
11C-Labeling of Aryl Ketones as Candidate Histamine Subtype-3 Receptor PET Radioligands through Pd(0)-Mediated 11C-Carbonylative Coupling



Abstract
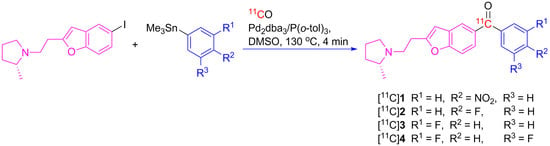
:
1. Introduction
2. Results and Discussion
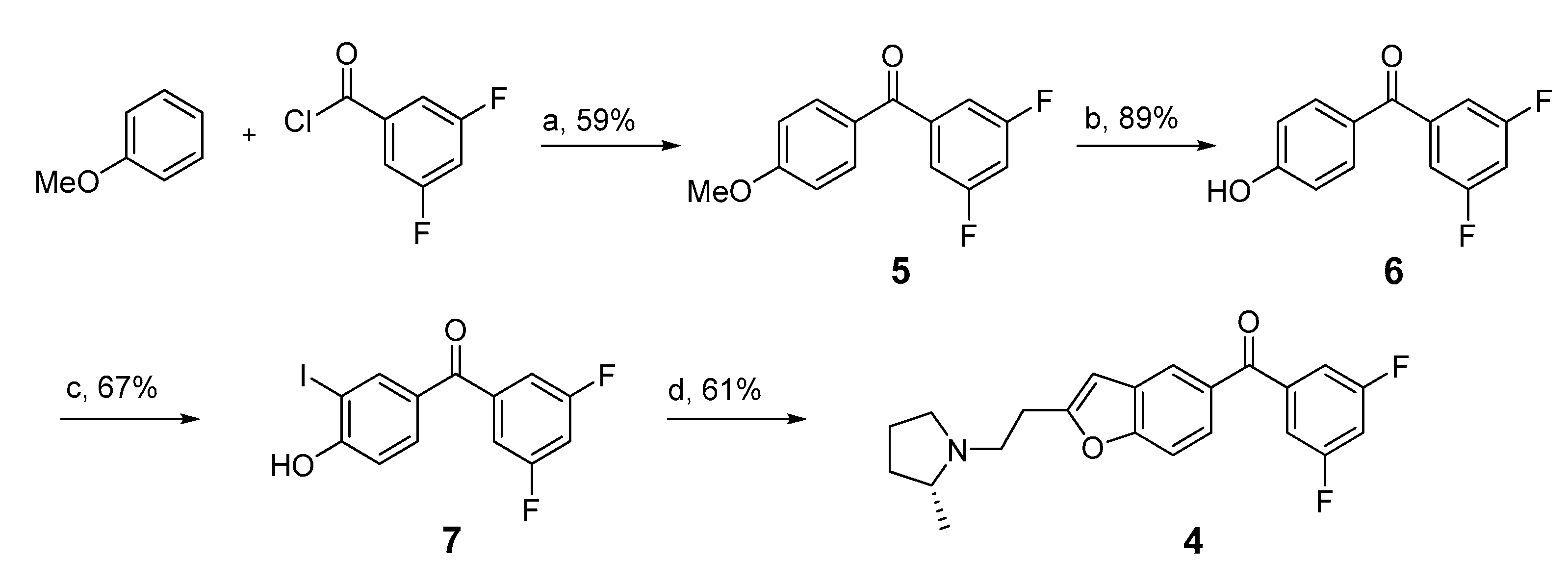
2.1. Lead Compound Selection and Synthesis of Non-Radioactive Standards
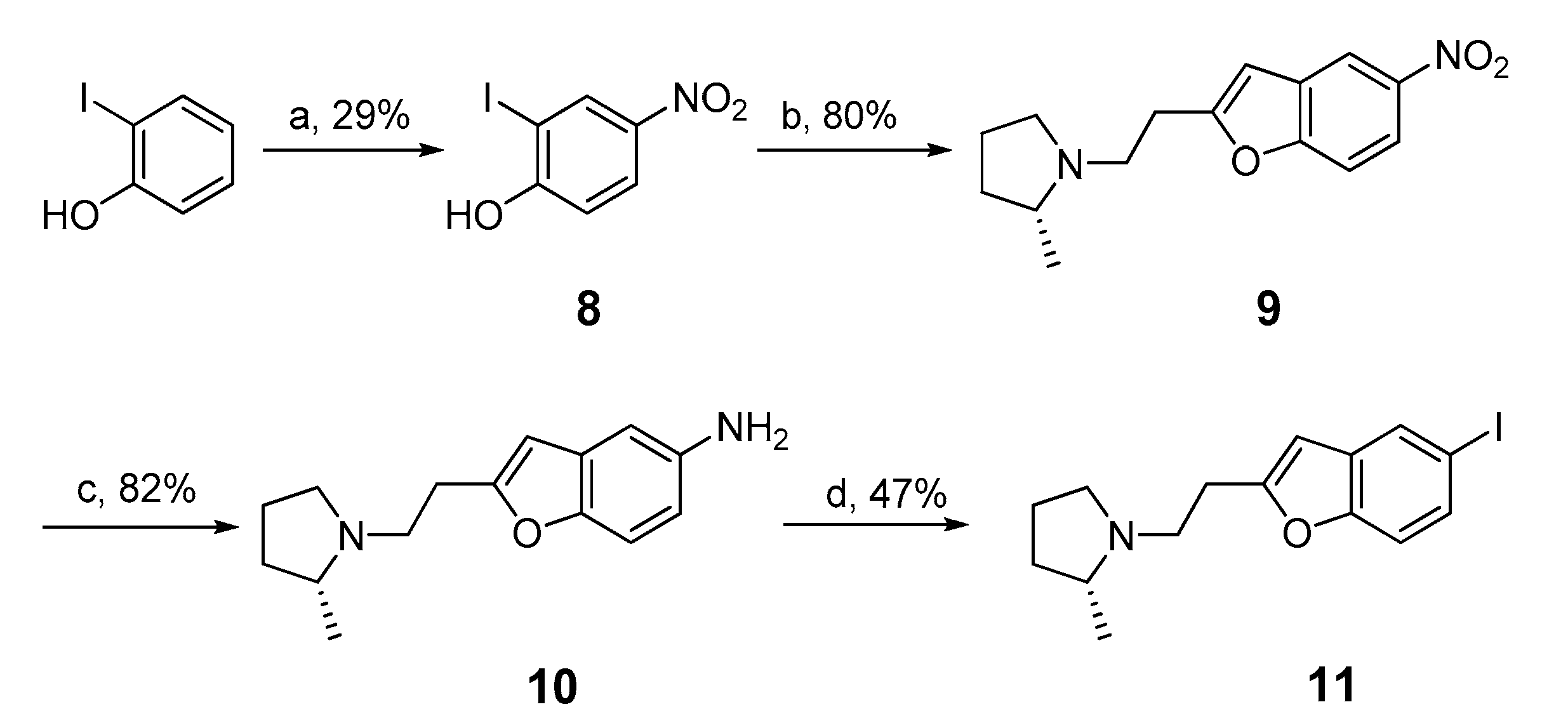
2.2. Synthesis of Iodo-Precursor (11) and Stannyl Derivatives (12–15)
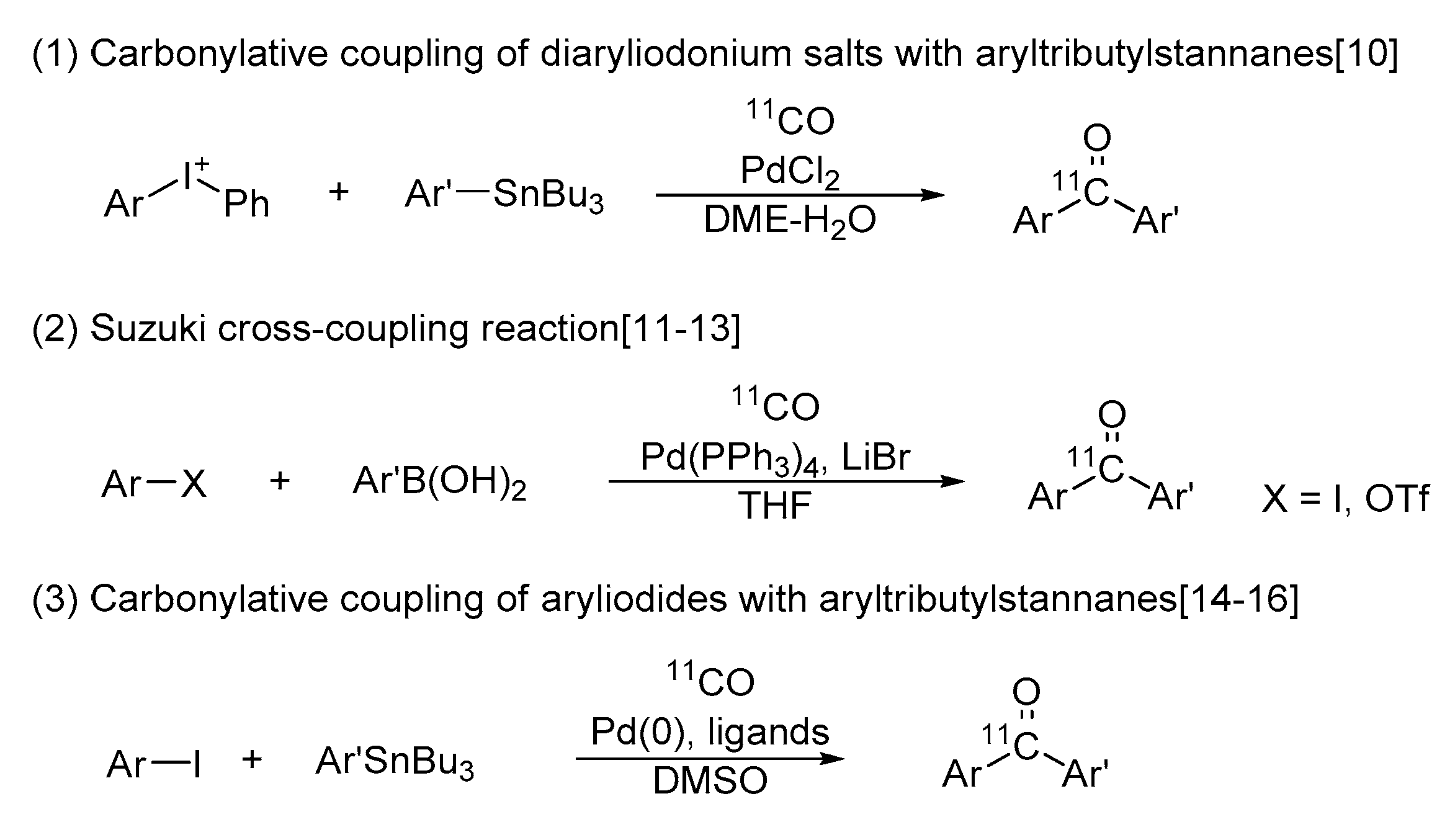
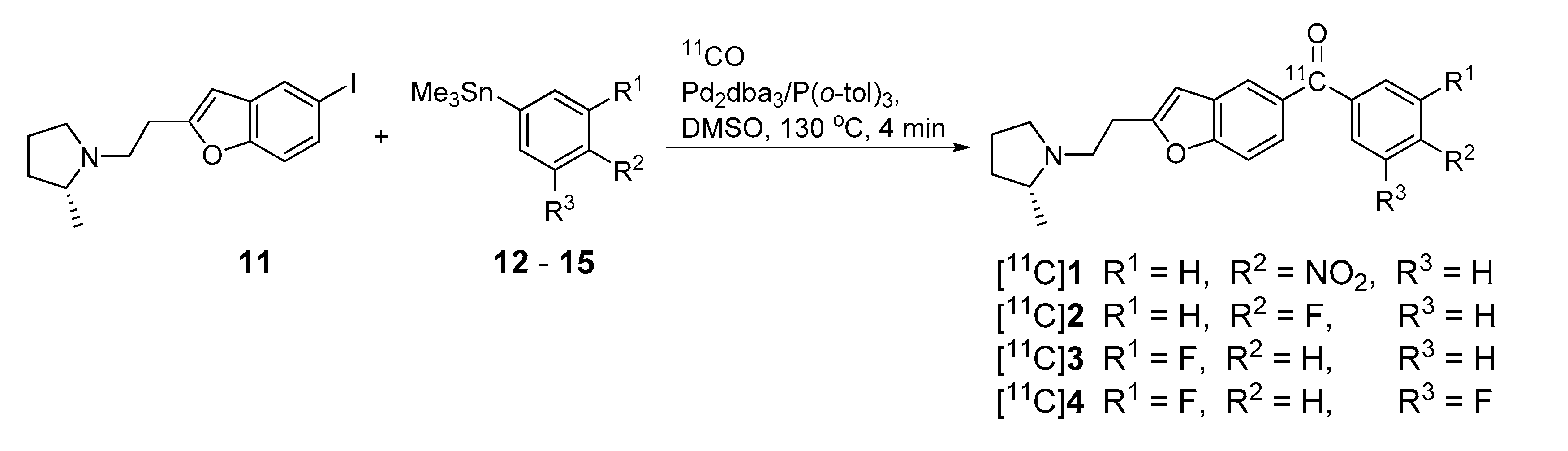
2.3. Radiosynthesis
3. Materials and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Esbenshade, T.A.; Browman, K.E.; Bitner, R.S.; Strakhova, M.; Cowart, M.D.; Brioni, J.D. The histamine H3 receptor: An attractive target for the treatment of cognitive disorders. Br. J. Pharm. 2008, 154, 1166–1181. [Google Scholar] [CrossRef] [PubMed]
- Arrang, J.M.; Garbarg, M.; Schwartz, J.C. Auto-inhibition of brain histamine-release mediated by a novel class (H-3) of histamine-receptor. Nature 1983, 302, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Raddatz, R.; Tao, M.; Hudkins, R.L. Histamine H3 antagonists for treatment of cognitive deficits in CNS diseases. Curr. Top. Med. Chem. 2010, 10, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Berlin, M.; Boyce, C.W.; de Lera Ruiz, M. Histamine H3 receptor as a drug discovery target. J. Med. Chem. 2011, 54, 26–53. [Google Scholar] [CrossRef] [PubMed]
- Plisson, C.; Gunn, R.N.; Cunningham, V.J.; Bender, D.; Salinas, C.A.; Medhurst, A.D.; Roberts, J.C.; Laruelle, M.; Gee, A.D. 11C-GSK189254: A selective radioligand for in vivo central nervous system imaging of H3 receptors by PET. J. Nuclear Med. 2009, 50, 2064–2072. [Google Scholar] [CrossRef] [PubMed]
- Hamill, T.G.; Sato, N.; Jitsuoka, M.; Tokita, S.; Sanabria, S.; Eng, W.; Ryan, C.; Krause, S.; Takenaga, N.; Patel, S.; et al. Inverse agonist histamine H3 receptor PET tracers labelled with carbon-11 or fluorine-18. Synapse 2009, 63, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Lu, S.; Liow, J.-S.; Zoghbi, S.S.; Jenko, K.J.; Clark, D.T.; Gladding, R.L.; Innis, R.B.; Pike, V.W. Radiosynthesis and evaluation of an 18F-labeled positron emission tomography (PET) radioligand for brain histamine subtype-3 receptors based on a nonimidazole 2-aminoethylbenzofuran chemotype. J. Med. Chem. 2012, 55, 2406–2415. [Google Scholar] [CrossRef] [PubMed]
- Schou, M.; Varnas, K.; Jureus, A.; Ahlgren, C.; Malmquist, J.; Haggkvist, J.; Tari, L.; Wesolowski, S.S.; Throner, S.R.; Brown, D.G.; et al. Discovery and preclinical validation of [11C]AZ13153556, a novel probe for the histamine type 3 receptor. ACS Chem. Neurosci. 2016, 7, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Funke, U.; Vugts, D.J.; Janssen, B.; Spaans, A.; Kruijer, P.S.; Lammertsma, A.A.; Perk, L.R.; Windhorst, A.D. 11C-labeled and 18F-labeled PET ligands for subtype-specific imaging of histamine receptors in the brain. J. Label Compd. Radiopharm. 2013, 56, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Al-Qahtani, M.H.; Pike, V.W. Palladium(II)-mediated 11C-carbonylative coupling of diaryliodonium salts with organostannanes—A new, mild and rapid synthesis of aryl [11C]ketones. J. Chem. Soc. Perkin Trans. 1 2000, 31, 1033–1036. [Google Scholar] [CrossRef]
- Dahl, K.; Itsenko, O.; Rahman, O.; Ulin, J.; Sjöberg, C.O.; Sandblom, P.; Larsson, L.A.; Schou, M.; Halldin, C. An evaluation of a high-pressure 11CO carbonylation apparatus. J. Label Compd. Radiopharm. 2015, 58, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Rahman, O.; Llop, J.; Långström, B. Organic bases as additives to improve the radiochemical yields of [C-11]ketones prepared by the Suzuki coupling reaction. Eur. J. Org. Chem. 2004, 12, 2674–2678. [Google Scholar] [CrossRef]
- Zeisler, S.K.; Nader, M.; Theobald, A.; Oberdorfer, F. Conversion of no-carrier-added [11C]carbon dioxide to [11C]carbon monoxide on molybdenum for the synthesis of 11C-labelled aromatic ketones. Appl. Radiat. Isot. 1997, 48, 1091–1095. [Google Scholar] [CrossRef]
- Karimi, F.; Barletta, J.; Långström, B. Palladium-mediated 11C-carbonylative cross-coupling of alkyl/aryl iodides with organostannanes: An efficient synthesis of unsymmetrical alkyl/aryl [11C-carbonyl]ketones. Eur. J. Org. Chem. 2005, 11, 2374–2378. [Google Scholar] [CrossRef]
- Al-Qahtani, M.H.; Pike, V.W. Rapid mild syntheses of [11C]benzophenones by Pd(0)-catalysed 11C-carbonylative coupling of iodoarenes with phenyltributylstannane in DME-water. J. Label. Compd. Radiopharm. 2000, 43, 825–835. [Google Scholar] [CrossRef]
- Siméon, F.G.; Lu, S.Y.; Culligan, W.J.; Jin., Y.Y.; Bao, X.F.; Pike, V.W. Generic syntheses of candidate 11C-labeled brain histamine subtype 3 receptor radioligands. J. Label. Compd. Radiopharm. 2013, 56, S78. [Google Scholar]
- Nader, M.W.; Oberdorfer, F. Syntheses of [carbonyl-11C]2-(2-benzoylphenoxy)-N-phenylacetamide from [11C]carbon monoxide by the Suzuki and the Stille reactions. Appl. Radiat. Isot. 2002, 57, 681–685. [Google Scholar] [CrossRef]
- Cowart, M.D.; Faghih, R.; Curtis, M.P.; Gfesser, G.A.; Bennani, Y.L.; Black, L.A.; Pan, L.; Marsh, K.C.; Sullivan, J.P.; Esbenshade, T.A.; et al. 4-(2-[2-(2(R)-methylpyrrolidin-1-yl)ethyl]benzofuran-5-yl)benzonitrile and related 2-aminoethylbenzofuran H-3 receptor antagonists potently enhance cognition and attention. J. Med. Chem. 2005, 48, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Pike, V.W. PET radiotracers: Crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol. Sci. 2009, 30, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Pike, V.W. Considerations in the development of reversibly binding PET radioligands for brain imaging. Curr. Med. Chem. 2016, 23, 1818–1869. [Google Scholar] [CrossRef] [PubMed]
- Chancellor, D.R.; Davies, K.E.; De Moor, O.; Dorgan, C.R.; Johnson, P.D.; Lambert, A.G.; Lawrence, D.; Lecci, C.; Maillol, C.; Middleton, P.J.; et al. Discovery of 2-arylbenzoxazoles as upregulators of utrophin production for the treatment of Duchenne Muscular Dystrophy. J. Med. Chem. 2011, 54, 3241–3250. [Google Scholar] [CrossRef] [PubMed]
- Kundu, N.G.; Pal, M.; Mahanty, J.S.; De, M. Palladium-catalysed heteroannulation with acetylenic compounds: Synthesis of benzofurans. J. Chem. Soc. Perkin Trans 1 1997, 19, 2815–2820. [Google Scholar] [CrossRef]
- Krasnokutskaya, E.A.; Semenischeva, N.I.; Filimonov, V.D.; Knochel, P. A new, one-step, effective protocol for the iodination of aromatic and heterocyclic compounds via aprotic diazotization of amines. Synth. Stuttg. 2007, 1, 81–84. [Google Scholar] [CrossRef]
- Bumagin, N.A.; Bumagina, I.G.; Beletskaya, I.P. Synthesis of aryltrimethyltins by the reaction of Me3SnSnMe3 with ArI, catalyzed by ligand-free palladium. Doklady Akademii Nauk SSSR 1984, 274, 53–55. [Google Scholar]
- Huang, H.H.; Hui, K.M. Organotin compounds. I. Electric dipole moments of some trimethylphenyltin derivatives. J. Organometal. Chem. 1966, 6, 504–514. [Google Scholar] [CrossRef]
- Eaborn, C.; Treverto, J.A.; Walton, D.R.M. Aromatic reactivity 34. Acid cleavage of (pentafluorophenyl)-trimethyl-stannane and –silane. J. Organometal. Chem. 1967, 9, 259–262. [Google Scholar] [CrossRef]
- Okitsu, T.; Ogasahara, M.; Wada, A. Convergent synthesis of Dronedarone, an antiarrhythmic agent. Chem. Pharm. Bull. 2016, 64, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Hong, J.S.; Morse, C.L.; Pike, V.W. Synthesis, structure-affinity relationships, and radiolabeling of selective high-affinity 5-HT4 receptor ligands as prospective imaging probes for positron emission tomography. J. Med. Chem. 2010, 53, 7035–7047. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Meng, H.; Jin, L.; Wang, S.; Tang, S.B.; Wang, X.; Mo, F.Y.; Zhang, Y.; Wang, J.B. Synthesis of Aryl Trimethylstannanes from Aryl Amines: A Sandmeyer-type stannylation reaction. Angew. Chem. Int. Ed. 2013, 52, 11581–11584. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.Y.; Hong, J.S.; Itoh, T.; Fujita, F.; Inoue, O.; Innis, R.B.; Pike, V.W. [carbonyl-11C]Benzyl acetate: Automated radiosynthesis via Pd-mediated [11C]carbon monoxide chemistry and PET measurement of brain uptake in monkey. J. Label. Compd. Radiopharm. 2010, 53, 548–551. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Ligand | Ar | Affinity (Ki, nM) | cLogD | |||
| H3 (lit) a | H3 b | H2 c | H1 c | |||
| 1 | 4-NO2-C6H4 | - | 2.1 | 706 | 1030 | 2.69 |
| 2 | 4-F-C6H4 | 0.27 | 4.2 | 923 | 7055 | 2.90 |
| 3 | 3-F-C6H4 | 0.10 | 4.9 | 1368 | >10,000 | 2.79 |
| 4 | 3,5-di-F-C6H3 | 0.09 | 2.9 | 630 | >10,000 | 2.75 |
| Radioligand | Yields a (%) | Molar activity at EOS b (GBq/µmol) | Prep-HPLC Method | tR (min) |
|---|---|---|---|---|
| [11C]1 | 4.9 (n = 10) | 293 ± 84 | A | 13.1 |
| [11C]2 | 6.8 (n = 5) | 115 ± 45 | A | 14.4 |
| [11C]3 | 6.1 (n = 4) | 420 ± 60 | B | 14.3 |
| [11C]4 | 8.6 (n = 3) | 398 ± 141 | B | 15.1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siméon, F.G.; Culligan, W.J.; Lu, S.; Pike, V.W. 11C-Labeling of Aryl Ketones as Candidate Histamine Subtype-3 Receptor PET Radioligands through Pd(0)-Mediated 11C-Carbonylative Coupling. Molecules 2017, 22, 792. https://doi.org/10.3390/molecules22050792
Siméon FG, Culligan WJ, Lu S, Pike VW. 11C-Labeling of Aryl Ketones as Candidate Histamine Subtype-3 Receptor PET Radioligands through Pd(0)-Mediated 11C-Carbonylative Coupling. Molecules. 2017; 22(5):792. https://doi.org/10.3390/molecules22050792
Chicago/Turabian StyleSiméon, Fabrice G., William J. Culligan, Shuiyu Lu, and Victor W. Pike. 2017. "11C-Labeling of Aryl Ketones as Candidate Histamine Subtype-3 Receptor PET Radioligands through Pd(0)-Mediated 11C-Carbonylative Coupling" Molecules 22, no. 5: 792. https://doi.org/10.3390/molecules22050792
APA StyleSiméon, F. G., Culligan, W. J., Lu, S., & Pike, V. W. (2017). 11C-Labeling of Aryl Ketones as Candidate Histamine Subtype-3 Receptor PET Radioligands through Pd(0)-Mediated 11C-Carbonylative Coupling. Molecules, 22(5), 792. https://doi.org/10.3390/molecules22050792