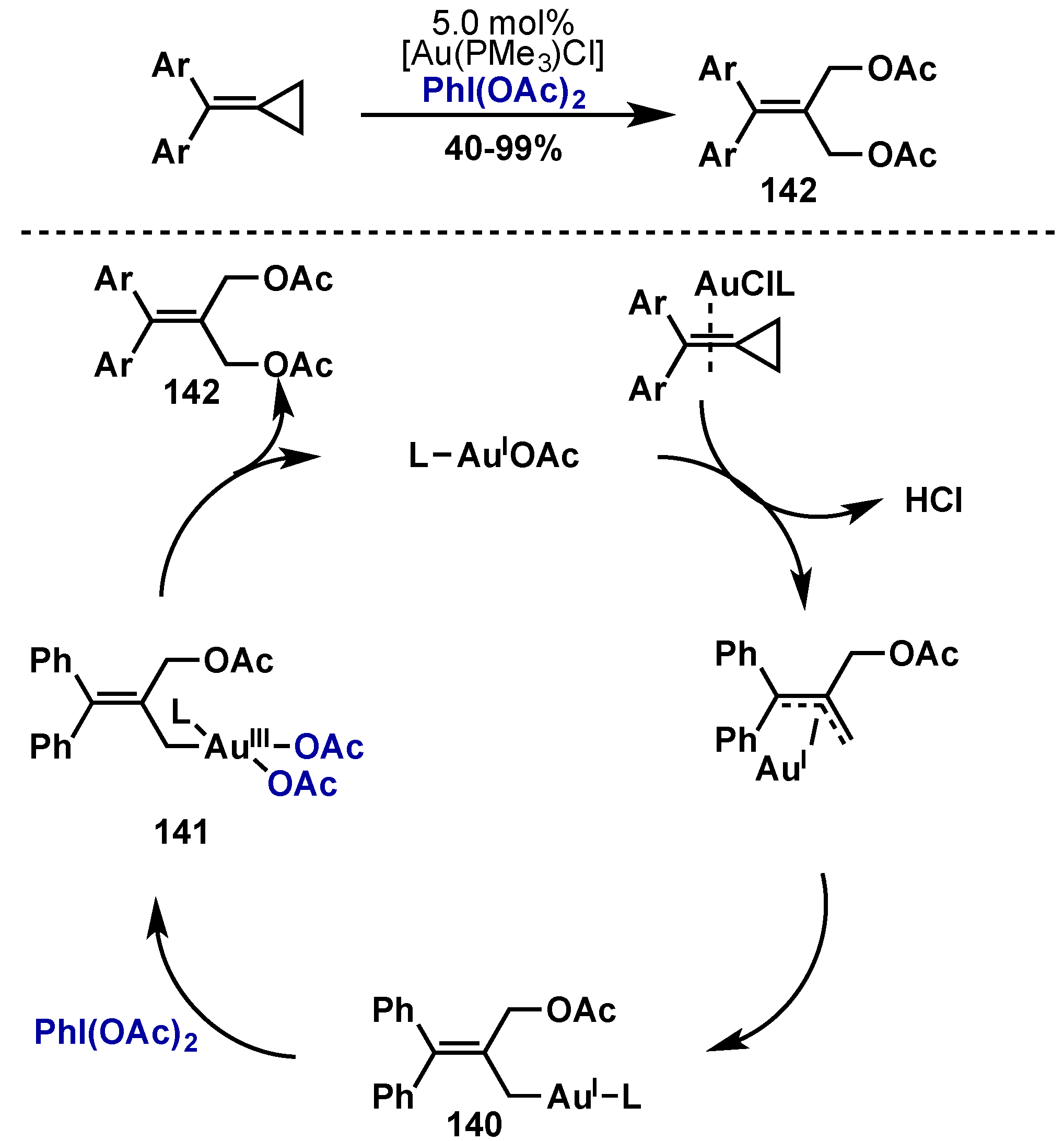
4. Gold
4.1. Introduction
4.2. Complex Synthesis and Characterization
4.3. Synthetic Applications
4.4. Conclusions
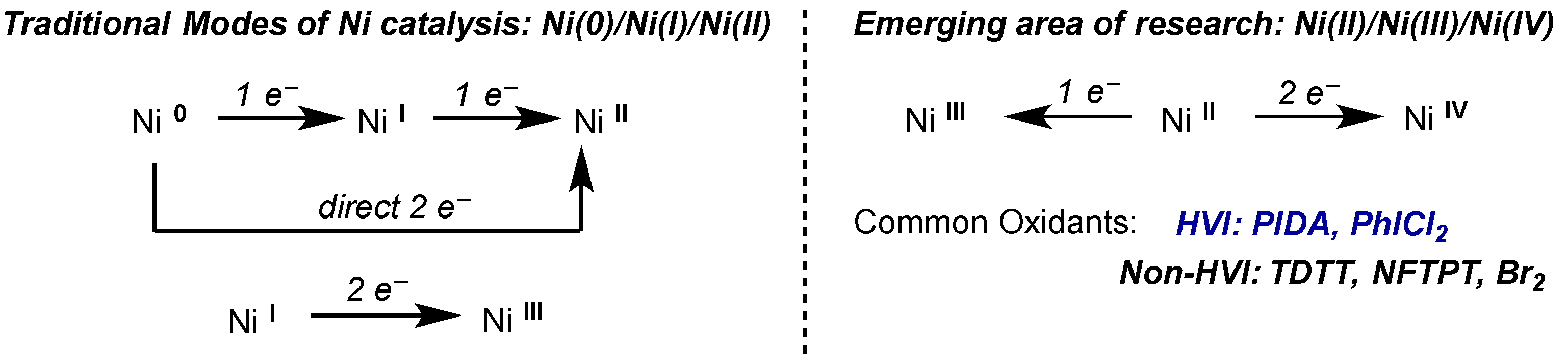
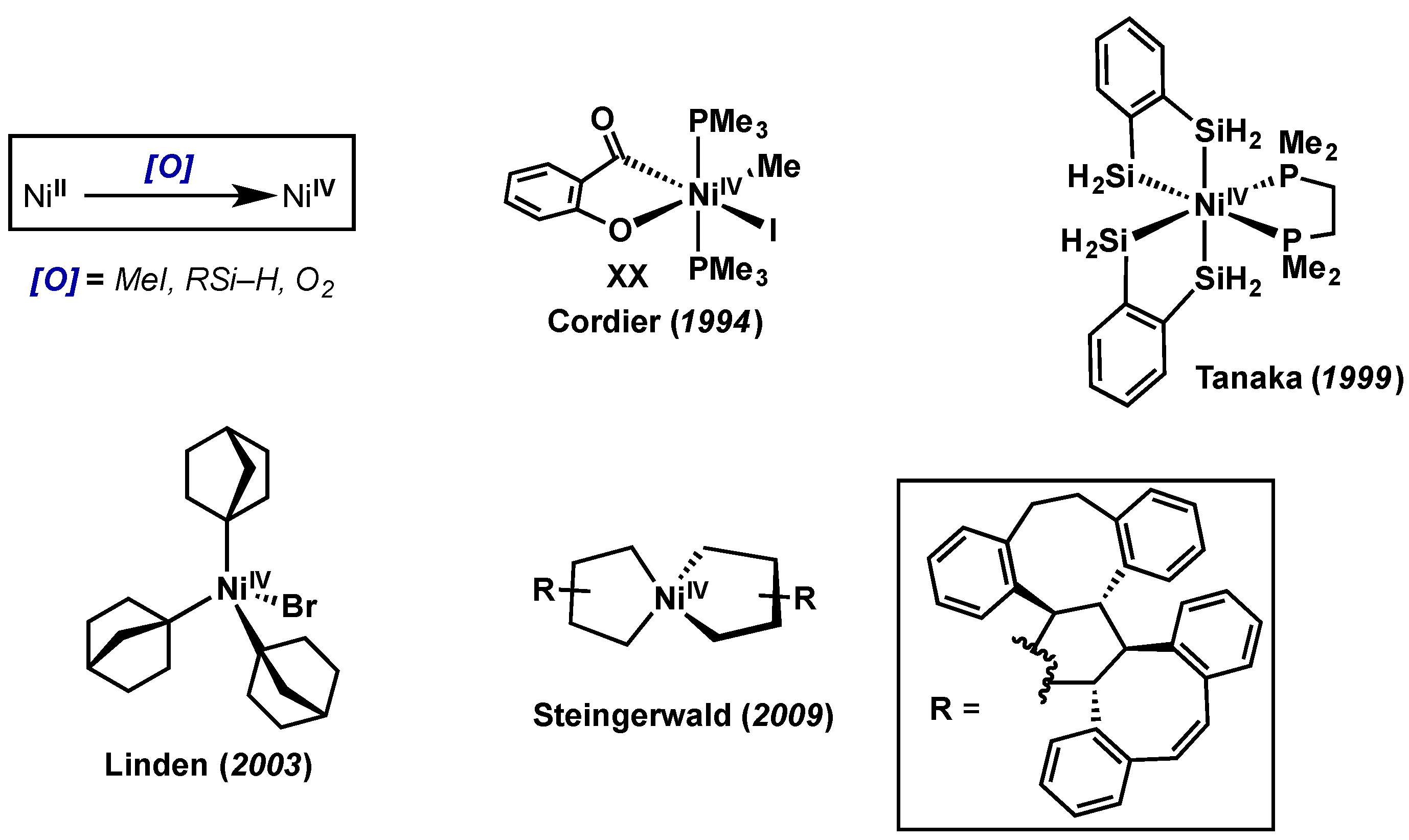
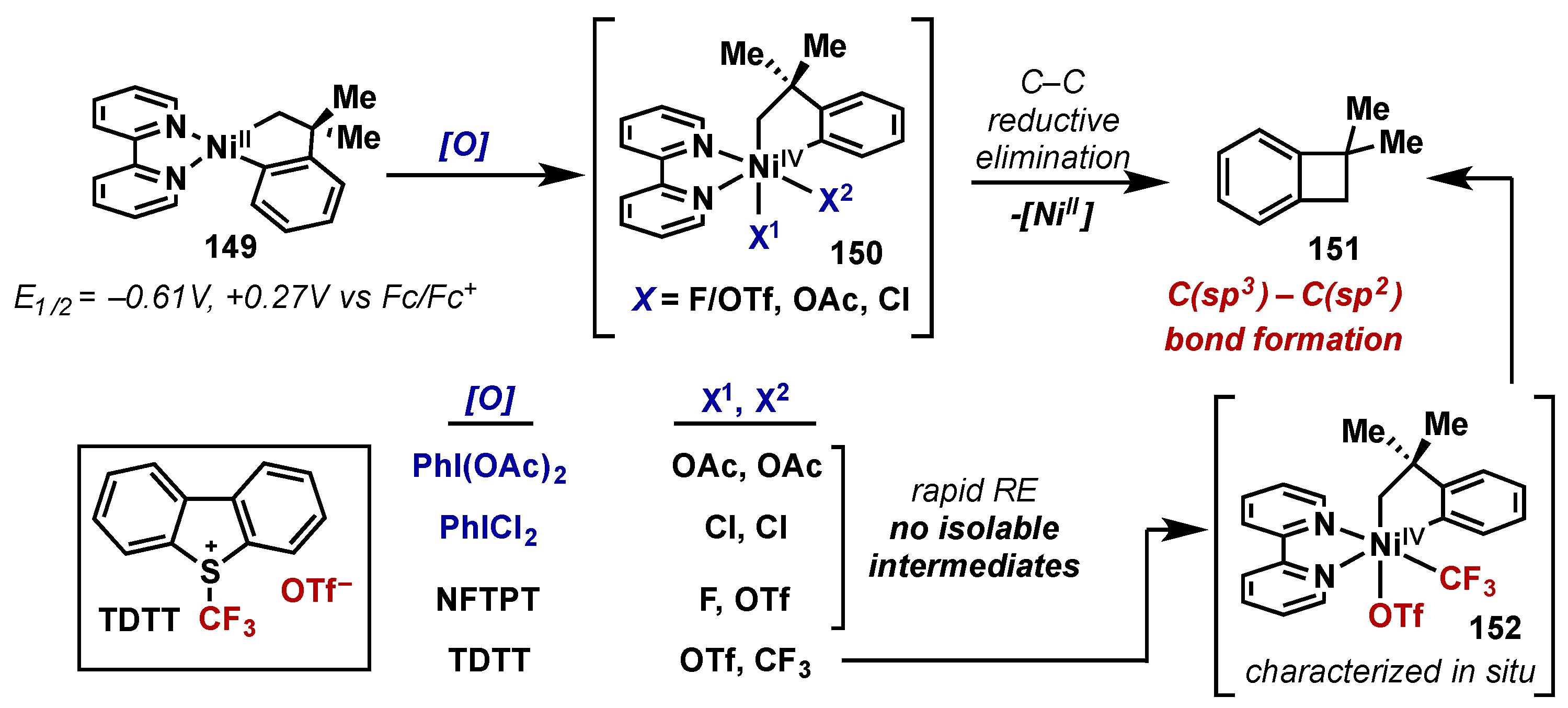
5. Nickel
5.1. Introduction
5.2. Stoichiometric Studies
5.3. Synthetic Applications
5.4. Conclusions
6. Copper
6.1. Introduction
6.2. Synthetic Applications
6.3. Conclusions
7. Miscellaneous High Valent Metal Complexes
7.1. Complex Synthesis and Isolation
7.2. Catalytic Applications
7.3. Conclusions
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hickman, A.J.; Sanford, M.S. High-valent organometallic copper and palladium in catalysis. Nature 2012, 484, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.W.; Sanford, M.S. Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef] [PubMed]
- Canty, A.J. Development of organopalladium(IV) chemistry: Fundamental aspects and systems for studies of mechanism in organometallic chemistry and catalysis. Acc. Chem. Res. 1992, 25, 83–90. [Google Scholar] [CrossRef]
- Xu, L.-M.; Li, B.-J.; Yang, Z.; Shi, Z.-J. Organopalladium(IV) chemistry. Chem. Soc. Rev. 2010, 39, 712–733. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, P.; Taylor, R.J.K.; Fairlamb, I.J.S. Emergence of Palladium(IV) Chemistry in Synthesis and Catalysis. Chem. Rev. 2010, 110, 824–889. [Google Scholar] [CrossRef] [PubMed]
- Dick, A.R.; Sanford, M.S. Transition metal catalyzed oxidative functionalization of carbon–hydrogen bonds. Tetrahedron 2006, 62, 2439–2463. [Google Scholar] [CrossRef]
- Topczewski, J.J.; Sanford, M.S. Carbon–hydrogen (C–H) bond activation at PdIV: A frontier in C–H functionalization catalysis. Chem. Sci. 2014, 6, 70–76. [Google Scholar] [CrossRef]
- Racowski, J.M.; Sanford, M.S. Carbon–Heteroatom Bond-Forming Reductive Elimination from Palladium(IV) Complexes. In Higher Oxidation State Organopalladium and Platinum Chemistry; Springer: Berlin/Heidelberg, Germany, 2011; Volume 35, pp. 61–84. [Google Scholar]
- Byers, P.K.; Canty, A.J.; Skelton, B.W.; White, A.H. The oxidative addition of lodomethane to [PdMe2(bpy)] and the X-ray structure of the organopalladium(IV) product fac-[PdMe3(bpy)1](bpy = 2,2′-bipyridyl). J. Chem. Soc. Chem. Commun. 1986, 1722–1724. [Google Scholar] [CrossRef]
- Yoneyama, T.; Crabtree, R.H. Pd(II) catalyzed acetoxylation of arenes with iodosyl acetate. J. Mol. Catal. A Chem. 1996, 108, 35–40. [Google Scholar] [CrossRef]
- Stock, L.M.; Tse, K.T.; Vorvick, L.J.; Walstrum, S.A. Palladium(II) acetate catalyzed aromatic substitution reaction. J. Org. Chem. 1981, 46, 1757–1759. [Google Scholar] [CrossRef]
- Dick, A.R.; Hull, K.L.; Sanford, M.S. A Highly Selective Catalytic Method for the Oxidative Functionalization of C–H Bonds. J. Am. Chem. Soc. 2004, 126, 2300–2301. [Google Scholar] [CrossRef] [PubMed]
- Kalyani, D.; Sanford, M.S. Regioselectivity in Palladium-Catalyzed C–H Activation/Oxygenation Reactions. Org. Lett. 2005, 7, 4149–4152. [Google Scholar] [CrossRef] [PubMed]
- Kalberer, E.W.; Whitfield, S.R.; Sanford, M.S. Application of recyclable, polymer-immobilized iodine(III) oxidants in catalytic C–H bond functionalization. J. Mol. Catal. A Chem. 2006, 251, 108–113. [Google Scholar] [CrossRef]
- Desai, L.V.; Hull, K.L.; Sanford, M.S. Palladium-Catalyzed Oxygenation of Unactivated sp3 C–H Bonds. J. Am. Chem. Soc. 2004, 126, 9542–9543. [Google Scholar] [CrossRef] [PubMed]
- Neufeldt, S.R.; Sanford, M.S. O-acetyl oximes as transformable directing groups for Pd-catalyzed C-H bond functionalization. Org. Lett. 2010, 12, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-H.; Hao, X.-S.; Wu, D.-F.; Yu, J.-Q. Palladium-catalyzed oxidation of Boc-protected N-methylamines with IOAc as the oxidant: A Boc-directed sp3 C-H bond activation. Org. Lett. 2006, 8, 3387–3390. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.K.; Sanford, M.S. Mechanism of the palladium-catalyzed arene C–H acetoxylation: A comparison of catalysts and ligand effects. J. Am. Chem. Soc. 2015, 137, 3109–3118. [Google Scholar] [CrossRef] [PubMed]
- Dick, A.R.; Kampf, J.W.; Sanford, M.S. Unusually Stable Palladium(IV) Complexes: Detailed Mechanistic Investigation of C–O Bond-Forming Reductive Elimination. J. Am. Chem. Soc. 2005, 127, 12790–12791. [Google Scholar] [CrossRef] [PubMed]
- Racowski, J.M.; Dick, A.R.; Sanford, M.S. Detailed study of C–O and C–C bond-forming reductive elimination from stable C2N2O2-ligated palladium(IV) complexes. J. Am. Chem. Soc. 2009, 131, 10974–10983. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.-F.; Li, Y.; Su, Y.-M.; Yin, F.; Wang, J.-Y.; Sheng, J.; Vora, H.U.; Wang, X.-S.; Yu, J.-Q. Pd(II)-catalyzed enantioselective C-H activation/C-O bond formation: Synthesis of chiral benzofuranones. J. Am. Chem. Soc. 2013, 135, 1236–1239. [Google Scholar] [CrossRef] [PubMed]
- Emmert, M.H.; Cook, A.K.; Xie, Y.J.; Sanford, M.S. Remarkably high reactivity of Pd(OAc)2/pyridine catalysts: Nondirected C-H oxygenation of arenes. Angew. Chem. Int. Ed. 2011, 50, 9409–9412. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-Y.; He, G.; Zhao, Y.; Wright, K.; Nack, W.A.; Chen, G. Efficient alkyl ether synthesis via palladium-catalyzed, picolinamide-directed alkoxylation of unactivated C(sp3)–H and C(sp2)–H bonds at remote positions. J. Am. Chem. Soc. 2012, 134, 7313–7316. [Google Scholar] [CrossRef] [PubMed]
- Shan, G.; Yang, X.; Zong, Y.; Rao, Y. An efficient palladium-catalyzed C-H alkoxylation of unactivated methylene and methyl groups with cyclic hypervalent iodine (i(3+)) oxidants. Angew. Chem. Int. Ed. 2013, 52, 13606–13610. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Stahl, S.S. Highly regioselective Pd-catalyzed intermolecular aminoacetoxylation of alkenes and evidence for cis-aminopalladation and SN2 C–O bond formation. J. Am. Chem. Soc. 2006, 128, 7179–7181. [Google Scholar] [CrossRef] [PubMed]
- Desai, L.V.; Sanford, M.S. Construction of tetrahydrofurans by PdII/PdIV-catalyzed aminooxygenation of alkenes. Angew. Chem. Int. Ed. Engl. 2007, 46, 5737–5740. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Song, D.; Dong, V.M. Palladium-Catalyzed Olefin Dioxygenation. J. Am. Chem. Soc. 2008, 130, 2962–2964. [Google Scholar] [CrossRef] [PubMed]
- Neufeldt, S.R.; Sanford, M.S. Asymmetric chiral ligand-directed alkene dioxygenation. Org. Lett. 2013, 15, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Pilarski, L.T.; Selander, N.; Böse, D.; Szabó, K.J. Catalytic allylic C-H acetoxylation and benzoyloxylation via suggested (eta(3)-allyl)palladium(IV) intermediates. Org. Lett. 2009, 11, 5518–5521. [Google Scholar] [CrossRef] [PubMed]
- Alam, R.; Pilarski, L.T.; Pershagen, E.; Szabó, K.J. Stereoselective intermolecular allylic C–H trifluoroacetoxylation of functionalized alkenes. J. Am. Chem. Soc. 2012, 134, 8778–8781. [Google Scholar] [CrossRef] [PubMed]
- Bigi, M.A.; White, M.C. Terminal Olefins to Linear α,β-Unsaturated Ketones: Pd(II)/Hypervalent Iodine Co-catalyzed Wacker Oxidation—Dehydrogenation. J. Am. Chem. Soc. 2013, 135, 7831–7834. [Google Scholar] [CrossRef] [PubMed]
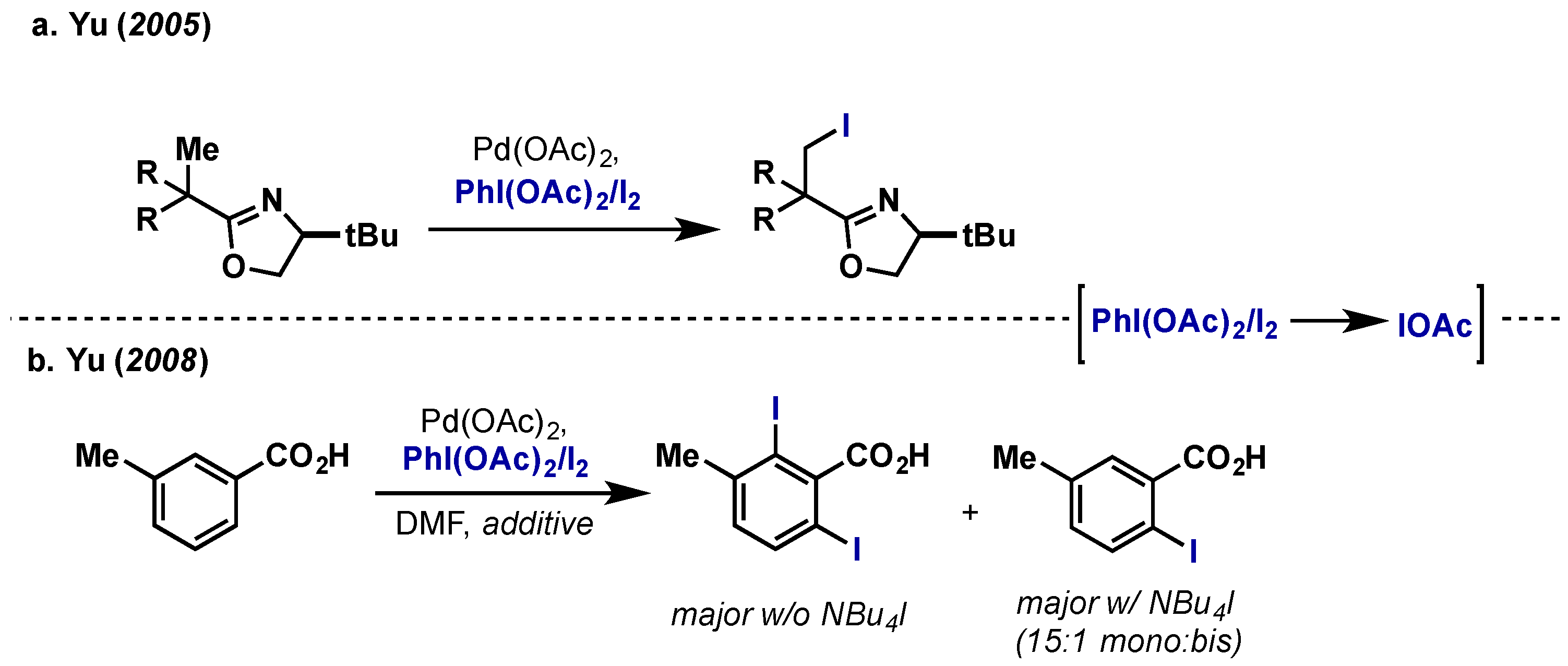
- Giri, R.; Chen, X.; Hao, X.-S.; Li, J.-J.; Liang, J.; Fan, Z.-P.; Yu, J.-Q. Catalytic and stereoselective iodination of prochiral C–H bonds. Tetrahedron Asymmetry 2005, 16, 3502–3505. [Google Scholar] [CrossRef]
- Giri, R.; Chen, X.; Yu, J.-Q. Palladium-catalyzed asymmetric iodination of unactivated C-H bonds under mild conditions. Angew. Chem. Int. Ed. Engl. 2005, 44, 2112–2115. [Google Scholar] [CrossRef]
- Mei, T.-S.; Giri, R.; Maugel, N.; Yu, J.-Q. Pd(II)-catalyzed monoselective ortho halogenation of C-H bonds assisted by counter cations: A complementary method to directed ortho lithiation. Angew. Chem. Int. Ed. 2008, 47, 5215–5219. [Google Scholar] [CrossRef] [PubMed]
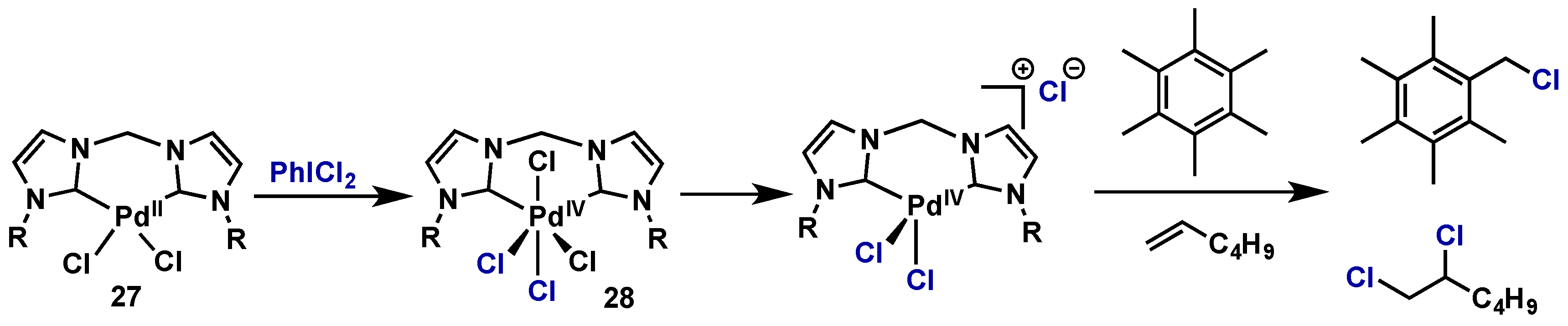
- McCall, A.S.; Wang, H.; Desper, J.M.; Kraft, S. Bis-N-heterocyclic carbene Palladium(IV) tetrachloride complexes: Synthesis, reactivity, and mechanisms of direct chlorinations and oxidations of organic substrates. J. Am. Chem. Soc. 2011, 133, 1832–1848. [Google Scholar] [CrossRef] [PubMed]
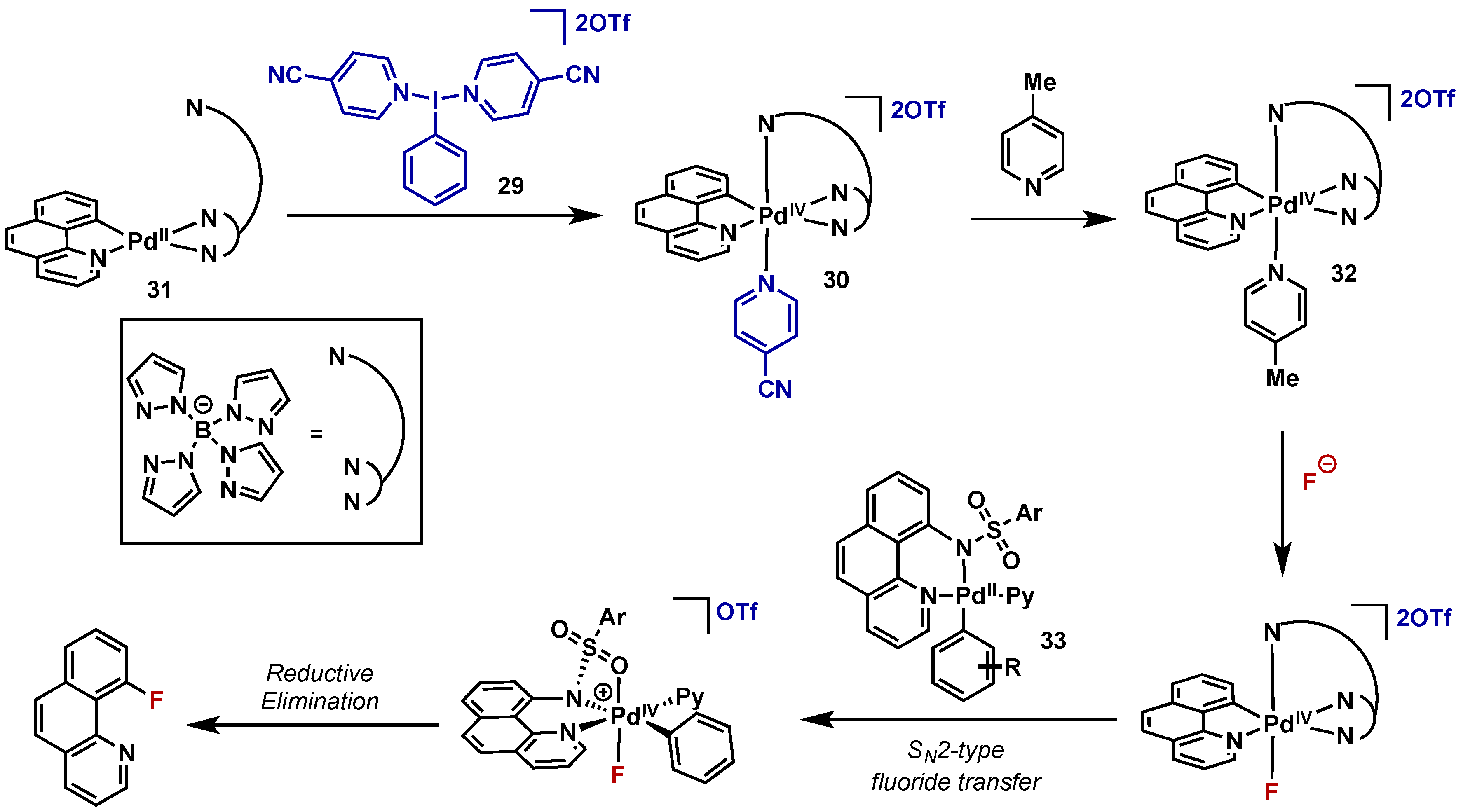
- Lee, E.; Kamlet, A.S.; Powers, D.C.; Neumann, C.N.; Boursalian, G.B.; Furuya, T.; Choi, D.C.; Hooker, J.M.; Ritter, T. A fluoride-derived electrophilic late-stage fluorination reagent for PET imaging. Science 2011, 334, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Furuya, T.; Kaiser, H.M.; Ritter, T. Palladium-mediated fluorination of arylboronic acids. Angew. Chem. Int. Ed. 2008, 47, 5993–5996. [Google Scholar] [CrossRef] [PubMed]
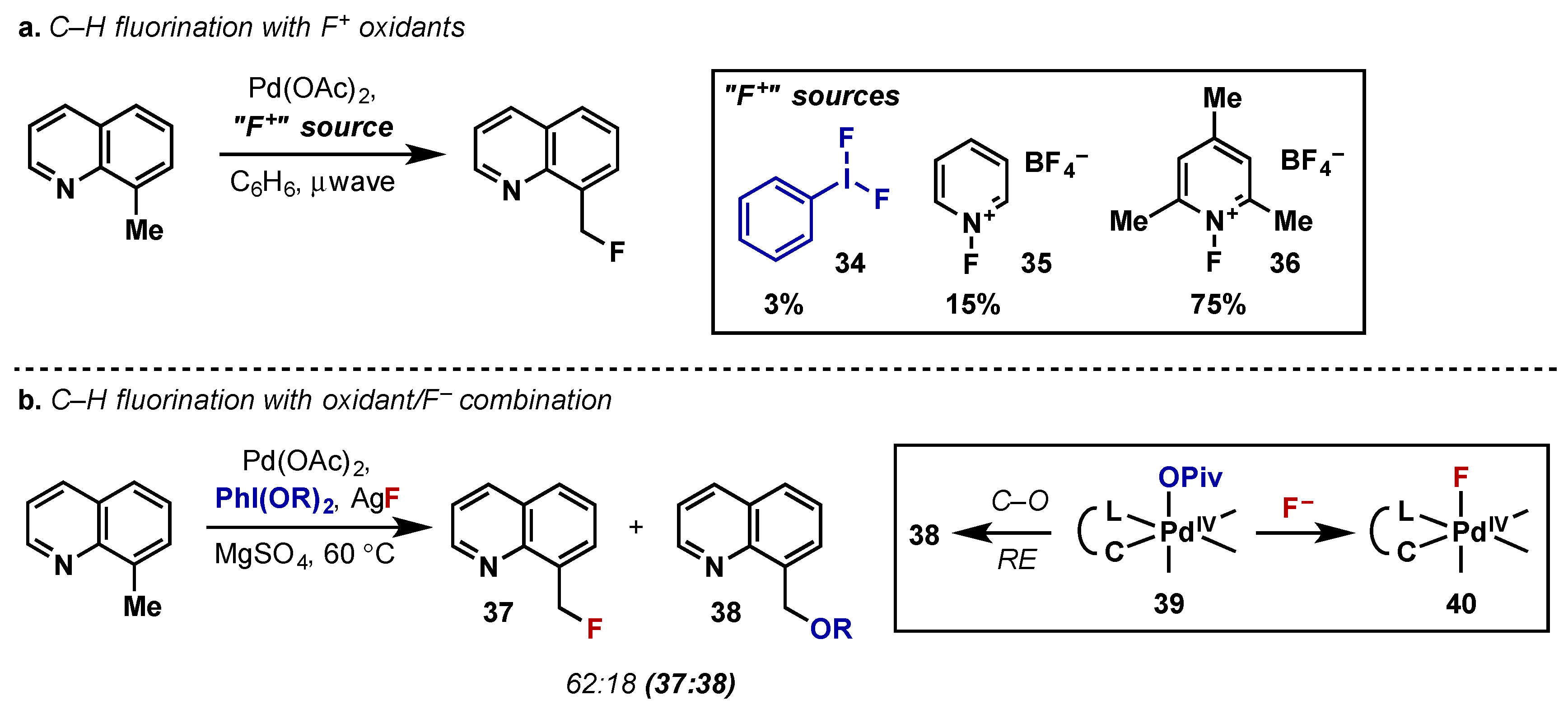
- Hull, K.L.; Anani, W.Q.; Sanford, M.S. Palladium-catalyzed fluorination of carbon-hydrogen bonds. J. Am. Chem. Soc. 2006, 128, 7134–7135. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Mei, T.-S.; Yu, J.-Q. Versatile Pd(OTf)2·2H2O-catalyzed ortho-fluorination using NMP as a promoter. J. Am. Chem. Soc. 2009, 131, 7520–7521. [Google Scholar] [CrossRef]
- McMurtrey, K.B.; Racowski, J.M.; Sanford, M.S. Pd-Catalyzed C–H Fluorination with Nucleophilic Fluoride. Org. Lett. 2012, 14, 4094–4097. [Google Scholar] [CrossRef] [PubMed]
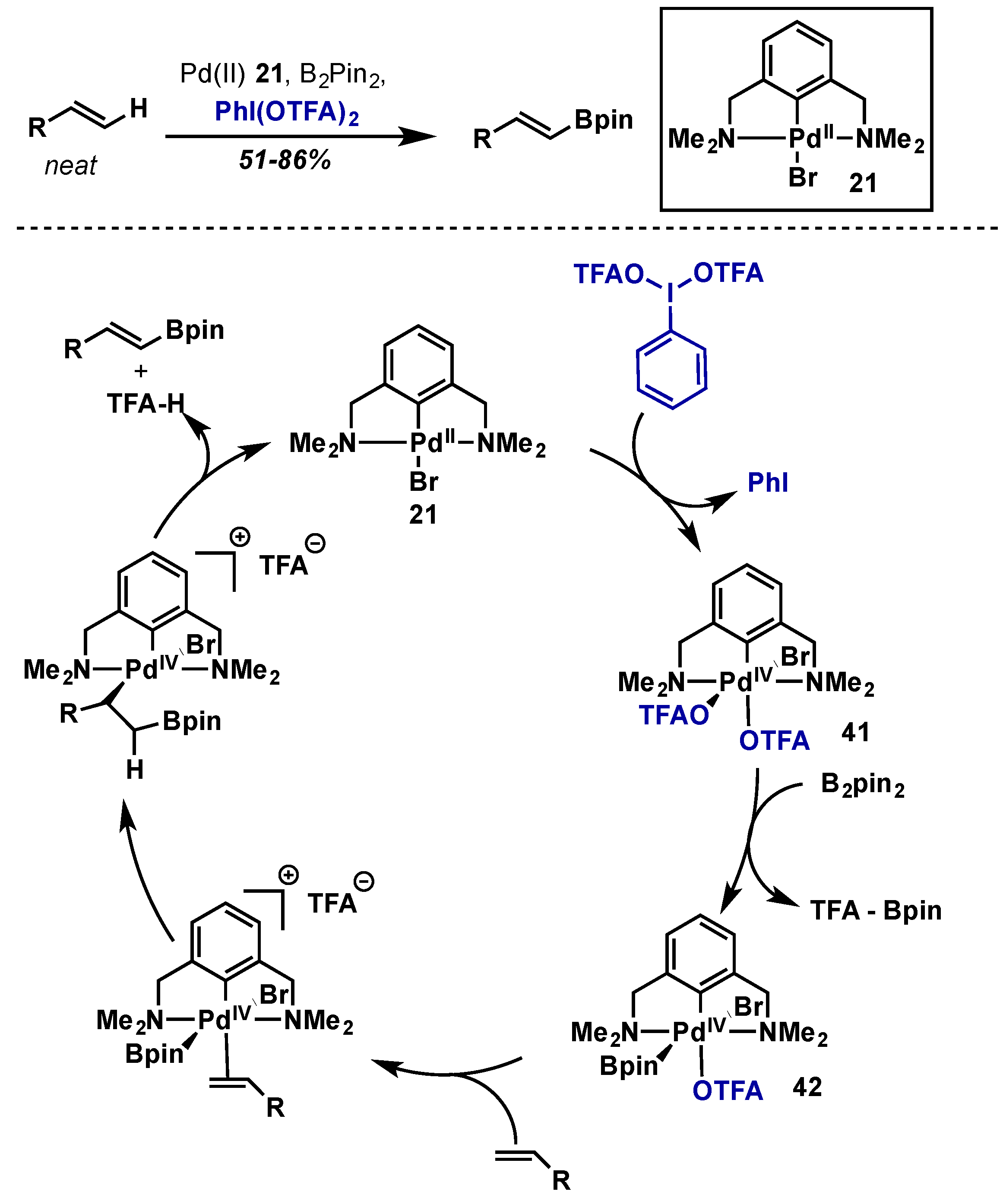
- Selander, N.; Willy, B.; Szabó, K.J. Selective C–H borylation of alkenes by palladium pincer complex catalyzed oxidative functionalization. Angew. Chem. Int. Ed. 2010, 49, 4051–4053. [Google Scholar] [CrossRef] [PubMed]
- Muñiz, K.; Martínez, C.; Iglesias, A. The Quest for Palladium-Catalysed Alkyl-Nitrogen Bond Formation. Chem. Rec. 2016, 16, 2561–2572. [Google Scholar] [CrossRef] [PubMed]
- Streuff, J.; Hövelmann, C.H.; Nieger, M.; Muñiz, K. Palladium(II)-catalyzed intramolecular diamination of unfunctionalized alkenes. J. Am. Chem. Soc. 2005, 127, 14586–14587. [Google Scholar] [CrossRef] [PubMed]
- Muñiz, K.; Hövelmann, C.H.; Streuff, J. Oxidative diamination of alkenes with ureas as nitrogen sources: Mechanistic pathways in the presence of a high oxidation state palladium catalyst. J. Am. Chem. Soc. 2008, 130, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, A.; Muñiz, K. Studies on Alkyl-Nitrogen Bond Formation via Reductive Elimination from Monomeric Palladium Complexes in High Oxidation State. Helv. Chim. Acta 2012, 95, 2007–2025. [Google Scholar] [CrossRef]
- Hövelmann, C.H.; Streuff, J.; Brelot, L.; Muñiz, K. Direct synthesis of bicyclic guanidines through unprecedented Palladium(II) catalysed diamination with copper chloride as oxidant. Chem. Commun. (Camb.) 2008, 2334–2336. [Google Scholar] [CrossRef] [PubMed]
- Welbes, L.L.; Lyons, T.W.; Cychosz, K.A.; Sanford, M.S. Synthesis of cyclopropanes via Pd(II/IV)-catalyzed reactions of enynes. J. Am. Chem. Soc. 2007, 129, 5836–5837. [Google Scholar] [CrossRef] [PubMed]
- Muñiz, K. Advancing palladium-catalyzed C-N bond formation: Bisindoline construction from successive amide transfer to internal alkenes. J. Am. Chem. Soc. 2007, 129, 14542–14543. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, A.; Pérez, E.G.; Muñiz, K. An Intermolecular Palladium-Catalyzed Diamination of Unactivated Alkenes. Angew. Chem. Int. Ed. 2010, 49, 8109–8111. [Google Scholar] [CrossRef] [PubMed]
- Muñiz, K.; Kirsch, J.; Chávez, P. Intermolecular Regioselective 1,2-Diamination of Allylic Ethers. Adv. Synth. Catal. 2011, 353, 689–694. [Google Scholar] [CrossRef]
- Martínez, C.; Muñiz, K. Palladium-catalyzed vicinal difunctionalization of internal alkenes: Diastereoselective synthesis of diamines. Angew. Chem. Int. Ed. 2012, 51, 7031–7034. [Google Scholar] [CrossRef] [PubMed]
- Ingalls, E.L.; Sibbald, P.A.; Kaminsky, W.; Michael, F.E. Enantioselective palladium-catalyzed diamination of alkenes using N-fluorobenzenesulfonimide. J. Am. Chem. Soc. 2013, 135, 8854–8856. [Google Scholar] [CrossRef] [PubMed]
- Sibbald, P.A.; Michael, F.E. Palladium-catalyzed diamination of unactivated alkenes using N-fluorobenzenesulfonimide as source of electrophilic nitrogen. Org. Lett. 2009, 11, 1147–1149. [Google Scholar] [CrossRef] [PubMed]
- Dick, A.R.; Remy, M.S.; Kampf, J.W.; Sanford, M.S. Carbon–Nitrogen Bond-Forming Reactions of Palladacycles with Hypervalent Iodine Reagents. Organometallics 2007, 26, 1365–1370. [Google Scholar] [CrossRef]
- Thu, H.-Y.; Yu, W.-Y.; Che, C.-M. Intermolecular amidation of unactivated sp2 and sp3 C-H bonds via palladium-catalyzed cascade C–H activation/nitrene insertion. J. Am. Chem. Soc. 2006, 128, 9048–9049. [Google Scholar] [CrossRef] [PubMed]
- Nadres, E.T.; Daugulis, O. Heterocycle synthesis via direct C–H/N–H coupling. J. Am. Chem. Soc. 2012, 134, 7–10. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Zhao, Y.; Zhang, S.; Lu, C.; Chen, G. Highly efficient syntheses of azetidines, pyrrolidines, and indolines via palladium catalyzed intramolecular amination of C(sp3)–H and C(sp2)–H bonds at γ and δ positions. J. Am. Chem. Soc. 2012, 134, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, A.; Alvarez, R.; de Lera, A.R.; Muñiz, K. Palladium-catalyzed intermolecular C(sp3)-H amidation. Angew. Chem. Int. Ed. 2012, 51, 2225–2228. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Peng, P.; Pi, S.-F.; Liang, Y.; Wang, N.-X.; Li, J.H. Sequential intermolecular aminopalladation/ortho-arene C-H activation reactions of N-phenylpropiolamides with phthalimide. Org. Lett. 2008, 10, 1179–1182. [Google Scholar] [CrossRef] [PubMed]
- Jordan-Hore, J.A.; Johansson, C.C.C.; Beck, E.M.; Gaunt, M.J. Oxidative Pd(II)-catalyzed C–H bond amination to carbazole at ambient temperature. J. Am. Chem. Soc. 2008, 130, 16184–16186. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewski, J.; Smalley, A.P.; Kabeshov, M.A.; Gaunt, M.J.; Lapkin, A.A. Continuous-Flow Synthesis and Derivatization of Aziridines through Palladium-Catalyzed C(sp3)–H Activation. Angew. Chem. Int. Ed. 2016, 55, 8878–8883. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Zhdankin, V.V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef] [PubMed]
- Canty, A.J.; Rodemann, T.; Skelton, B.W.; White, A.H. Access to Alkynylpalladium(IV) and -Platinum(IV) Species, Including Triorgano(diphosphine)metal(IV) Complexes and the Structural Study of an Alkynyl(pincer)platinum(IV) Complex, Pt(O2CArF)I(C⋮CSiMe3)(NCN) (ArF = 4-CF3C6H4, NCN = [2,6-(dimethylaminomethyl)phenyl-N,C,N]-). Organometallics 2006, 23, 3466–3473. [Google Scholar]
- Canty, A.J.; Patel, J.; Rodemann, T.; Ryan, J.H.; Skelton, B.W.; White, A.H. Reactivity of Diaryliodine(III) Triflates toward Palladium(II) and Platinum(II): Reactions of C(sp2)–I Bonds to Form Arylmetal(IV) Complexes; Access to Dialkyl(aryl)metal(IV), 1,4-Benzenediyl-Bridged Platinum(IV), and Triphenylplatinum(IV) Species; and Structural Studies of Platinum(IV) Complexes. Organometallics 2004, 23, 3466–3473. [Google Scholar]
- Bayler, A.; Canty, A.J.; Ryan, J.H.; Skelton, B.W.; White, A.H. Arylation of palladium(II) and platinum(II) by diphenyliodonium triflate to form metal(IV) species, and a structural analysis of an isomer of PtIMe2Ph(bpy) (bpy = 2,2′-bipyridine). Inorg. Chem. Commun. 2000, 3, 575–578. [Google Scholar] [CrossRef]
- Canty, A.J.; Rodemann, T. Entry to alkynylplatinum(IV) chemistry using hypervalent iodine(III) reagents, and the synthesis of triphenyl{4,4′-bis(tert-butyl)-2,2′-bipyridine}iodoplatinum(IV). Inorg. Chem. Commun. 2003, 6, 1382–1384. [Google Scholar] [CrossRef]
- Kalyani, D.; Deprez, N.R.; Desai, L.V.; Sanford, M.S. Oxidative C–H activation/C–C bond forming reactions: Synthetic scope and mechanistic insights. J. Am. Chem. Soc. 2005, 127, 7330–7331. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.M.; Sanford, M.S. Palladium-catalyzed C-H arylation of 2,5-substituted pyrroles. Org. Lett. 2011, 13, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Deprez, N.R.; Sanford, M.S. Synthetic and mechanistic studies of Pd-catalyzed C-H arylation with diaryliodonium salts: Evidence for a bimetallic high oxidation state Pd intermediate. J. Am. Chem. Soc. 2009, 131, 11234–11241. [Google Scholar] [CrossRef] [PubMed]
- Canty, A.J.; Ariafard, A.; Sanford, M.S.; Yates, B.F. Mechanism of Pd-Catalyzed Ar–Ar Bond Formation Involving Ligand-Directed C–H Arylation and Diaryliodonium Oxidants: Computational Studies of Orthopalladation at Binuclear Pd(II) Centers, Oxidation To Form Binuclear Palladium(III) Species, and Ar···Ar Reductive Coupling. Organometallics 2013, 32, 544–555. [Google Scholar]
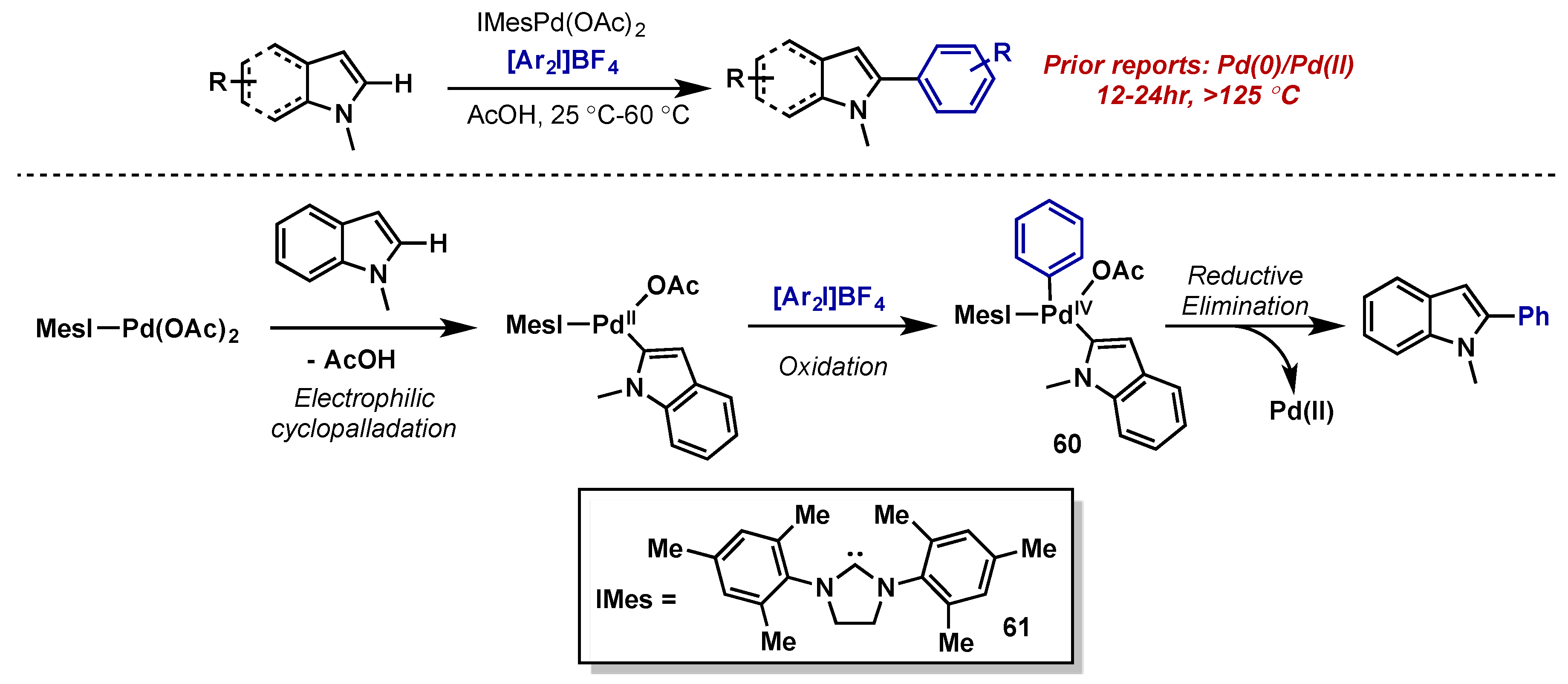
- Deprez, N.R.; Kalyani, D.; Krause, A.; Sanford, M.S. Room temperature palladium-catalyzed 2-arylation of indoles. J. Am. Chem. Soc. 2006, 128, 4972–4973. [Google Scholar] [CrossRef] [PubMed]
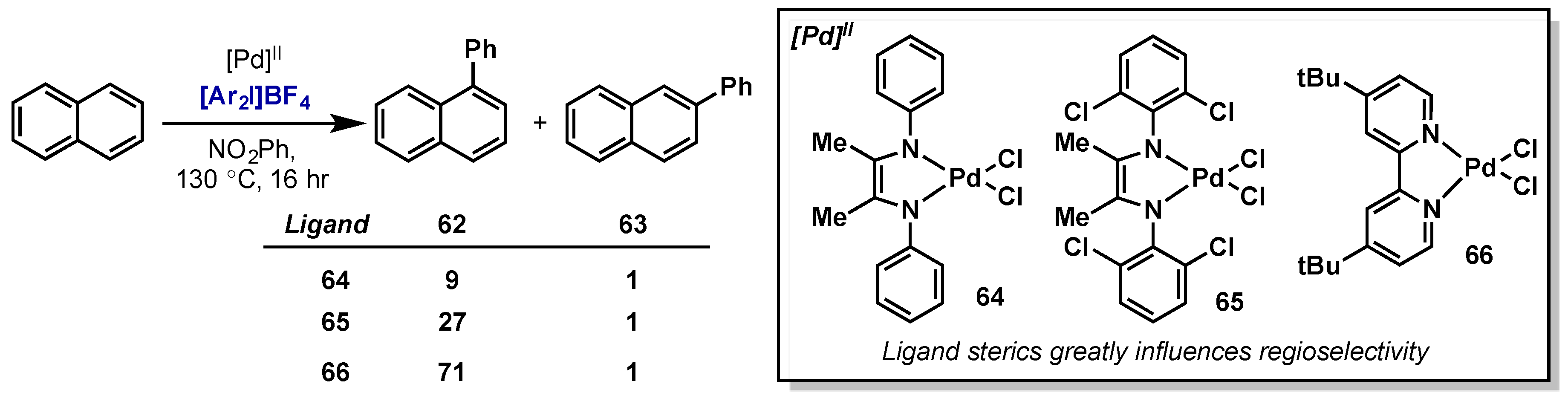
- Hickman, A.J.; Sanford, M.S. Catalyst Control of Site Selectivity in the Pd II/IV-Catalyzed Direct Arylation of Naphthalene. ACS Catal. 2011, 1, 170–174. [Google Scholar] [CrossRef]
- Tolnai, G.L.; Ganss, S.; Brand, J.P.; Waser, J. C2-Selective Direct Alkynylation of Indoles. Org. Lett. 2012, 15, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.J.; Senecal, T.D.; Kinzel, T.; Zhang, Y.; Watson, D.A.; Buchwald, S.L. The Palladium-Catalyzed Trifluoromethylation of Aryl Chlorides. Science 2010, 328, 1679–1681. [Google Scholar] [CrossRef] [PubMed]
- Grushin, V.V.; Marshall, W.J. Facile Ar–CF3 Bond Formation at Pd. Strikingly Different Outcomes of Reductive Elimination from [(Ph3P)2Pd(CF3)Ph] and [(Xantphos)Pd(CF3)Ph]. J. Am. Chem. Soc. 2006, 128, 12644–12645. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Truesdale, L.; Yu, J.-Q. Pd(II)-Catalyzed ortho-Trifluoromethylation of Arenes Using TFA as a Promoter. J. Am. Chem. Soc. 2010, 132, 3648–3649. [Google Scholar] [CrossRef] [PubMed]
- Ball, N.D.; Kampf, J.W.; Sanford, M.S. Aryl–CF3 Bond-Forming Reductive Elimination from Palladium(IV). J. Am. Chem. Soc. 2010, 132, 2878–2879. [Google Scholar] [CrossRef] [PubMed]
- Ball, N.D.; Gary, J.B.; Ye, Y.; Sanford, M.S. Mechanistic and computational studies of oxidatively-induced aryl-CF3 bond-formation at Pd: Rational design of room temperature aryl trifluoromethylation. J. Am. Chem. Soc. 2011, 133, 7577–7584. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Chen, S.; Zhen, X.; Liu, G. Palladium-Catalyzed Oxidative Trifluoromethylation of Indoles at Room Temperature. Chem. Eur. J. 2011, 17, 6039–6042. [Google Scholar] [CrossRef] [PubMed]
- Powers, D.C.; Benitez, D.; Tkatchouk, E.; Goddard, W.A.; Ritter, T. Bimetallic reductive elimination from dinuclear Pd(III) complexes. J. Am. Chem. Soc. 2010, 132, 14092–14103. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Ball, N.D.; Kampf, J.W.; Sanford, M.S. Oxidation of a cyclometalated Pd(II) dimer with “CF3+”: Formation and reactivity of a catalytically competent monomeric Pd(IV) aquo complex. J. Am. Chem. Soc. 2010, 132, 14682–14687. [Google Scholar] [CrossRef] [PubMed]
- Powers, D.C.; Lee, E.; Ariafard, A.; Sanford, M.S.; Yates, B.F.; Canty, A.J.; Ritter, T. Connecting binuclear Pd(III) and mononuclear Pd(IV) chemistry by Pd-Pd bond cleavage. J. Am. Chem. Soc. 2012, 134, 12002–12009. [Google Scholar] [CrossRef] [PubMed]
- Cotton, F.A.; Gu, J.; Murillo, C.A.; Timmons, D.J. The First Dinuclear Complex of Palladium(III). J. Am. Chem. Soc. 1998, 120, 13280–13281. [Google Scholar] [CrossRef]
- Cotton, F.A.; Koshevoy, I.O.; Lahuerta, P.; Murillo, C.A.; Sanaú, M.; Ubeda, M.A.; Zhao, Q. High Yield Syntheses of Stable, Singly Bonded Pd26+ Compounds. J. Am. Chem. Soc. 2006, 128, 13674–13675. [Google Scholar] [CrossRef] [PubMed]
- Powers, D.C.; Geibel, M.A.L.; Klein, J.E.M.N.; Ritter, T. Bimetallic palladium catalysis: Direct observation of Pd(III)-Pd(III) intermediates. J. Am. Chem. Soc. 2009, 131, 17050–17051. [Google Scholar] [CrossRef] [PubMed]
- Gol’dshleger, N.F.; Eskova, V.V.; Shilov, A.E.; Shteinman, A.A. Alkane Reactions in Solutions of Chloride Complexes of Platinum. Zh. Fiz. Khim. 1972, 46, 1353. [Google Scholar]
- Byers, P.K.; Canty, A.J.; Honeyman, T. Conformational studies in Palladium(IV) and Platinum(IV) chemistry. Crystal structure of the 1,1-bis(pyrazol-1-yl)-ethane complex fac-PtIMe3[(pz)2CHMe-N,N′]. J. Orgmet. Chem. 1992, 433, 223–229. [Google Scholar] [CrossRef]
- Canty, A.J. Organopalladium and platinum chemistry in oxidising milieu as models for organic synthesis involving the higher oxidation states of palladium. Dalton Trans. 2009, 10409–10417. [Google Scholar] [CrossRef] [PubMed]
- Labinger, J.A. Platinum-Catalyzed C–H Functionalization. Chem. Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Dick, A.R.; Kampf, J.W.; Sanford, M.S. Platinum Model Studies for Palladium-Catalyzed Oxidative Functionalization of C–H Bonds. Organometallics 2005, 24, 482–485. [Google Scholar] [CrossRef]
- Whitfield, S.R.; Sanford, M.S. Reactions of Platinum(II) Complexes with Chloride-Based Oxidants: Routes to Pt(III) and Pt(IV) Products. Organometallics 2008, 27, 1683–1689. [Google Scholar] [CrossRef]
- Mamtora, J.; Crosby, S.H.; Newman, C.P.; Clarkson, G.J.; Rourke, J.P. Platinum(IV) Complexes: C–H Activation at Low Temperatures. Organometallics 2008, 27, 5559–5565. [Google Scholar] [CrossRef]
- Corbo, R.; Georgiou, D.C.; Wilson, D.J.D.; Dutton, J.L. Reactions of [PhI(pyridine)2]2+ with model Pd and Pt II/IV redox couples. Inorg. Chem. 2014, 53, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Lanci, M.P.; Remy, M.S.; Lao, D.B.; Sanford, M.S.; Mayer, J.M. Modulating Sterics in Trimethylplatinum(IV) Diimine Complexes To Achieve C–C Bond-Forming Reductive Elimination. Organometallics 2011, 30, 3704–3707. [Google Scholar] [CrossRef]
- Mutule, I.; Suna, E.; Olofsson, K.; Pelcman, B. Catalytic direct acetoxylation of indoles. J. Org. Chem. 2009, 74, 7195–7198. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.M.; Hickman, A.J.; Sanford, M.S. Platinum-catalyzed C–H arylation of simple arenes. J. Am. Chem. Soc. 2013, 135, 15710–15713. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K. Gold-catalyzed organic reactions. Chem. Rev. 2007, 107, 3180–3211. [Google Scholar] [CrossRef]
- Pflästerer, D.; Hashmi, A.S.K. Gold catalysis in total synthesis—Recent achievements. Chem. Soc. Rev. 2016, 45, 1331–1367. [Google Scholar] [CrossRef] [PubMed]
- Fürstner, A.; Davies, P.W. Catalytic carbophilic activation: Catalysis by platinum and gold pi acids. Angew. Chem. Int. Ed. Engl. 2007, 46, 3410–3449. [Google Scholar] [CrossRef] [PubMed]
- Wegner, H.A.; Auzias, M. Gold for C-C coupling reactions: A Swiss-Army-knife catalyst? Angew. Chem. Int. Ed. 2011, 50, 8236–8247. [Google Scholar] [CrossRef] [PubMed]
- Komiya, S.; Kochi, J.K. Electrophilic Cleavage of Organogold Complexes with Acids. The Mechanism of the Reductive Elimination of Dialkyl(aniono)gold. J. Am. Chem. Soc. 1976, 98, 7599–7607. [Google Scholar] [CrossRef]
- Hopkinson, M.N.; Gee, A.D.; Gouverneur, V. AuI/AuIII Catalysis: An Alternative Approach for C–C Oxidative Coupling. Chem. Eur. J. 2011, 17, 8248–8262. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Horibe, T.; Jacobsen, C.B.; Toste, F.D. Stable Gold(III) catalysts by oxidative addition of a carbon-carbon bond. Nature 2015, 517, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Wolf, W.J.; Winston, M.S.; Toste, F.D. Exceptionally fast carbon-carbon bond reductive elimination from Gold(III). Nat. Chem. 2014, 6, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Winston, M.S.; Wolf, W.J.; Toste, F.D. Halide-Dependent Mechanisms of Reductive Elimination from Gold(III). J. Am. Chem. Soc. 2015, 137, 7921–7928. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K.; Blanco, M.C.; Fischer, D.; Bats, J.W. Gold Catalysis: Evidence for the In-situ Reduction of Gold(III) During the Cyclization of Allenyl Carbinols. Eur. J. Org. Chem. 2006, 2006, 1387–1389. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Ramamurthi, T.D.; Rominger, F. Synthesis, structure and reactivity of organogold compounds of relevance to homogeneous gold catalysis. J. Organomet. Chem. 2009, 694, 592–597. [Google Scholar] [CrossRef]
- Bennett, M.A.; Hockless, D.C.R.; Rae, A.D.; Welling, L.; Willis, A.C. Carbon−Carbon Coupling in Dinuclear Cycloaurated Complexes Containing Bridging 2-(Diphenylphosphino)phenyl or 2-(Diethylphosphino)phenyl. Role of the Axial Ligand and the Fine Balance between Gold(II)–Gold(II) and Gold(I)–Gold(III). Organometallics 2000, 20, 79–87. [Google Scholar] [CrossRef]
- Zamora, F.; Amo-Ochoa, P.; Fischer, B.; Schimanski, A.; Lippert, B. 5,5′-Diuracilyl Species from Uracil and. Angew. Chem. Int. Ed. 1999, 38, 2274–2275. [Google Scholar] [CrossRef]
- Fuchita, Y.; Utsunomiya, Y.; Yasutake, M. Synthesis and reactivity of Arylgold(III) complexes from aromatic hydrocarbons via C–H bond activation. J. Chem. Soc. Dalton Trans. 2001, 2330–2334. [Google Scholar] [CrossRef]
- Constable, E.C.; Sousa, L.R. Metal-ion dependent reactivity of 2-(2′-thienyl)pyridine (Hthpy). J. Organomet. Chem. 1992, 427, 125–139. [Google Scholar] [CrossRef]
- Huynh, H.V.; Guo, S.; Wu, W. Detailed Structural, Spectroscopic, and Electrochemical Trends of Halido Mono- and Bis(NHC) Complexes of Au(I) and Au(III). Organometallics 2013, 32, 4591–4600. [Google Scholar] [CrossRef]
- Pažický, M.; Loos, A.; Ferreira, M.J.; Serra, D.; Vinokurov, N.; Rominger, F.; Jäkel, C.; Hashmi, A.S.K.; Limbach, M. Synthesis, Reactivity, and Electrochemical Studies of Gold(I) and Gold(III) Complexes Supported by N-Heterocyclic Carbenes and Their Application in Catalysis. Organometallics 2010, 29, 4448–4458. [Google Scholar] [CrossRef]
- Gaillard, S.; Nun, P.; Slawin, A.M.Z.; Nolan, S.P. Expeditious Synthesis of [Au(NHC)(L)]+ (NHC = N-Heterocyclic Carbene; L = Phosphine or NHC) Complexes. Organometallics 2010, 29, 5402–5408. [Google Scholar] [CrossRef]
- Hofer, M.; Nevado, C. Unexpected Outcomes of the Oxidation of (Pentafluorophenyl)triphenylphosphanegold(I). Eur. J. Inorg. Chem. 2012, 2012, 1338–1341. [Google Scholar] [CrossRef]
- Ghidiu, M.J.; Pistner, A.J.; Yap, G.P.A.; Lutterman, D.A.; Rosenthal, J. Thermal versus Photochemical Reductive Elimination of Aryl Chlorides from NHC–Gold Complexes. Organometallics 2013, 32, 5026–5029. [Google Scholar] [CrossRef]
- Orbisaglia, S.; Jacques, B.; Braunstein, P.; Hueber, D.; Pale, P.; Blanc, A.; de Frémont, P. Synthesis, Characterization, and Catalytic Activity of Cationic NHC Gold(III) Pyridine Complexes. Organometallics 2013, 32, 4153–4164. [Google Scholar] [CrossRef]
- Corbo, R.; Pell, T.P.; Stringer, B.D.; Hogan, C.F.; Wilson, D.J.D.; Barnard, P.J.; Dutton, J.L. Facile formation of homoleptic Au(III) trications via simultaneous oxidation and ligand delivery from [PhI(pyridine)2](2+). J. Am. Chem. Soc. 2014, 136, 12415–12421. [Google Scholar] [CrossRef] [PubMed]
- Roşca, D.-A.; Smith, D.A.; Bochmann, M. Cyclometallated Gold(III) hydroxides as versatile synthons for Au-N, Au-C complexes and luminescent compounds. Chem. Commun. (Camb.) 2012, 48, 7247–7249. [Google Scholar] [CrossRef] [PubMed]
- Kar, A.; Mangu, N.; Kaiser, H.M.; Beller, M.; Tse, M.K. A general gold-catalyzed direct oxidative coupling of non-activated arenes. Chem. Commun. (Camb.) 2008, 106, 386–388. [Google Scholar] [CrossRef]
- Kar, A.; Mangu, N.; Kaiser, H.M.; Tse, M.K. Gold-catalyzed direct oxidative coupling reactions of non-activated arenes. J. Organomet. Chem. 2009, 694, 524–537. [Google Scholar] [CrossRef]
- Cambeiro, X.C.; Boorman, T.C.; Lu, P.; Larrosa, I. Redox-controlled selectivity of C–H activation in the oxidative cross-coupling of arenes. Angew. Chem. Int. Ed. 2013, 52, 1781–1784. [Google Scholar] [CrossRef] [PubMed]
- Ball, L.T.; Lloyd-Jones, G.C.; Russell, C.A. Gold-catalyzed direct arylation. Science 2012, 337, 1644–1648. [Google Scholar] [CrossRef] [PubMed]
- Hofer, M.; Nevado, C. Cross-coupling of arene–Gold(III) complexes. Tetrahedron 2013, 69, 5751–5757. [Google Scholar] [CrossRef]
- Haro, T.D.; Nevado, C. Gold-Catalyzed Ethynylation of Arenes. J. Am. Chem. Soc. 2010, 132, 1512–1513. [Google Scholar] [CrossRef] [PubMed]
- Brand, J.P.; Charpentier, J.; Waser, J. Direct alkynylation of indole and pyrrole heterocycles. Angew. Chem. Int. Ed. 2009, 48, 9346–9349. [Google Scholar] [CrossRef] [PubMed]
- Brand, J.P.; Li, Y.; Waser, J. Gold-Catalyzed Alkynylation: Acetylene-Transfer instead of Functionalization. Isr. J. Chem. 2013, 53, 901–910. [Google Scholar] [CrossRef]
- Brand, J.P.; Waser, J. Direct alkynylation of thiophenes: Cooperative activation of TIPS-EBX with gold and Brønsted acids. Angew. Chem. Int. Ed. 2010, 49, 7304–7307. [Google Scholar] [CrossRef] [PubMed]
- Brand, J.P.; Waser, J. Para-Selective Gold-Catalyzed Direct Alkynylation of Anilines. Org. Lett. 2012, 14, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Brand, J.P.; Waser, J. Gold-catalyzed regioselective synthesis of 2- and 3-alkynyl furans. Angew. Chem. Int. Ed. Engl. 2013, 52, 6743–6747. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, A.; Muñiz, K. Oxidative Interception of the Hydroamination Pathway: A Gold-Catalyzed Diamination of Alkenes. Chem. Eur. J. 2009, 15, 10563–10569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-H.; Dai, L.-Z.; Shi, M. C(sp3)–C(sp3) Bond Breaking in Methylenecyclopropanes Involving a AuI/AuIII Catalytic Cycle. Eur. J. Org. Chem. 2010, 2010, 5454–5459. [Google Scholar] [CrossRef]
- Ananikov, V.P. Nickel: The “Spirited Horse” of Transition Metal Catalysis. ACS Catal. 2015, 5, 1964–1971. [Google Scholar] [CrossRef]
- Klein, H.-F.; Bickelhaupt, A.; Jung, T.; Cordier, G. Syntheses and Properties of the First Octahedral Diorganonickel(IV) Compounds. Organometallics 1994, 13, 2557–2559. [Google Scholar] [CrossRef]
- Shimada, S.; Rao, M.; Tanaka, M. Reaction of 1,2-Disilylbenzene with Bis[1,2-bis(dimethylphosphino)ethane]nickel(0). Isolation and Characterization of the First Silylnickel(IV) Complex—Organometallics (ACS Publications). Organometallics 1999, 18, 291–293. [Google Scholar] [CrossRef]
- Dimitrov, V.; Linden, A. A pseudotetrahedral, high-oxidation-state organonickel compound: Synthesis and structure of bromotris(1-norbornyl)nickel(IV). Angew. Chem. Int. Ed. Engl. 2003, 42, 2631–2633. [Google Scholar] [CrossRef] [PubMed]
- Carnes, M.; Buccella, D.; Chen, J.Y.-C.; Ramirez, A.P.; Turro, N.J.; Nuckolls, C.; Steigerwald, M. A stable tetraalkyl complex of nickel(IV). Angew. Chem. Int. Ed. 2009, 48, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Higgs, A.T.; Zinn, P.J.; Simmons, S.J.; Sanford, M.S. Oxidatively Induced Carbon–Halogen Bond-Forming Reactions at Nickel. Organometallics 2009, 28, 6142–6144. [Google Scholar] [CrossRef]
- Higgs, A.T.; Zinn, P.J.; Sanford, M.S. Synthesis and Reactivity of NiII(Phpy)2 (Phpy = 2-Phenylpyridine). Organometallics 2010, 29, 5446–5449. [Google Scholar] [CrossRef]
- Camasso, N.M.; Sanford, M.S. Design, synthesis, and carbon-heteroatom coupling reactions of organometallic Nickel(IV) complexes. Science 2015, 347, 1218–1220. [Google Scholar] [CrossRef] [PubMed]
- Bour, J.R.; Camasso, N.M.; Sanford, M.S. Oxidation of Ni(II) to Ni(IV) with Aryl Electrophiles Enables Ni-Mediated Aryl-CF3 Coupling. J. Am. Chem. Soc. 2015, 137, 8034–8037. [Google Scholar] [CrossRef] [PubMed]
- Meucci, E.A.; Camasso, N.M.; Sanford, M.S. An Organometalllic Ni(IV) Complex That Participates in Competing Transmetalation and C(sp2)–O Bond-Forming Reductive Elimination Reactions. Organometallics 2017, 36, 247–250. [Google Scholar] [CrossRef]
- Martinez, G.E.; Ocampo, C.; Park, Y.J.; Fout, A.R. Accessing Pincer Bis(carbene) Ni(IV) Complexes from Ni(II) via Halogen and Halogen Surrogates. J. Am. Chem. Soc. 2016, 138, 4290–4293. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.J.; Powers, D.C.; Maher, A.G.; Anderson, B.L.; Hadt, R.G.; Zheng, S.-L.; Chen, Y.-S.; Nocera, D.G. Trap-Free Halogen Photoelimination from Mononuclear Ni(III) Complexes. J. Am. Chem. Soc. 2015, 137, 6472–6475. [Google Scholar] [CrossRef] [PubMed]
- Yokota, A.; Aihara, Y.; Chatani, N. Nickel(II)-Catalyzed Direct Arylation of C–H Bonds in Aromatic Amides Containing an 8-Aminoquinoline Moiety as a Directing Group. J. Org Chem. 2014, 79, 11922–11932. [Google Scholar] [CrossRef] [PubMed]
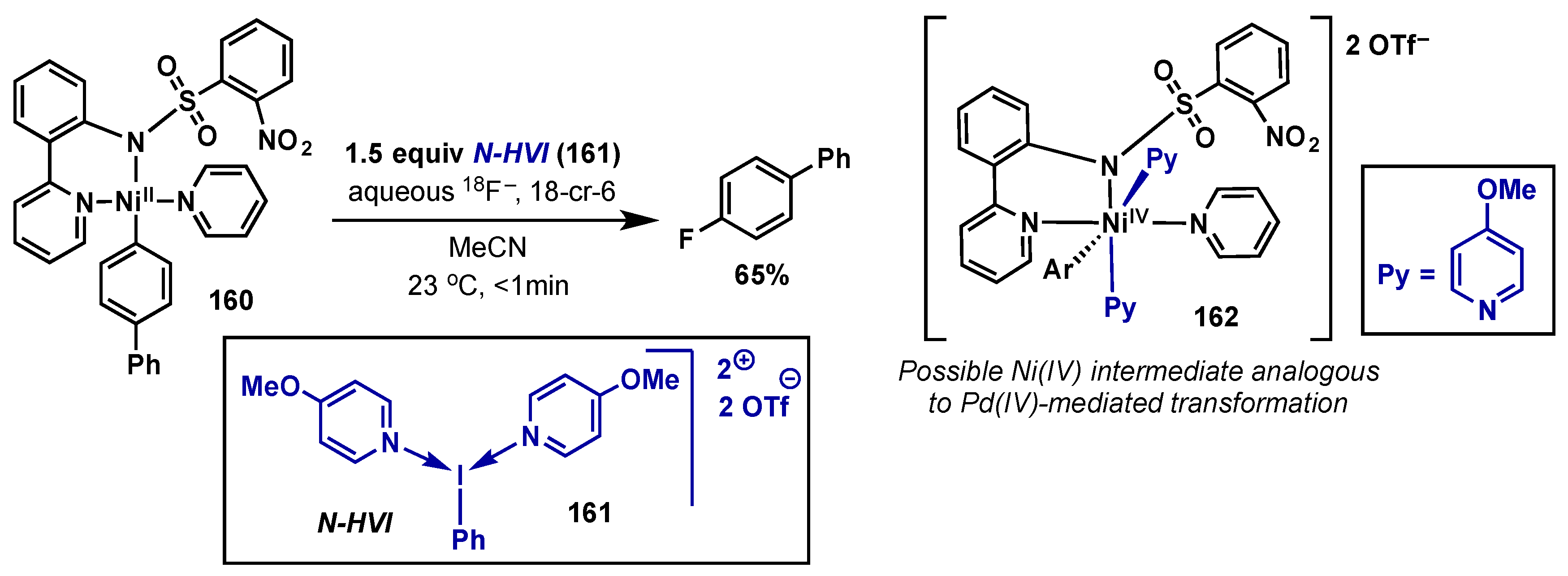
- Lee, E.; Hooker, J.M.; Ritter, T. Nickel-Mediated Oxidative Fluorination for PET with Aqueous [18F] Fluoride. J. Am. Chem. Soc. 2012, 134, 17456–17458. [Google Scholar] [CrossRef] [PubMed]
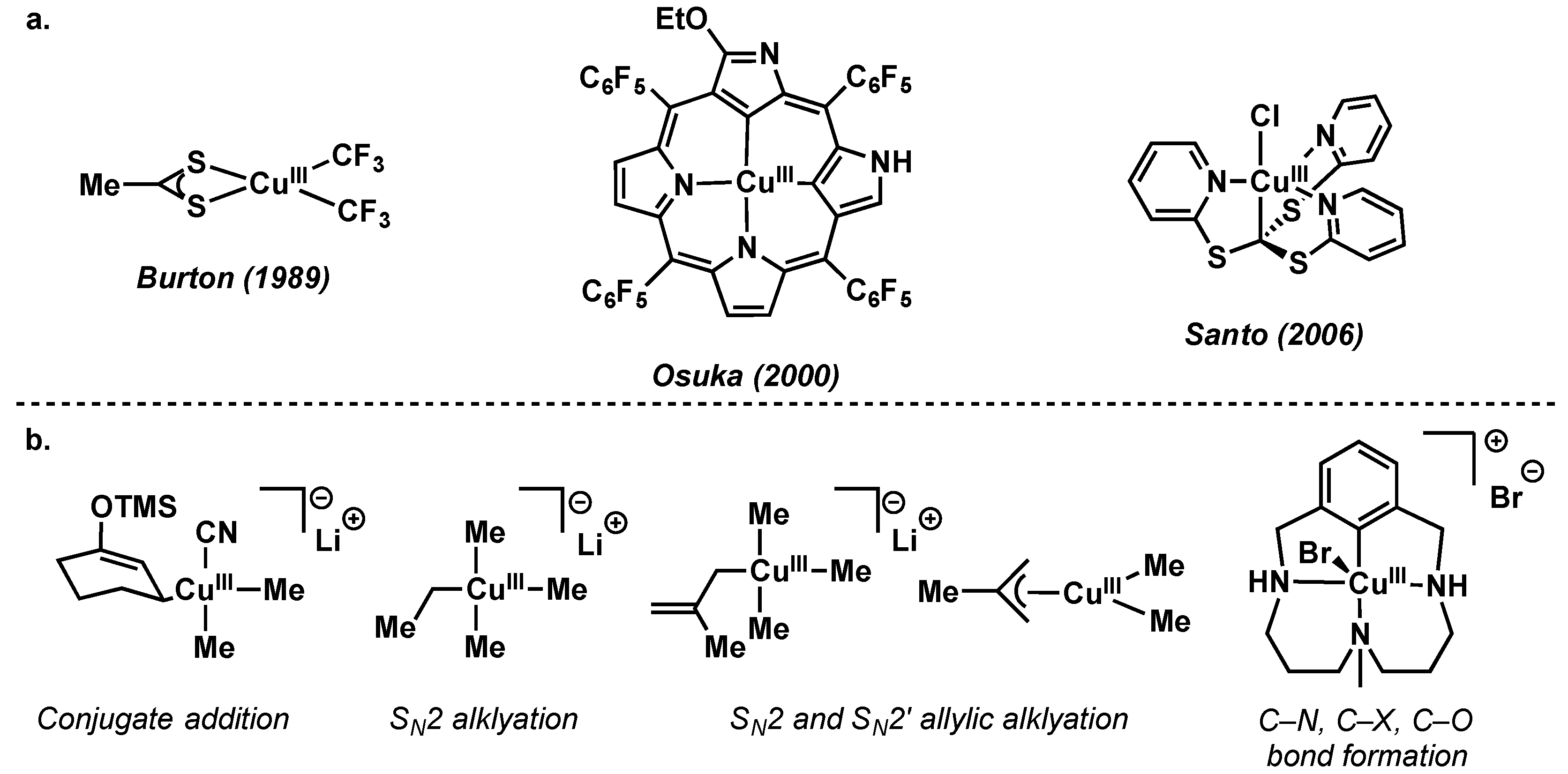
- Willert-Porada, M.A.; Burton, D.J.; Baenziger, N.C. Synthesis and X-ray structure of bis(trifluoromethyl)(N, N-diethyldithiocarbamato)-copper; a remarkably stable perfluoroalkylcopper(III) complex. J. Chem. Soc. Chem. Commun. 1989, 1633–1634. [Google Scholar] [CrossRef]
- Furuta, H.; Maeda, H.; Osuka, A. Doubly N-Confused Porphyrin: A New Complexing Agent Capable of Stabilizing Higher Oxidation States. J. Am. Chem. Soc. 2000, 122, 803–807. [Google Scholar] [CrossRef]
- Santo, R.; Miyamoto, R.; Tanaka, R.; Nishioka, T.; Sato, K.; Toyota, K.; Obata, M.; Yano, S.; Kinoshita, I.; Ichimura, A.; et al. Diamagnetic-paramagnetic conversion of tris(2-pyridylthio)methylcopper(III) through a structural change from trigonal bipyramidal to octahedral. Angew. Chem. Int. Ed. Engl. 2006, 45, 7611–7614. [Google Scholar] [CrossRef] [PubMed]
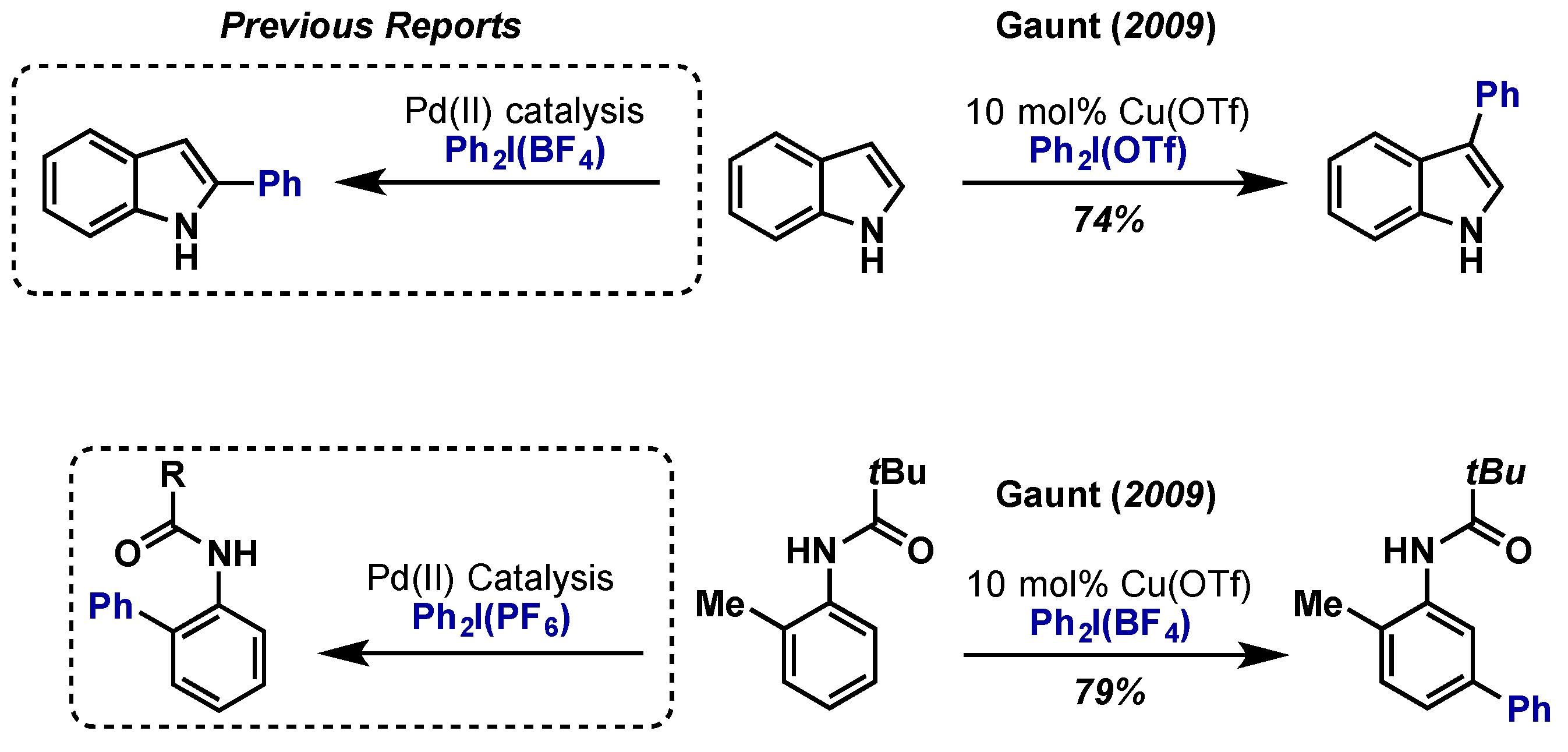
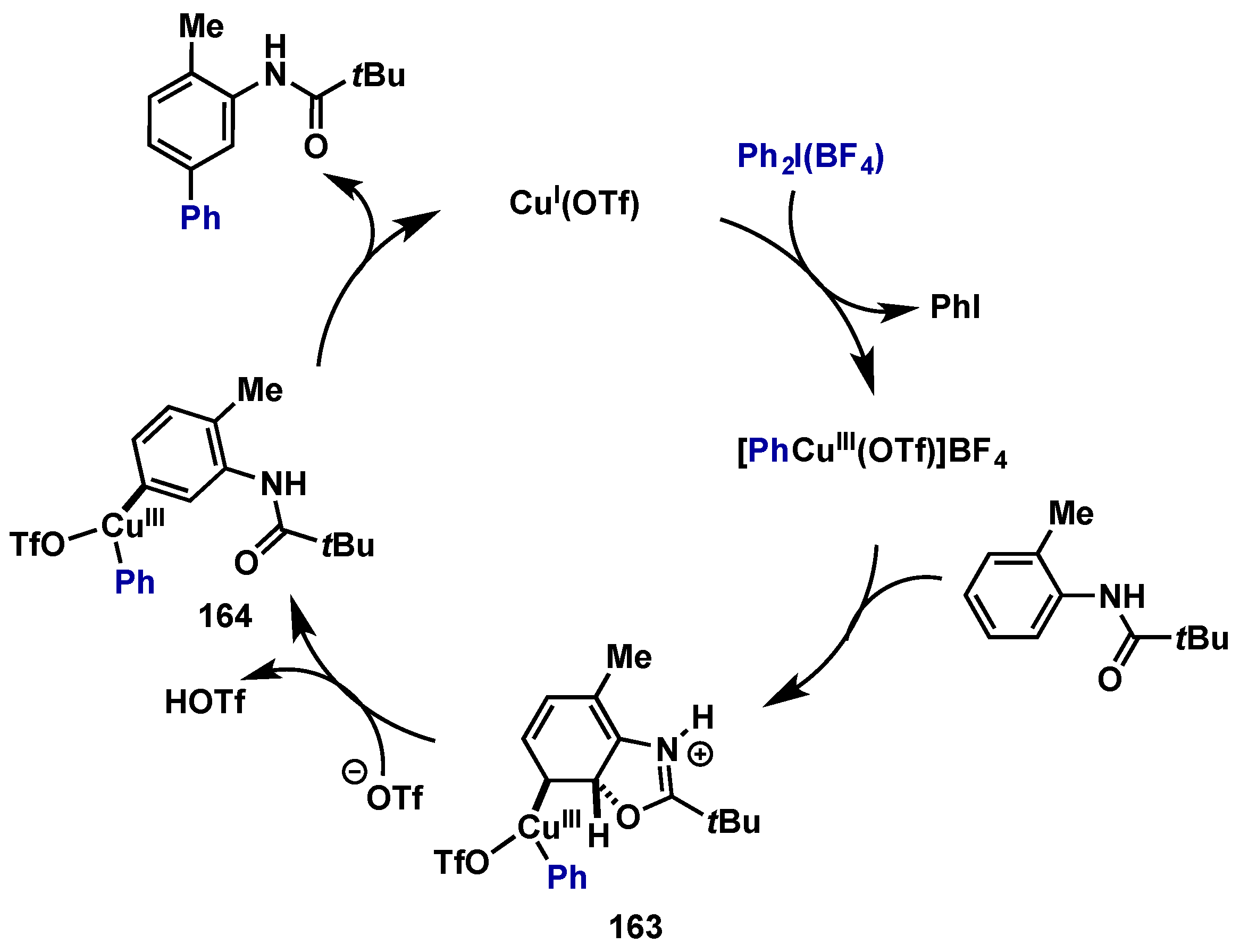
- Phipps, R.J.; Gaunt, M.J. A meta-selective copper-catalyzed C–H bond arylation. Science 2009, 323, 1593–1597. [Google Scholar] [CrossRef] [PubMed]
- Phipps, R.J.; Grimster, N.P.; Gaunt, M.J. Cu(II)-catalyzed direct and site-selective arylation of indoles under mild conditions. J. Am. Chem. Soc. 2008, 130, 8172–8174. [Google Scholar] [CrossRef] [PubMed]
- Parsons, A.T.; Buchwald, S.L. Copper-Catalyzed Trifluoromethylation of Unactivated Olefins. Angew. Chem. Int. Ed. 2011, 50, 9120–9123. [Google Scholar] [CrossRef] [PubMed]
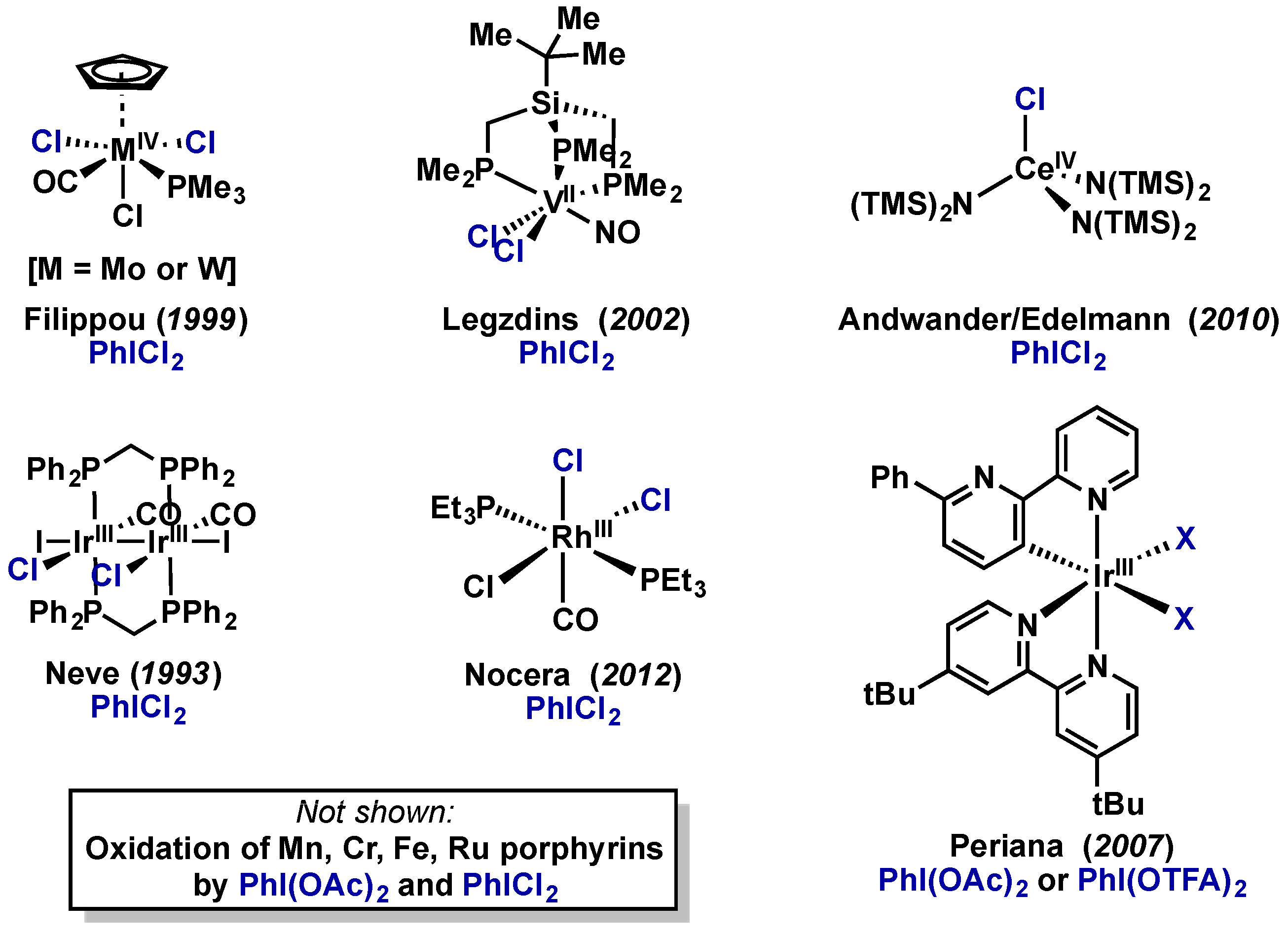
- Filippou, A.C.; Winter, J.G.; Kociok-Köhn, G.; Troll, C. Electron-Rich Trichlorogermyl Complexes of Molybdenum and Tungsten Bearing a Cyclopentadienyl Ligand: Synthesis, Crystal Structures, and Cyclic Voltammetric Studies. Organometallics 1999, 18, 2649–2659. [Google Scholar] [CrossRef]
- Hayton, T.W.; Legzdins, P.; Patrick, B.O. Differing reactivities of (trimpsi)M(CO)(2)(NO) complexes [M = V, Nb, Ta; trimpsi = (t)BuSi(CH(2)PMe(2))(3)] with halogens and halogen sources. Inorg. Chem. 2002, 41, 5388–5396. [Google Scholar] [CrossRef] [PubMed]
- Dröse, P.; Crozier, A.R.; Lashkari, S.; Gottfriedsen, J.; Blaurock, S.; Hrib, C.G.; Maichle-Mössmer, C.; Schädle, C.; Anwander, R.; Edelmann, F.T. Facile access to tetravalent cerium compounds: One-electron oxidation using iodine(III) reagents. J. Am. Chem. Soc. 2010, 132, 14046–14047. [Google Scholar] [CrossRef] [PubMed]
- Crispini, A.; Ghedini, M.; Neve, F. Synthesis and structure of the dimer Ir2Cl2I2(CO)2(μ-dppm )2]. Inorg. Chim. Acta 1993, 209, 235–237. [Google Scholar] [CrossRef]
- Teets, T.S.; Nocera, D.G. Oxygen reduction reactions of monometallic rhodium hydride complexes. Inorg. Chem. 2012, 51, 7192–7201. [Google Scholar] [CrossRef] [PubMed]
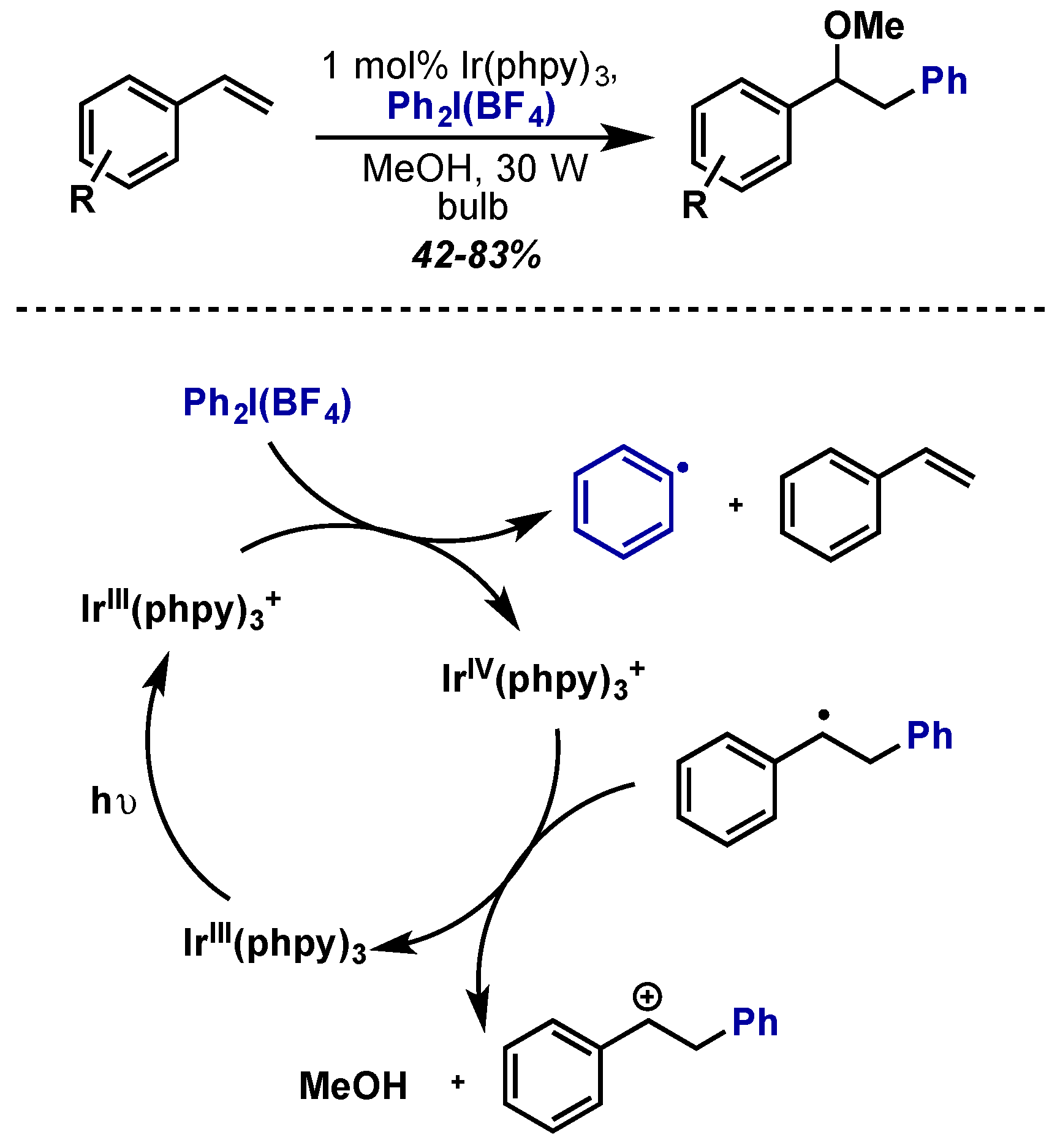
- Young, K.J.H.; Mironov, O.A.; Periana, R.A. Stoichiometric Oxy Functionalization and CH Activation Studies of Cyclometalated Iridium(III) 6-Phenyl-2,2′-Bipyridine Hydrocarbyl Complexes. Organometallics 2007, 26, 2137–2140. [Google Scholar] [CrossRef]
- Li, Z. Oxidation of hydrocarbons with iodobenzene diacetate catalyzed by manganese(III) porphyrins in a room temperature ionic liquid. J. Mol. Catal. A Chem. 2004, 214, 95–101. [Google Scholar] [CrossRef]
- Karimipour, G.R.; Shadegan, H.A.; Ahmadpour, R. Clean and highly selective oxidation of alcohols by the PhI(OAc)2/Mn(TPP)CN/Im catalytic system. J. Chem. Res. 2007, 2007, 252–256. [Google Scholar] [CrossRef]
- In, J.-H.; Park, S.-E.; Song, R.; Nam, W. Iodobenzene diacetate as an efficient terminal oxidant in Iron(III) porphyrin complex-catalyzed oxygenation reactions. Inorg. Chim. Acta 2003, 343, 373–376. [Google Scholar] [CrossRef]
- Fumagalli, G.; Boyd, S.; Greaney, M.F. Oxyarylation and aminoarylation of styrenes using photoredox catalysis. Org. Lett. 2013, 15, 4398–4401. [Google Scholar] [CrossRef] [PubMed]
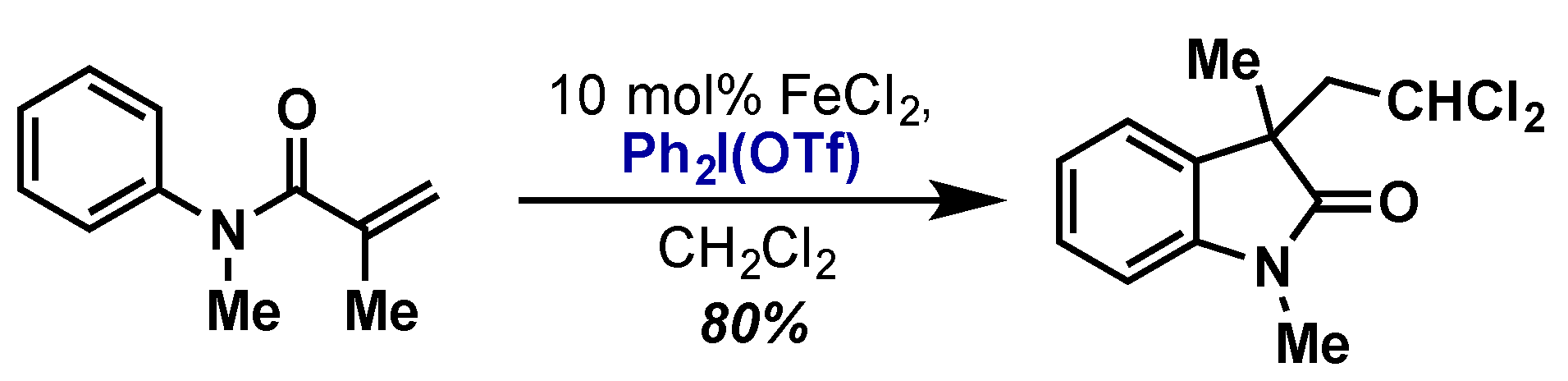
- Lu, M.-Z.; Loh, T.-P. Iron-catalyzed cascade carbochloromethylation of activated alkenes: Highly efficient access to chloro-containing oxindoles. Org. Lett. 2014, 16, 4698–4701. [Google Scholar] [CrossRef] [PubMed]









































































© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sousa e Silva, F.C.; Tierno, A.F.; Wengryniuk, S.E. Hypervalent Iodine Reagents in High Valent Transition Metal Chemistry. Molecules 2017, 22, 780. https://doi.org/10.3390/molecules22050780
Sousa e Silva FC, Tierno AF, Wengryniuk SE. Hypervalent Iodine Reagents in High Valent Transition Metal Chemistry. Molecules. 2017; 22(5):780. https://doi.org/10.3390/molecules22050780
Chicago/Turabian StyleSousa e Silva, Felipe Cesar, Anthony F. Tierno, and Sarah E. Wengryniuk. 2017. "Hypervalent Iodine Reagents in High Valent Transition Metal Chemistry" Molecules 22, no. 5: 780. https://doi.org/10.3390/molecules22050780
APA StyleSousa e Silva, F. C., Tierno, A. F., & Wengryniuk, S. E. (2017). Hypervalent Iodine Reagents in High Valent Transition Metal Chemistry. Molecules, 22(5), 780. https://doi.org/10.3390/molecules22050780