
Exploring Anti-Prion Glyco-Based and Aromatic Scaffolds: A Chemical Strategy for the Quality of Life



Abstract
:1. Introduction
2. Synthesis and Mode of Action of Glyco-Based and Aromatic Scaffolds with anti-Prion Effects
2.1. Glyco-Based Molecular Entities
2.2. Aromatic Scaffolds with Nitrogen Containing Six-Membered Ring Heterocycles
2.2.1. Pyridine Dicarbonitrile Derivatives
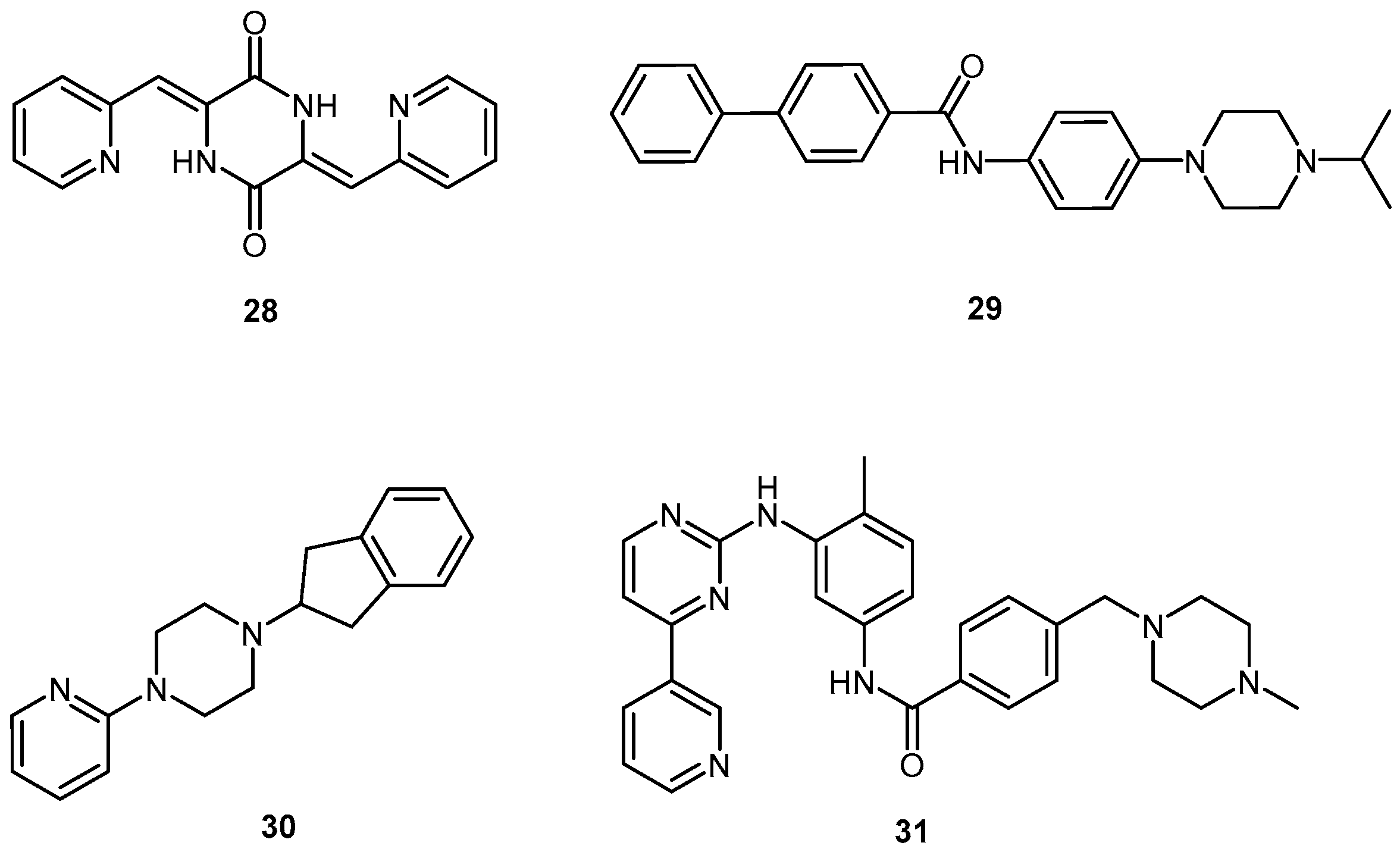
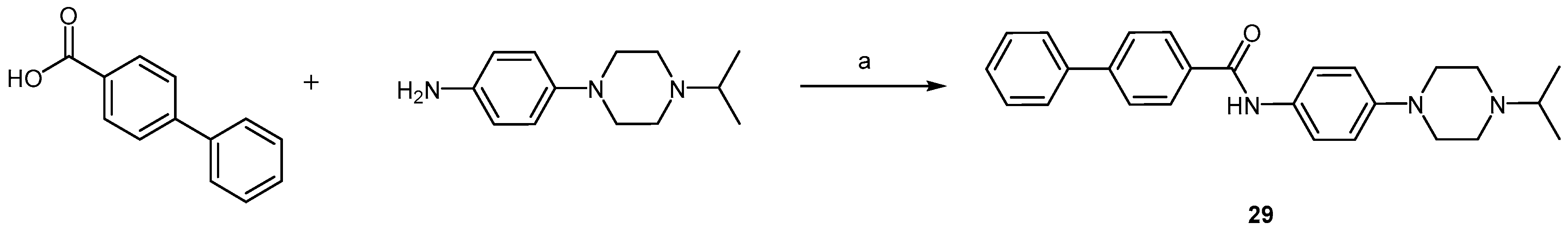
2.2.2. Piperazine Derivatives
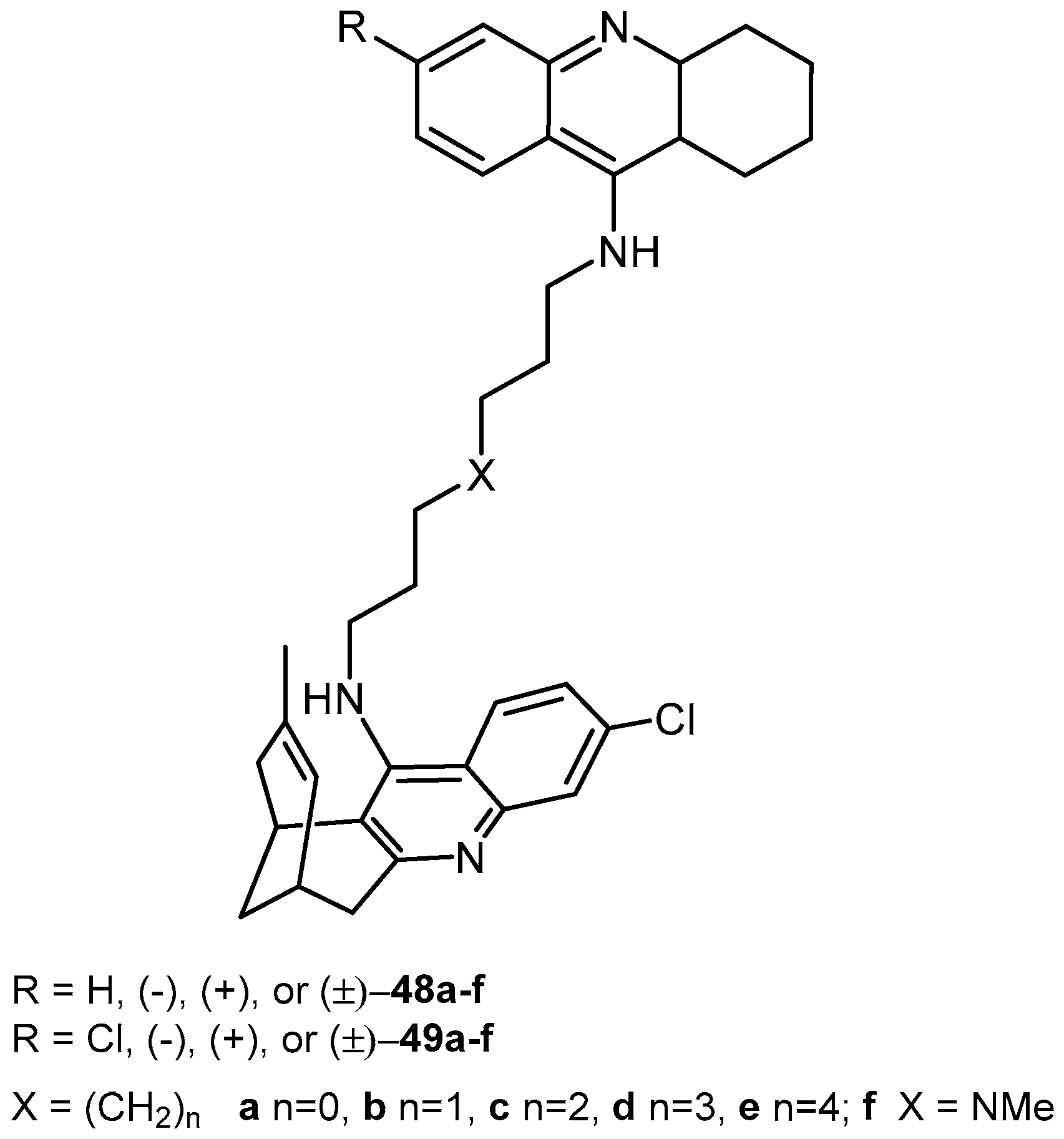
2.2.3. Acridine Derivatives
2.3. Aromatic Scaffolds Bearing Five-Membered Ring Heterocycles as Core Structure
2.3.1. Diphenylpyrazole and Analogs
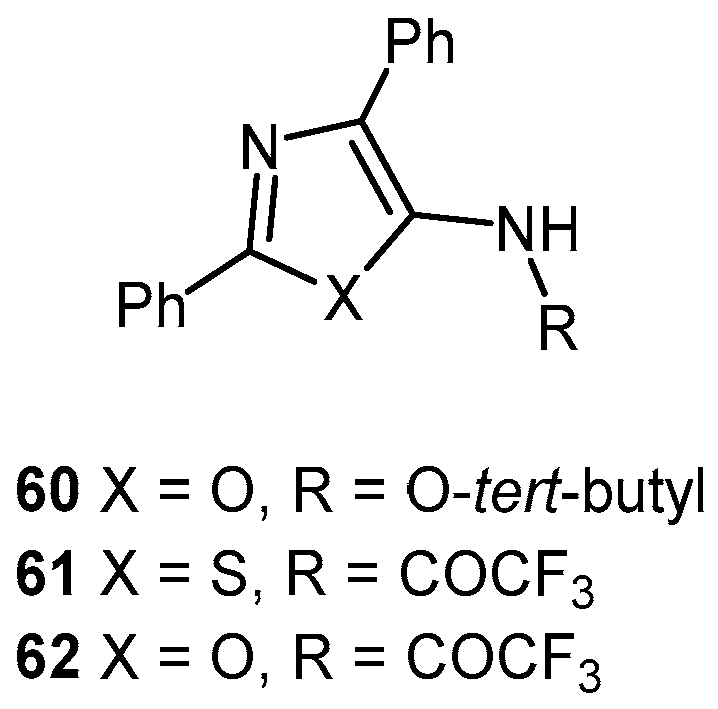
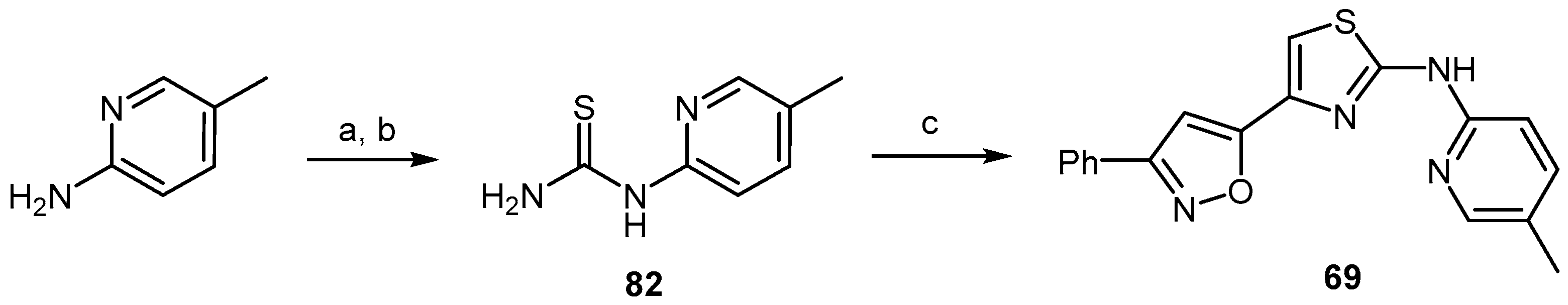
2.3.2. Thiazolamines and Oxazolamines
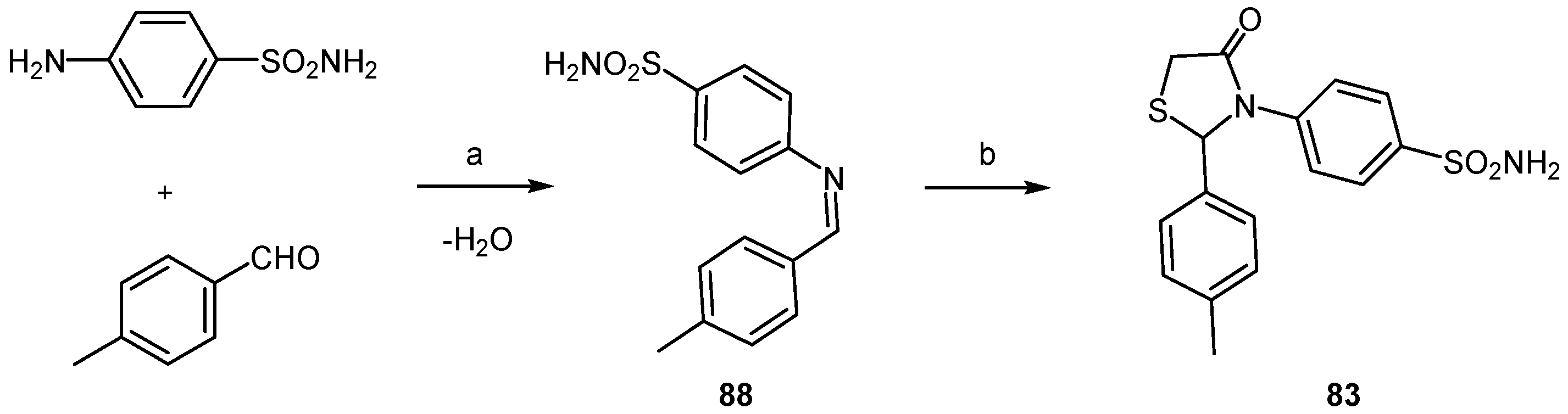
2.3.3. Thiazolidines
2.3.4. Polythiophenes
2.3.5. Carbazoles
2.3.6. GN8 and Derivatives
2.4. Aryl Scaffolds Linked to Nitrogen Containing Functional Groups
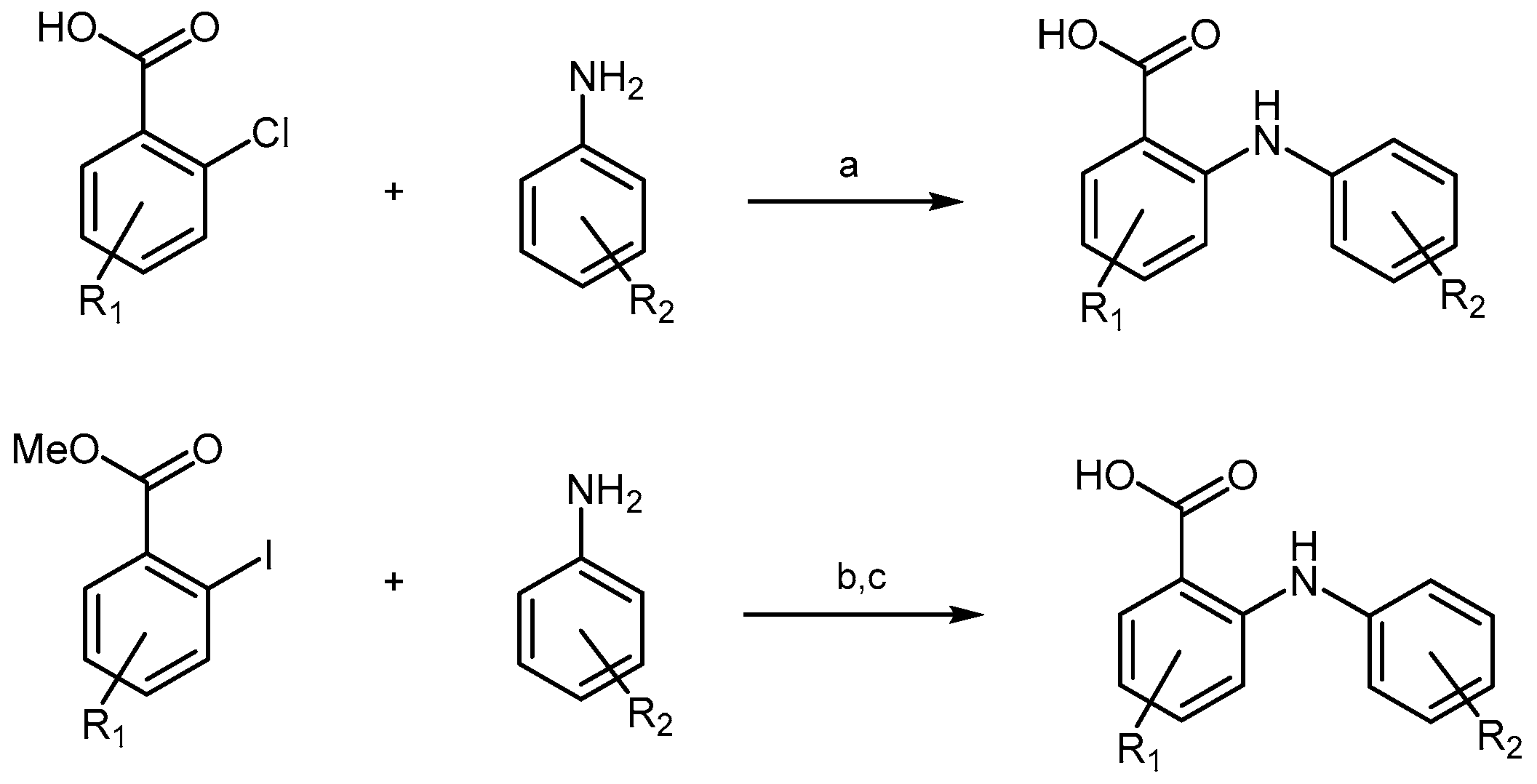
2.4.1. Congo Red Analogs
2.4.2. Sulfonated Arylamides
2.5. Polyphenols
3. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Aguzzi, A.; Heikenwalder, M.; Polumenidou, M. Insights into prion strains and neurotoxicity. Nat. Rev. Mol. Cell Biol. 2007, 8, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Choi, H.S.; Park, J.H.; Kim, M.J.; Lee, H.G.; Petersen, R.B.; Kim, Y.S.; Park, J.B.; Choi, E.K. Regulation of RhoA activity by the cellular prion protein. Cell Death Dis. 2017, 8, e2668. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Yang, Y.; Wang, T.; Kouadir, M.; Zhao, D.; Hu, S. Cellular prion protein promotes neuronal differentiation of adipose-derived stem cells by ppregulating miRNA-124. J. Mol. Neurosci. 2016, 59, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Slapšak, U.; Salzano, G.; Amin, L.; Abskharon, R.N.; Ilc, G.; Zupančič, B.; Biljan, I.; Plavec, J.; Giachin, G.; Legname, G. The N terminus of the prion protein mediates functional interactions with the neuronal cell adhesion molecule (NCAM) fibronectin domain. Biol. Chem. 2016, 291, 21857–21868. [Google Scholar] [CrossRef] [PubMed]
- Soto, G.; Satani, N. The intricate mechanisms of neurodegeneration in prion diseases. Trends Mol. Med. 2012, 17, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Bradley, R.; Liberski, P.P. Bovine spongiform encephalopathy (BSE): The end of the beginning or the beginning of the end? Folia Neuropathol. 2004, 42, 55–68. [Google Scholar] [PubMed]
- Cordeiro, Y.; Ferreira, N.C. New approaches for the selection and evaluation of anti-prion organic compounds. Mini Rev. Med. Chem. 2015, 15, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.A.; Striebel, J.F.; Rangel, A.; Woods, T.; Phillips, K.; Peterson, K.E.; Race, B.; Chesebro, B. Prion strain differences in accumulation of PrPSc on neurons and glia are associated with similar expression profiles of neuroinflammatory genes: Comparison of three prion strains. PLoS Pathog. 2016, 12, e1005551. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.; Abid, K.; Soto, S. The prion strain phenomenon: Molecular basis and unprecedented features. Biochim. Biophys. Acta 2007, 1772, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Kocisko, D.A.; Engel, A.L.; Harbuck, K.; Arnold, K.M.; Olsen, E.A.; Raymond, L.D.; Vilette, D.; Caughey, B. Comparison of protease-resistant prion protein inhibitors in cell cultures infected with two strains of mouse and sheep scrapie. Neurosci. Lett. 2005, 388, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Snow, A.D.; Wight, T.N.; Nochlin, Y.; Koike, K.; Kimata, S.J.; De Armond, S.B.; Prusiner, S.B. Immunolocalization of heparin sulfate proteoglycans to the prion protein amyloid plaques of Gerstmann-Straussler syndrome, Creutzfeldt-Jakob disease and scrapie. Lab. Investig. 1990, 63, 601–611. [Google Scholar] [PubMed]
- Horonchik, L.; Tzaban, S.; Ben-Zaken, O.; Yedidia, Y.; Rouvinski, A.; Papy-Garcia, D.; Barritault, D.; Vlodavsky, I.; Taraboulos, A. Heparan sulfate is a cellular receptor for purified infectious prions. J. Biol. Chem. 2005, 280, 17062–17067. [Google Scholar] [CrossRef] [PubMed]
- Capila, I.; Linhardt, R.J. Heparin-protein interactions. Angew. Chem. Int. Ed. Engl. 2002, 41, 391–412. [Google Scholar] [CrossRef]
- Caughey, B.; Brown, K.; Raymond, G.J.; Katzenstein, G.E.; Thresher, W. Binding of the protease-sensitive form of prion protein PrP to sulfated glycosaminoglycan and congo red [corrected]. J. Virol. 1994, 68, 2135–2141. [Google Scholar] [PubMed]
- Warner, R.G.; Hundt, C.; Weiss, S.; Turnbull, J.E. Identification of the heparan sulfate binding sites in the cellular prion protein. J. Biol. Chem. 2002, 277, 18421–18430. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.; Wandosell, F.; Colaco, C.; Avila, J. Sulphated glycosaminoglycans prevent the neurotoxicity of a human prion protein fragment. Biochem. J. 1998, 335, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Ouidja, M.-O.; Petit, E.; Kerros, M.-E.; Ikeda, Y.; Morin, C.; Carpentier, G.; Barritault, D.; Bruge’re-Picoux, J.; Deslys, J.-P.; Adjou, K.; et al. Structure-activity studies of heparan mimetic polyanions for anti-prion therapies. Biochem. Biophys. Res. Commun. 2007, 363, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Kirby, L.; Birkett, C.R.; Rudyk, H.; Gilbert, I.H.; Hope, J. In vitro cell-free conversion of bacterial recombinant PrP to PrPres as a model for conversion. J. Gen. Virol 2003, 84, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Teruya, K.; Wakao, M.; Sato, M.; Hamanaka, T.; Nishizawa, K.; Funayama, Y.; Sakasegawa, Y.; Suda, Y.; Doh-ura, K. Heparinase I-specific disaccharide unit of heparin is a key structure but insufficient for exerting anti-prion activity in prion-infected cells. Biochem. Biophys. Res. Commun. 2015, 460, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Wakao, M.; Deguchi, H.; Mawatari, A.; Sobel, M.; Suda, Y. Towards the assembly of heparin and heparan sulfate oligosaccharide libraries: efficient synthesis of uronic acid and disaccaride building blocks. Tetrahedron 2010, 66, 3951–3962. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Baron, G.S. Prions and their partners in crime. Nature 2006, 443, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Raymond, G.J. Sulfate polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J. Virol. 1993, 67, 643–650. [Google Scholar] [PubMed]
- Gabizon, R.; Meiner, Z.; Halimi, M.; Ben-Sasson, S.A. Heparin-like molecules bind differentially to prion-proteins and change their intracellular metabolic fate. J. Cell Physiol. 1993, 157, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Brown, K.; Raymond, G.J.; Katzenstein, G.E.; Thresher, W. Binding of the protease-sensitive form of PrP (prion protein) to sulfated glycosaminoglycan and congo red. J. Virol. 1994, 68, 4107, (Erratum of [15]). [Google Scholar]
- Yamaguchi, S.; Nishida, Y.; Sasaki, K.; Kambara, K.; Kim, C.-L.; Ishiguro, N.; Nagatsuka, T.; Uzawa, T.; Horiuchi, M. Inhibition of PrPSc formation by synthetic O-sulfated glycopyranosides and their polymers. Biochem. Biophys. Res. Commun. 2006, 349, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Nishida, Y.; Uzawa, H.; Kobayashia, K. N-Acetyl-6-sulfo-d-glucosamine as a Promising Mimic of N-Acetyl Neuraminic Acid. Bioorg. Med. Chem.Lett. 2003, 13, 2821–2823. [Google Scholar] [CrossRef]
- Nishizawa, K.; Oguma, A.; Kawata, S.Y.; Teruya, K.; Dohura, K. Efficacy and Mechanism of a Glycoside Compound Inhibiting Abnormal Prion Protein Formation in Prion-Infected Cells: Implications of Interferon and Phosphodiesterase 4D-Interacting Protein. J. Virol. 2014, 88, 4083–4099. [Google Scholar] [CrossRef] [PubMed]
- Charvériat, M.; Reboul, M.; Wang, Q.; Picoli, C.; Lenuzza, N.; Montagnac, A.; Nhiri, N.; Jacquet, E.; Guèritte, F.; Lallemand, J.-Y.; et al. New inhibitors of prion replication that target the amyloid precucrsor. J. Gen. Vir. 2009, 90, 1294–1301. [Google Scholar]
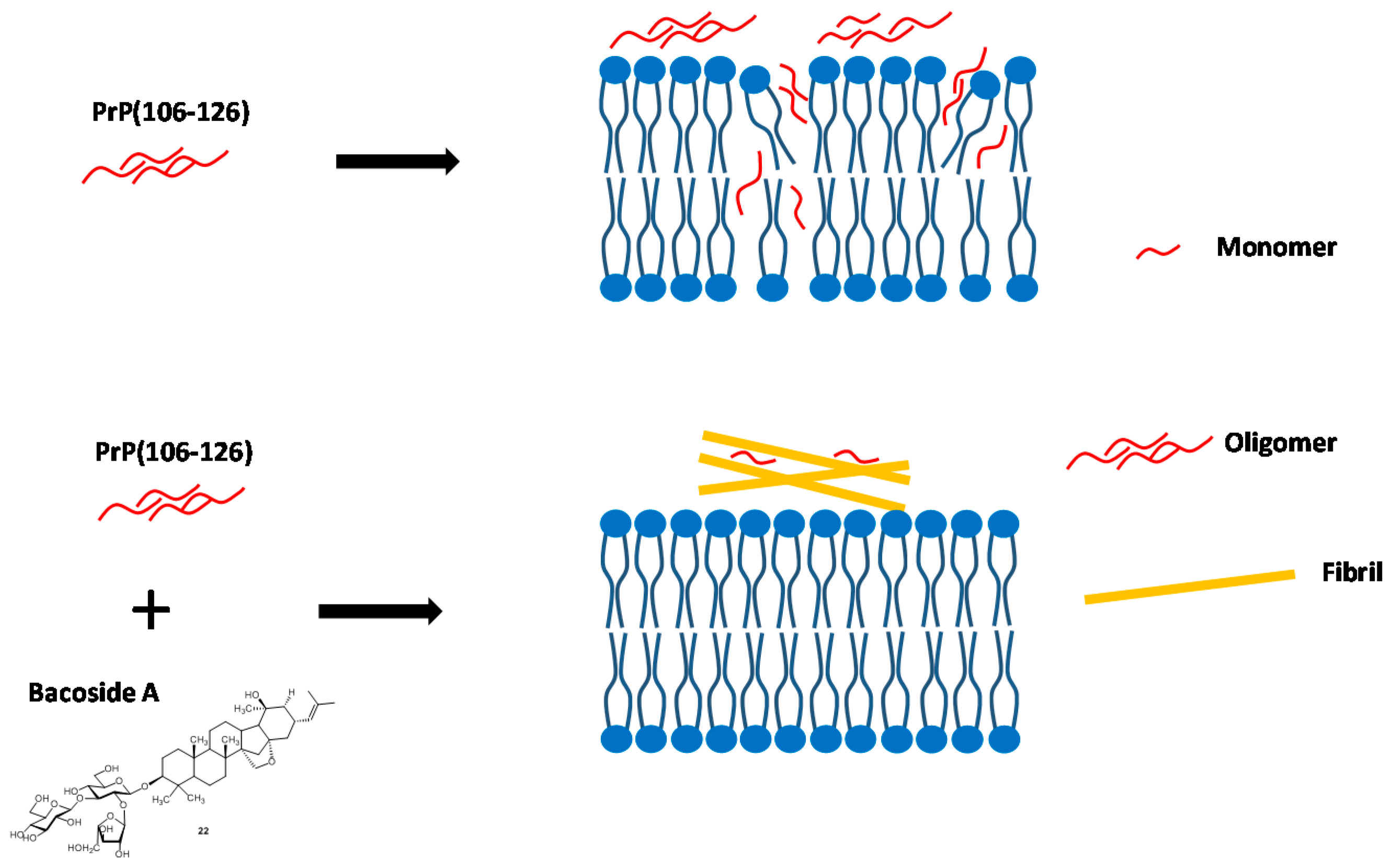
- Malishev, R.; Nandi, S.; Kolusheva, S.; Shaham-Niv, S.; Gazit, E.; Jelinek, R. Bacoside-A, an anti-amyloid natural substance, inhibits membrane disruption by the amyloidogenic determinant of prion protein through accelerating fibril formation. Biochim. Biophis. Acta 2016, 1858, 2208–2214. [Google Scholar] [CrossRef] [PubMed]
- Limpeanchob, S.; Jaipan, S.; Rattanakaruna, W.; Phrompittayarat, K. Ingkaninan. Neuroprotective effect of Bacopa monnieri on beta-amyloid-induced cell death in primary cortical culture. J. Ethnopharmacol. 2008, 120, 112–117. [Google Scholar]
- Bammidi, S.R.; Volluri, S.S.; Chippada, S.C.; Avanigadda, S.; Vangalapati, M. A review on pharmacological studies of Bacopa monniera. J. Chem. Biol. Phys. Sci. 2001, 1, 250. [Google Scholar]
- Apetz, N.; Munch, G.; Govindaraghavan, S.; Gyengesi, E. Natural Compounds and Plant Extracts as Therapeutics against Chronic Inflammation in Alzheimer’s Disease—A Translational Perspective, CNS & Neurological Disorders-Drug Targets (Formerly Current Drug Targets-CNS & Neurological Disorders). CNS Neurol. Disord. Drug Targets 2014, 13, 1175–1191. [Google Scholar] [PubMed]
- Holcomb, L.A.; Dhanasekaran, M.; Hitt, A.-R.; Young, K.A.; Riggs, M.; Manyam, B.V. Bacopa monniera extract reduces amyloid levels in PSAPP mice. J. Alzheimers Dis. 2006, 9, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Miura, M.; Yoda, M.; Takaku, N.; Hirose, T.; Takeuchi, H. Clustered negative charges on the lipid membrane surface induce β-sheet formation of prion protein fragment 106–126. Biochemistry 2007, 46, 11589–11597. [Google Scholar] [CrossRef] [PubMed]
- Tagliavini, F.; Prelli, F.; Verga, L.; Giaccone, G.; Sarma, R.; Gorevic, P.; Ghetti, B.; Passerini, F.; Ghibaudi, E.; Forloni, G. Synthetic peptides homologous to prion protein residues 106–147 form amyloid-like fibrils in vitro. Proc. Natl. Acad. Sci. USA 1993, 90, 9678–9682. [Google Scholar] [CrossRef] [PubMed]
- Perrier, V.; Wallace, A.C.; Kaneko, K.; Safar, J.; Prusiner, S.B.; Cohen, F.E. Mimicking dominant negative inhibition of prion replication through structure-based drug design. Proc. Natl. Acad. Sci. USA 2000, 97, 6073–6078. [Google Scholar] [CrossRef] [PubMed]
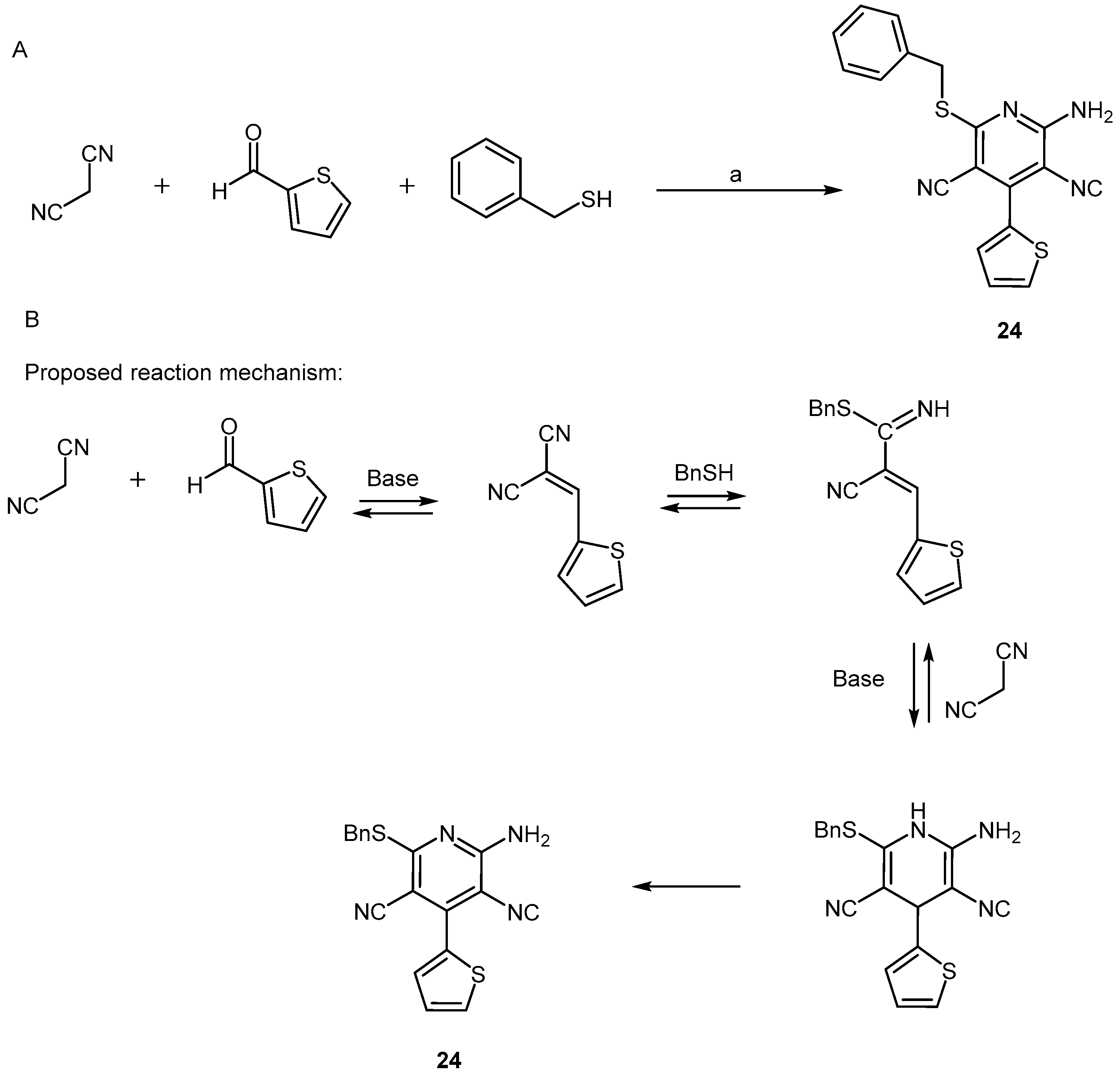
- May, B.C.H.; Zorn, J.A.; Wiktop, J.; Sherrill, J.; Wallace, A.C.; Legname, G.; Prusiner, S.B.; Cohen, F.E. Structure-activity relationship study of prion inhibition by 2-aminopyridine-3,5-dicarbonitrile-based compounds: Parallel synthesis, bioactivity and in vitro pharmacokinetics. J. Med. Chem. 2007, 50, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Reddy, T.R.K.; Mutter, R.; Heal, W.; Guo, K.; Gillet, V.J.; Pratt, S.; Chen, B. Library design, synthesis and screening: Pyridine dicarbonitriles as potential prion disease therapeutics. J. Med. Chem. 2006, 49, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Niida, A.; Tanigaki, H.; Inokuchi, E.; Sasaki, Y.; Oishi, S.; Ohno, H.; Tamamura, H.; Wang, Z.; Peiper, S.C.; Kitaura, K.; et al. Stereoselective synthesis of 3,6-disubstituted-3,6-dihydropyridin-2-ones as potential diketopiperazine mimetics using organocopper-mediated anti-SN2‘ reactions and their use in the preparation of low-molecule CXCR4 antagonists. J. Org. Chem. 2006, 71, 3942–3951. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Ai Tran, H.N.; Staderini, M.; Monaco, A.; López-Cobeñas, A.; Bongarzone, S.; Biarnés, X.; López-Alvarado, P.; Cabezas, N.; Caramelli, M.; et al. Discovery of a class of diketopiperazines as antiprion compounds. ChemMedChem 2010, 5, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rao, S.; Gever, J.R.; Widjaja, K.; Prusiner, S.B.; Silber, B.M. Towards optimization of arylamides as novel, potent, and brain-penetrant antiprion lead compounds. ACS Med. Chem. Lett. 2013, 4, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Liedel, F.; Eiden, M.; Geissen, M.; Hirschberger, T.; Tavan, P.; Giese, A.; Kretzschmar, H.A.; Schätzl, H.; Groschup, M.H. Piperazine derivatives inhibit PrP/PrPres propagation in vitro and in vivo. Biochem. Biophys. Res. Commun. 2014, 445, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Ertmer, A.; Gilch, S.; Yun, S.W.; Flechsig, E.; Klebl, B.; Stein-Gerlach, M.; Klein, M.A.; Schätzl, H.M. The tyrosine kinase inhibitor STI571 induces cellular clearance of PrPSc in prion-infected cells. J. Biol. Chem. 2004, 279, 41918–41927. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, J.; Caravatti, G.; Mett, H.; Meyer, T.; Müller, M.; Lydon, N.B.; Fabbro, D. Phenylamino-pyrimidine (PAP) derivatives: A new class of potent and selective inhibitors of protein kinase C (PKC). Arch. Pharm. (Weinheim) 1996, 329, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Loiseleur, O.; Kaufmann, D.; Abel, S.; Buerger, H.M.; Meisenbach, M.; Schmitz, B.; Sedelmeier, G. N-phenyl-2-pyrimidine-amine Derivatives. PCT EP Appl. WO2003066613 A1, 14 August 2003. [Google Scholar]
- Kompella, A.; Bhujanga, R.A.K.S.; Venkaiah, C.N.; Srinivas, R. Process for the Preparation of the Anti-Cancer Drug Imatinib and Its Analoges. PCT IN Appl. WO2004108699 A1, 16 December 2004. [Google Scholar]
- Liu, Y.F.; Wang, C.L.; Bai, Y.J.; Jiao, J.P.; Qi, X.L. A facile total synthesis of imatinib base and its analoges. Org. Process Res. Dev. 2008, 12, 490–495. [Google Scholar] [CrossRef]
- Korth, C.; May, B.C.H.; Cohen, F.E.; Prusiner, S.B. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc. Natl. Acad. Sci. USA 2001, 98, 9836–9841. [Google Scholar] [CrossRef] [PubMed]
- Csuk, R.; Barthel, A.; Raschke, C. Convenient access to substituted acridines by a Buchwald–Hartwig amination. Tetrahedron 2004, 60, 5737–5750. [Google Scholar] [CrossRef]
- Hassan, J.; Sevigon, M.; Gozzi, C.; Schulz, E.; Lemaire, M. Aryl−Aryl Bond Formation One Century after the Discovery of the Ullmann Reaction. Chem. Rev. 2002, 102, 1359–1469. [Google Scholar] [CrossRef] [PubMed]
- Vogtherr, M.; Grimme, S.; Elshorst, B.; Jacobs, D.M.; Fiebig, K.; Griesinger, K.; Zahn, R. Antimalarial Drug Quinacrine Binds to C-Terminal Helix of Cellular Prion Protein. J. Med. Chem. 2003, 46, 3563–3564. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Okochi, H.; May, B.C.H.; Legname, G.; Prusiner, S.B. Quinacrine is mainly metabolized to mono-desethyl quinacrine by CYP3A4/5 and its brain accumulation is limited by P-glycoprotein. Drug Metab. Dispos. 2006, 34, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Ghaemmaghami, S.; Ahn, M.; Lessard, P.; Giles, K.; Legname, G.; DeArmond, G.J.; Prusiner, S.B. Continuous quinacrine treatment results in the formation of drug-resistant prions. PLoS Pathog. 2009, 5, e1000673. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Sakasegawa, Y.; Doh-ura, K.; Go, M.L. Anti-prion activities and drug-like potential of functionalized quinacrine analogs with basic phenyl residues at the 9-amino position. Eur. J. Med. Chem. 2011, 46, 2917–2929. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.T.; Lee, C.-Y.; Teruya, K.; Ong, W.-Y.; Doh-ura, K.; Go, M.-L. Anti-prion activity of functionalized 9-aminoacridines related to quinacrine. Bioorg. Med. Chem. 2008, 16, 6737–6746. [Google Scholar] [CrossRef] [PubMed]
- May, B.C.H.; Fafarman, A.T.; Hong, S.B.; Rogers, M.; Deady, L.W.; Stanley, B.; Prusiner, S.B.; Cohen, F.E. Potent inhibition of scrapie prion replication in cultured cells by bis-acridines. Proc. Natl. Acad. Sci. USA 2003, 100, 3416–3421. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Tewari, S.; Chauhan, P.M.S.; Puri, S.K.; Bhaduri, A.P.; Pandey, V.C. Synthesis of bisquinolines and their in vitro ability to produce methemoglobin in canin hemosylate. Bioorg. Med. Chem. Lett. 1999, 9, 653–658. [Google Scholar] [CrossRef]
- Coste, J.; Frerot, E.; Jouin, P. Coupling N-Methylated Amino Acids Using PyBroP and PyCloP Halogenophosphonium Salts: Mechanism and Fields of Application. J. Org. Chem. 1994, 59, 2437–2446. [Google Scholar] [CrossRef]
- Bongarzone, S.; Tran, H.N.A.; Cavalli, A.; Roberti, M.; Carloni, P.; Giuseppe Legname, G.; Bolognesi, M.L. Parallel synthesis, evaluation, and preliminary structure-activity relationship of 2,5-diamino-1,4-benzoquinones as a novel class of bivalent anti-prion compound. J. Med. Chem. 2010, 53, 8197–8201. [Google Scholar] [CrossRef] [PubMed]
- Galdeano, C.; Viayna, E.; Sola, I.; Formosa, X.; Camps, P.; Badia, A.; Clos, M.V.; Relat, J.; Ratia, M.; Bartolini, M.; et al. Huprine-tacrine heterodimers as anti-amyloidogenic compounds of potential interest against Alzheimer´s and prion diseases. J. Med. Chem. 2012, 55, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Gregor, V.E.; Emmerling, M.R.; Lee, C.; Moore, C.J. The synthesis and in vitro acetylcholinesterase and butyrylcholinesterase inhibitory activity of tacrine (Cognex®) derivatives. Bioorg. Med. Chem. Lett. 1992, 2, 861–864. [Google Scholar] [CrossRef]
- Pang, Y.-P.; Quiram, P.; Jelacic, T.; Hong, F.; Brimijoin, S. Highly potent, selective, and low cost bis-tetrahydroaminacrine inhibitors of acetylcholinesterase. J. Biol. Chem. 1996, 271, 23646–23649. [Google Scholar] [PubMed]
- Camps, P.; Contreras, J.; Font-Bardia, M.; Morral, J.; Muñoz-Torrero, D.; Solans, X. Enantioselective synthesis of tacrine-huperzine A hybrids. Preparative chiral MPLC separation of their racemic mixtures and absolute configuration assignments by X-ray diffraction analysis. Tetrahedron Asymmetry 1998, 9, 835–849. [Google Scholar] [CrossRef]
- Collins, S.J.; Lewis, V.; Brazier, M.; Hill, A.F.; Fletcher, A.; Masters, C.L. Quinacrine does not prolong survival in a murine Creutzfeldt-Jakob disease model. Ann. Neurol. 2002, 52, 503–508. [Google Scholar]
- Cope, H.; Mutter, R.; Heal, W.; Pascoe, C.; Brown, P.; Pratt, S.; Chen, B. Synthesis and SAR study of acridine, 2-methylquinoline and 2-phenylquinazoline analogs as anti-prion agents. Eur. J. Med. Chem. 2006, 41, 1124–1143. [Google Scholar] [CrossRef] [PubMed]
- Macedo, B.; Kaschula, C.H.; Hunter, R.; van der Merwe, J.D.; Silva, J.L.; Egan, T.J.; Cordeiro, Y. Synthesis and anti-prion activity evaluation of aminoquinoline analogues. Eur. J. Med. Chem. 2010, 45, 5468. [Google Scholar] [CrossRef] [PubMed]
- Egan, T.J.; Hunter, R.; Kaschula, C.H.; Marqes, H.M.; Misplon, A.; Walden, J. Structure-function relationships in aminoquinolines: Effect of amino and chloro groups on quinoline-hematin complex formation, inhibition of beta-hematin formation, and antiplasmodial activity. J. Med. Chem. 2000, 43, 283. [Google Scholar] [CrossRef] [PubMed]
- Kocisko, D.A.; Caughey, B. Mefloquine, an antimalarial drug with antiprion activity in vitro lacks activity in vivo. J. Virol. 2006, 80, 1044–1046. [Google Scholar] [CrossRef] [PubMed]
- Leidel, F.; Eiden, M.; Geissen, M.; Kretzschmar, H.A.; Giese, A.; Hirschberger, T.; Tavan, P.; Schätzl, H.M.; Groschup, M.H. Diphenylpyrazole-derived compounds increase survival time of mice after prion infection. Antimicrob. Agents Chemother. 2011, 55, 4774–4781. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Ryazanov, S.; Leonov, A.; Levin, J.; Shi, S.; Schmidt, F.; Prix, C.; Pan-Montojo, F.; Bertsch, U.; Mitteregger-Kretzschmar, G.; et al. Anle138b: A novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol. 2013, 125, 795–813. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L.; Kayed, R. Therapeutic approaches against common structural features of toxic oligomers shared by multiple amyloidogenic proteins. Biochem. Pharmacol. 2014, 88, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.; Schmidt, F.; Boehm, C.; Prix, C.; Bötzel, K.; Ryazanov, S.; Leonov, A.; Griesinger, C.; Giese, A. The oligomer modulator anle138b inhibits disease progression in a Parkinson mouse model even with treatment started after disease onset. Acta Neuropathol. 2014, 127, 779–780. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Krauss, S.; Shi, S.; Ryazanov, S.; Steffen, J.; Miklitz, C.; Leonov, A.; Kleinknecht, A.; Göricke, B.; Weishaupt, J.H.; et al. Reducing tau aggregates with anle138b delays disease progression in a mouse model of tauopathies. Acta Neuropathol. 2015, 130, 619–631. [Google Scholar] [CrossRef] [PubMed]
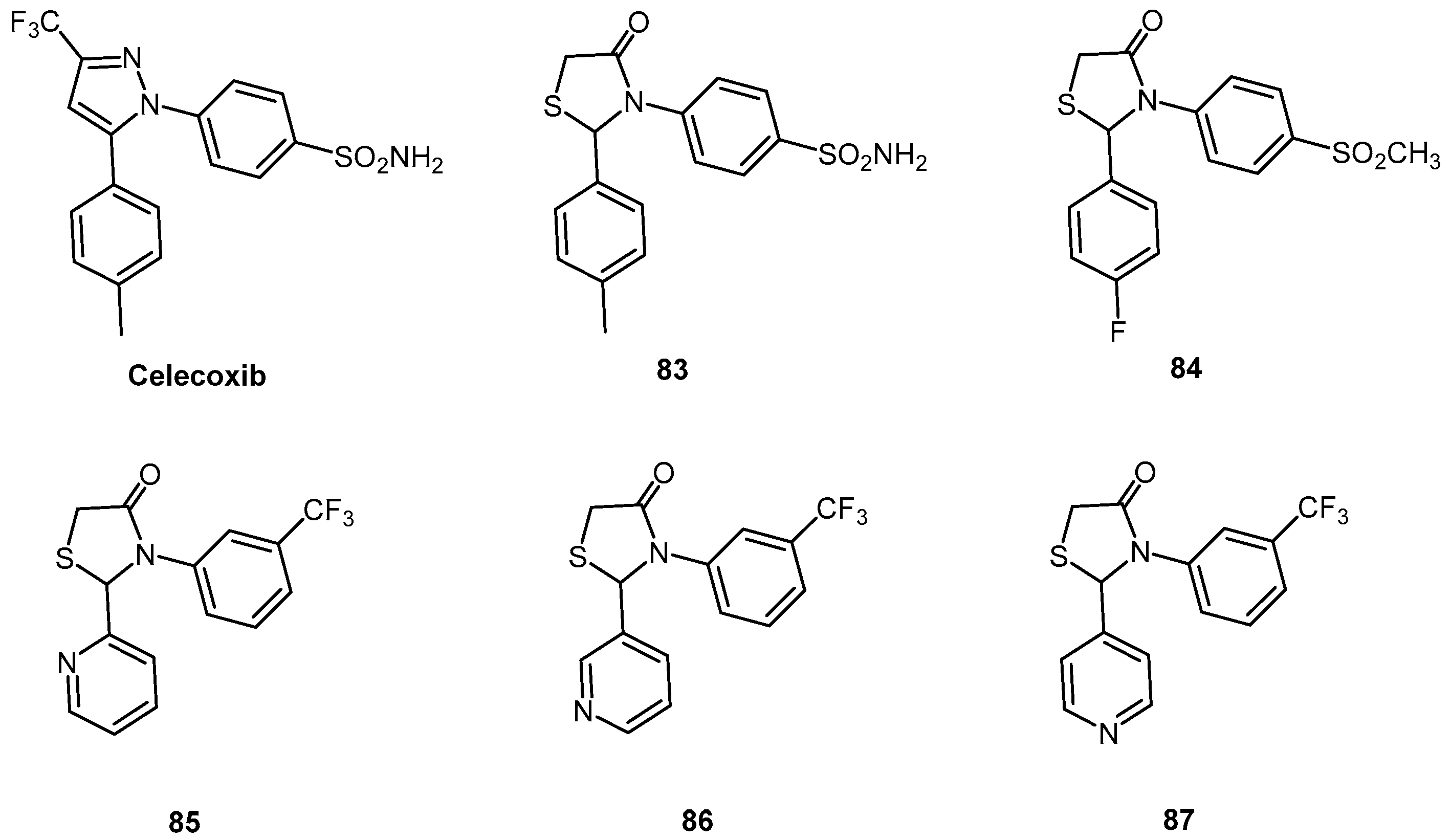
- Villa, V.; Thellung, S.; Corsaro, A.; Novelli, F.; Tasso, B.; Colucci-D’Amato, L.; Gatta, E.; Tonelli, M.; Florio, T. Celecoxib inhibits prion protein 90–231-mediatedpro-inflammatory responses in microglial cells. Mol. Neurobiol. 2016, 53, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.D.; Talley, J.J.; Bertenshaw, S.R.; Carter, J.S.; Collins, P.W.; Docter, S.; Graneto, M.J.; Lee, L.F.; Malecha, J.W.; Miyashiro, J.M.; et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: Identification of 4-[-5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, celecoxib). J. Med. Chem. 1997, 40, 1347–1365. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, K.; Ishida, H.; Omovskaya, O.; Dudley, M.; Rleger, R.; Watkins, W.J.; Zhang, J.Z.; Renau, T.E.; Lee, V.J.; Ota, T.; et al. Drug Discharging Pump Inhibitor. Japan Patent JP 2002322054 A, 8 November 2002. [Google Scholar]
- Shimada, Y. Thiazolideneacetanilide Photographic Color Couplers. Japan Patent JP 02113070 A, 25 April 1990. [Google Scholar]
- Heal, W.; Thompson, M.J.; Mutter, R.; Cope, H.; Louth, J.C.; Chen, B. Library Synthesis and Screening: 2,4-Diphenylthiazoles and 2,4-Diphenyloxazoles as Potential Novel Prion Disease Therapeutics. J. Med. Chem. 2007, 50, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.J.; Heal, W.; Chen, B. Synthesis of 5-aminothiazoles as building blocks for library synthesis. Tetrahedron Lett. 2006, 47, 2361–2364. [Google Scholar] [CrossRef]
- Ghaemmaghami, S.; May, B.C.H.; Renslo, A.R.; Prusiner, S.B. Discovery of 2-aminothiazoles as potent antiprion compounds. J. Virol. 2010, 84, 3408–3412. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Godoy, A.; Gever, J.; Fife, K.L.; Silber, B.M.; Prusiner, S.B.; Renslo, A.R. 2-Aminothiazoles as Therapeutic Leads for Prion Diseases. J. Med. Chem. 2011, 54, 1010–1021. [Google Scholar] [CrossRef] [PubMed]
- Ghaemmaghami, S.; Russo, M.; Renslo, A.R. Successes and Challenges in Phenotype-Based Lead Discovery for Prion Diseases. J. Med. Chem. 2014, 57, 6919–6929. [Google Scholar] [CrossRef] [PubMed]
- Teruya, K.; Kawagoe, K.; Kimura, T.; Chen, C.J.; Sakasegawa, Y.; Doh-ura, K. Amyloidophilic compounds for prion diseases. Infect. Disord. Drug Targets 2009, 9, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.B.; Lu, D.; Geva, M.; Watts, J.C.; Bhardwaj, S.; Oehler, A.; Renslo, A.R.; DeArmond, S.J.; Prusiner, S.B.; Giles, K. Drug resistance confounding prion therapeutics. Proc. Natl. Acad. Sci. USA 2013, 110, E4160–E4169. [Google Scholar] [CrossRef] [PubMed]
- Baral, P.K.; Swayampakula, M.; Rout, M.K.; Kav, N.N.; Spyracopoulos, L.; Aguzzi, A.; James, M.N. Structural basis of prion inhibition by phenothiazine compounds. Structure 2014, 22, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Minagar, A.; Shapshak, P.; Fujimura, R.; Ownby, R.; Heyes, M.; Eisdorfer, C. Therole of macrophage/microglia and astrocytes in the pathogenesis of threeneurologic disorders: HIV-associated dementia, Alzheimer disease, andmultiple sclerosis. J. Neurol. Sci. 2002, 202, 13–23. [Google Scholar] [CrossRef]
- Tribouillard-Tanvier, D.; Race, B.; Striebel, J.F.; Carroll, J.A.; Phillips, K.; Chesebro, B. Early cytokine elevation, PrPres deposition and gliosis in mousescrapie: No effect on disease by deletion of cytokine genes, IL-12p40 andIL-12p35. J. Virol. 2012, 86, 10377–10383. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Wang, Q.; Hand, T.; Wu, L.; Breyer, R.M.; Montine, T.J.; Andreasson, K. Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage andamyloid burden in a model of Alzheimer’s disease. J. Neurosci. 2005, 25, 10180–10187. [Google Scholar] [CrossRef] [PubMed]
- Montine, T.J.; Sidell, K.R.; Crews, B.C.; Markesbery, W.R.; Marnett, L.J.; Roberts, L.J., 2nd; Morrow, J.D. Elevated CSF prostaglandin E2 levels in patients withprobable AD. Neurology 1999, 53, 1495–1498. [Google Scholar] [CrossRef] [PubMed]
- Eikelenboom, P.; Bate, C.; Van Gool, W.A.; Hoozemans, J.J.; Rozemuller, J.M.; Veerhuis, R.; Williams, A. Neuroinflammation in Alzheimer’s disease and prion disease. Glia 2002, 40, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Kempster, S.; Williams, A. Prostaglandin D2 mediates neuronaldamage by amyloid-beta or prions which activates microglial cells. Neuropharmacology 2006, 50, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Villaa, V.; Thellung, S.; Bajetto, A.; Gatta, E.; Robello, M.; Novelli, F.; Tasso, B.; Tonelli, M.; Florio, T. Novel celecoxib analogues inhibit glial production of prostaglandinE2, nitric oxide, and oxygen radicals reverting the neuroinflammatoryresponses induced by misfolded prion protein fragment 90–231 or lipopolysaccharide. Pharmacol. Res. 2016, 113, 500–514. [Google Scholar] [CrossRef] [PubMed]
- Vazzana, I.; Terranova, E.; Mattioli, F.; Sparatore, F. Aromatic Schiff bases and 2,3-disubstituted-1,3-thiazolidin-4-one derivatives as anti-inflammatory agents. Arkivoc 2004, V, 364–374. [Google Scholar]
- Herrmann, U.S.; Schütz, A.K.; Shirani, H.; Huang, D.; Saban, D.; Nuvolone, M.; Li, B.; Ballmer, B.; Åslund, A.K.O.; Mason, J.J.; et al. Structure-based drug design identifies polythiophenes as anti-prion compounds. Sci. Transl. Med. 2015, 7, 299ra123. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.A.; Shirani, H.; Andreas Åslund, K.O.; Marcus Back, M.; Haroutunian, V.; Gandy, S.; Nilsson, K.P.R. Pentameric Thiophene-Based Ligands that Spectrally Discriminate Amyloid-b and Tau Aggregates Display Distinct Solvatochromism and Viscosity-Induced Spectral Shifts. Chem. Eur. J. 2014, 20, 12537–12543. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Hosokawa-Muto, J.; Asami, K.; Murai, T.; Kuwata, K. Synthesis of 9-substituted 2,3,4,9-tetrahydro-1H-carbazole derivatives and evaluation of their anti-prion activity in TSE-infected cells. Eur. J. Med. Chem. 2011, 46, 5675–5679. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, K.; Nishida, N.; Matsumoto, T.; Kamatari, Y.O.; Hosokawa-Muto, J.; Kodama, K.; Nakamura, H.K.; Kimura, K.; Kawasaki, M.; Takakura, Y.; et al. Hot spots in prion protein for pathogenic conversion. Proc. Natl. Acad. Sci. USA 2007, 104, 11921–11926. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Hosokawa-Muto, J.; Kamatari, Y.O.; Kuwata, K. Synthesis of GN8 derivatives and evaluation of their antiprion activity in TSE-infected cells. Bioorg. Med. Chem. Lett. 2011, 21, 1502–1507. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B.; McKinley, M.P.; Bowman, K.A.; Bendheim, P.E.; Groth, D.F.; Glenner, C.G. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell 1983, 35, 349–358. [Google Scholar] [CrossRef]
- Caspi, S.; Halimi, M.; Yanai, A.; Sasson, S.B.; Taraboulos, A.; Gabizon, R. The anti-prion activity of congo red. Putative mechanism. J. Biol. Chem. 1998, 273, 3484–3489. [Google Scholar] [CrossRef] [PubMed]
- Klunk, W.E.; Debnath, M.L.; Pettergrew, J.W. Development of small-molecule probes for the beta-amyloid protein of Alzheimer’s disease. Neurobiol. Aging 1994, 15, 691–698. [Google Scholar] [CrossRef]
- Klunk, W.E.; Debnath, M.L.; Koros, A.M.C.; Pettergrew, J.W. Chrysamine-G, a lipophilic analog of Congo red, inhibits A beta-induced toxicity in PC12 cells. Life Sci. 1998, 63, 1807–1814. [Google Scholar] [CrossRef]
- Talaska, G. Aromatic amines and human urinary bladder cancer: Exposure sources and epidemiology. J. Environ. Sci. Health 2003, 21, 29–43. [Google Scholar] [CrossRef] [PubMed]
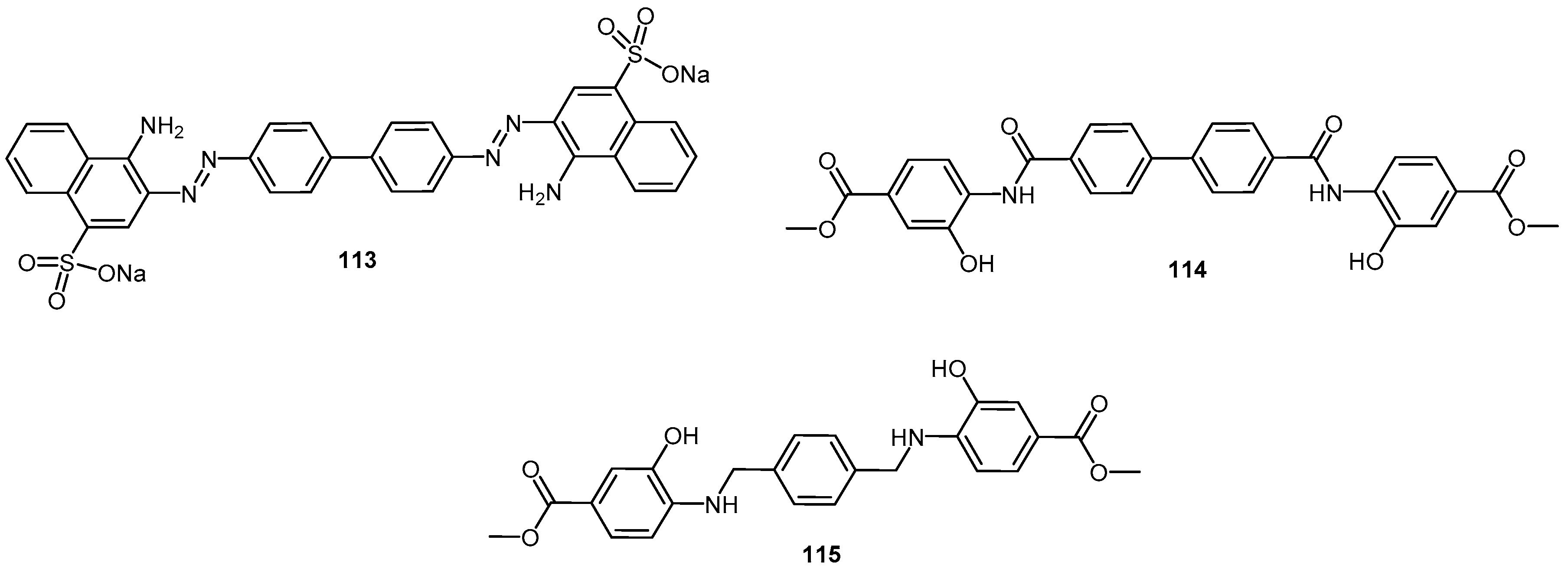
- Rudyk, H.; Vasiljevic, S.; Hennion, R.M.; Birkett, C.R.; Hope, J.; Gilbert, I.H. Screening Congo Red and its analogs for their ability to prevent the formation of PrPres in scrapie-infected cells. J. Gen. Virol. 2000, 81, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Sellarajah, S.; Lekishvili, T.; Bowring, C.; Thompsett, A.R.; Rudyk, H.; Birekett, C.R.; Brown, D.R.; Gilbert, I.H. Synthesis of analogs of congo red and evaluation of their anti-prion activity. J. Med. Chem. 2004, 47, 5515–5534. [Google Scholar] [CrossRef] [PubMed]
- Rudyk, H.; Knaggs, M.H.; Vasiljevic, S.; Hope, J.; Birkett, C.; Gilbert, I.H. Synthesis and evaluation of analogs of Congo red as potential compounds against transmissible spongiform encephalopathies. Eur. J. Med. Chem. 2003, 38, 567–579. [Google Scholar] [CrossRef]
- Webb, S.; Lekishvili, T.; Loeschner, C.; Sellarajah, S.; Prelli, F.; Wisniewski, T.; Gilbert, I.H.; Brown, D.R. Mechanistic insights into the cure of prion disease by novel antiprion compounds. J. Virol. 2007, 81, 10729–10741. [Google Scholar] [CrossRef] [PubMed]
- Dressel, J.; Oesper, R. The discovery of germanin by Oskar dressel and richard kothe. J. Chem. Educ. 1961, 38, 620–621. [Google Scholar] [CrossRef]
- Nunziante, M.; Kehler, C.; Maas, E.; Kassack, M.U.; Groschup, M.; Schatzl, H.M. Charged bipolar suramin derivatives induce aggregation of the prion protein at the cell surface and inhibit PrPSc replication. J. Cell Sci. 2005, 118, 4959–4973. [Google Scholar] [CrossRef] [PubMed]
- Kiachopoulos, S.; Heske, J.; Tatzelt, J.; Winklhofer, K.F. Misfolding of the prion protein at the plasma membrane induces endocytosis, intracellular retention and degradation. Traffic 2004, 5, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Gilch, S.; Winklhofer, K.F.; Groschup, M.H.; Nunziante, M.; Lucassen, R.; Spielhaupter, C.; Muranyi, W.; Riesner, D.; Tatzelt, J.; Schatzl, H.M. Intracellular re-routing of prion protein prevents propagation of PrPSc and delays onset of prion disease. EMBO J. 2001, 20, 3957–3966. [Google Scholar] [CrossRef] [PubMed]
- Ullmann, H.; Meis, S.; Hongwiset, D.; Marzian, C.; Wiese, M.; Nickel, P.; Communi, D.; Boeynaems, J.-M.; Wolf, C.; Hausmann, R.; et al. Synthesis and Structure−Activity Relationships of Suramin-Derived P2Y11 Receptor Antagonists with Nanomolar Potency. J. Med. Chem. 2005, 48, 7040–7048. [Google Scholar] [CrossRef] [PubMed]
- Rice-Evans, C.A.; Miller, N.J.; Paganga, G. Structure antioxidant activity relationships of flavonoids and phenoliacids. Free Radic. Biol. Med. 1996, 20, 933–956. [Google Scholar] [CrossRef]
- Pandey, K.B.; Rizvi, S.I. Plant polyphenols as dietary antioxidants in human health and disease. Oxid. Med. Cell Longev. 2009, 2, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-S.; Quon, M.J.; Kim, J. New insights into the mechanisms of polyphenols beyond antioxidant properties; lessons from the green tea polyphenol, epigallocatechin 3-gallate. Redox Biol. 2014, 2, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Perron, N.R.; Brumaghim, J.L. A review of the antioxidant mechanisms of polyphenol compounds related to iron binding. Cell Biochem. Biophys. 2009, 53, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, P.; Ghiselli, A.; Buchetti, B. Polyphenols synergisticall inhibit oxidative stress in subjects given red and white wine. Atherosclerosis 2006, 188, 77–83. [Google Scholar] [CrossRef] [PubMed]
- González, R.; Ballester, I.; López-Posadas, R.; Suárez, M.D.; Zarzuelo, A.; Martínez-Augustin, O.; Sánchez de Medina, F. Effects of flavonoids and other polyphenols on inflammation. Crit. Rev. Food Sci. Nutr. 2011, 51, 331–362. [Google Scholar] [CrossRef] [PubMed]
- Sergent, T.; Piront, N.; Meurice, J.; Toussaint, O.; Schneider, Y.-J. Anti-inflammatory effects of dietary phenolic compounds in an in vitro model of inflamed human intestinal epithelium. Chem.-Biol. Int. 2010, 188, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Yoshiike, Y.; Takashima, A.; Hasegawa, K.; Naiki, H.; Yamada, M. Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols in vitro: Implications for the prevention and therapeutics of Alzheimer’s disease. J. Neurochem. 2003, 87, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Porzoor, A.; Alford, B.; Hügel, H.M.; Grando, D.; Caine, J.; Macreadie, I. Anti-Amyloidogenic Properties of Some Phenolic Compounds. Biomolecules 2015, 5, 505–527. [Google Scholar] [CrossRef] [PubMed]
- Viviane, L.; Ngoungoure, N.; Schluesener, J.; Moundipa, P.F.; Schluesener, H. Natural polyphenols binding to amyloid: A broad class of compounds to treat different human amyloid diseases. Mol. Nutr. Food Res. 2015, 59, 8–20. [Google Scholar]
- Wright, B.; Moraes, L.A.; Kemp, C.F. A structural basis for the inhibition of collagen-stimulated platelet function by quercetin and structurally related flavonoids. Br. J. Pharmacol. 2010, 159, 1312–1325. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Moro, S.; Manthey, J.A.; West, P.L.; Ji, X.-D. Interactions of flavones and other phytochemicals with adenosine receptors. Adv. Exp. Med. Biol. 2002, 505, 163–171. [Google Scholar] [PubMed]
- Pawlikowska-Pawlega, B.; Gruszecki, W.I.; Misiak, L. Modification of membranes by quercetin, a naturally occurring flavonoid, via its incorporation in the polar head group. Biochim. Biophys. Acta Biomembr. 2007, 1768, 2195–2204. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Hernández, J.; Zaldívar-Machorro, V.J.; Villanueva-Porras, D.; Vega-Ávila, E.; Chavarría, A. Hindawi Publishing Corporation. Oxid. Med. Cell. Long 2016, 8378613. [Google Scholar] [CrossRef]
- Rauter, A.P.; Branco, I.; Lopes, R.G.; Justin, J.; Silva, F.V.M.; Noronha, J.P. A new lupene triterpenetriol and anticholinesterase activity of Salvia sclareoides. Fitoterapia 2007, 78, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.; Araújo, M.E.; Branco, I.; Meireles, M.; Almeida, J.; Sepulveda, C.; Neng, N.; Nogueira, J.; Jacob, P.; Goulart, M.; et al. Evaluation of plant extracts against prion diseases and cancer. In Proceedings of the 42nd IUPAC Congress—Chemistry Solutions, Session for Chemistry for Health—Chemistry in the Food Chain and Health, Glasgow, UK, 2–7 August 2009. [Google Scholar]
- Rauter, A.P.; Dias, C.; Martins, A.; Branco, I.; Neng, N.R.; Nogueira, J.M.; Goulart, M.; Filipa, V.M.; Silva, F.V.M.; Justino, J.; et al. Non-toxic Salvia sclareoides Brot. extracts as a source of functional food ingredients: Phenolic profile, antioxidant activity and prion binding properties. Food Chem. 2012, 132, 1930–1935. [Google Scholar]
- Airoldi, C.; Sironi, E.; Dias, C.; Marcelo, F.; Martins, A.; Rauter, A.P.; Nicotra, F.; Jimenez-Barbero, J. Natural compounds against Alzheimer’s disease: Molecular recognition of Aβ1–42 peptide by Salvia sclareoides extract and its major component, rosmarinic acid, as investigated by NMR. Chem. Asian J. 2013, 8, 596. [Google Scholar] [CrossRef] [PubMed]
- Marcelo, F.; Dias, C.; Martins, A.; Madeira, P.J.; Jorge, T.M.; Helena Florencio, M.H.; Cañada, F.J.; Cabrita, E.J.; Jiménez-Barbero, J.; Rauter, A.P. Molecular Recognition of Rosmarinic Acid from Salvia sclareoides Extracts by Acetylcholinesterase: A New Binding Site Detected by NMR Spectroscopy. Chem. Eur. J. 2013, 19, 6641–6649. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Sagal, J.P.; Colombres, M. Acetylcholinesterase interaction with Alzheimer amyloid beta. Subcell. Biochem. 2005, 38, 299–317. [Google Scholar] [PubMed]
- Gasperini, L.; Legname, G. Prion Protein and Aging. Front Cell Dev Biol. 2014, 2, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Kocisko, D.A.; Baron, G.S.; Rubenstein, R.; Chen, J.; Kuizon, S.; Caughey, B. New Inhibitors of Scrapie-Associated Prion Protein Formation in a Library of 2,000 Drugs and Natural Products. J. Virol. 2003, 77, 10288–10294. [Google Scholar] [CrossRef] [PubMed]
- Doh-Ura, K.; Iwaki, T.; Caughey, B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J. Virol. 2000, 74, 4894–4897. [Google Scholar] [CrossRef] [PubMed]
- Taraboulos, A.; Scott, M.; Semenov, M.; Avrahami, D.; Laszlo, L.; Prusiner, S.B.; Avrahami, D. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J. Cell Biol. 1995, 129, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Miesbauer, M.; Olschewski, D.; Seidel, R.; Riemer, C.; Smale, L.; Brumm, L.; Levy, M.; Gazit, E.; Oesterhelt, D.; et al. Green tea extracts interfere with the stress-protective activity of PrPC and the formation of PrPSc. J. Neurochem. 2008, 107, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Suganuma, M.; Okabe, S.; Oniyama, M.; Tada, Y.; Ito, H.; Fujiki, H. Wide distribution of [3H](-)-epigallocatechin gallate, a cancer preventive tea polyphenol, in mouse tissue. Carcinogenesis 1998, 19, 1771–1776. [Google Scholar] [CrossRef] [PubMed]
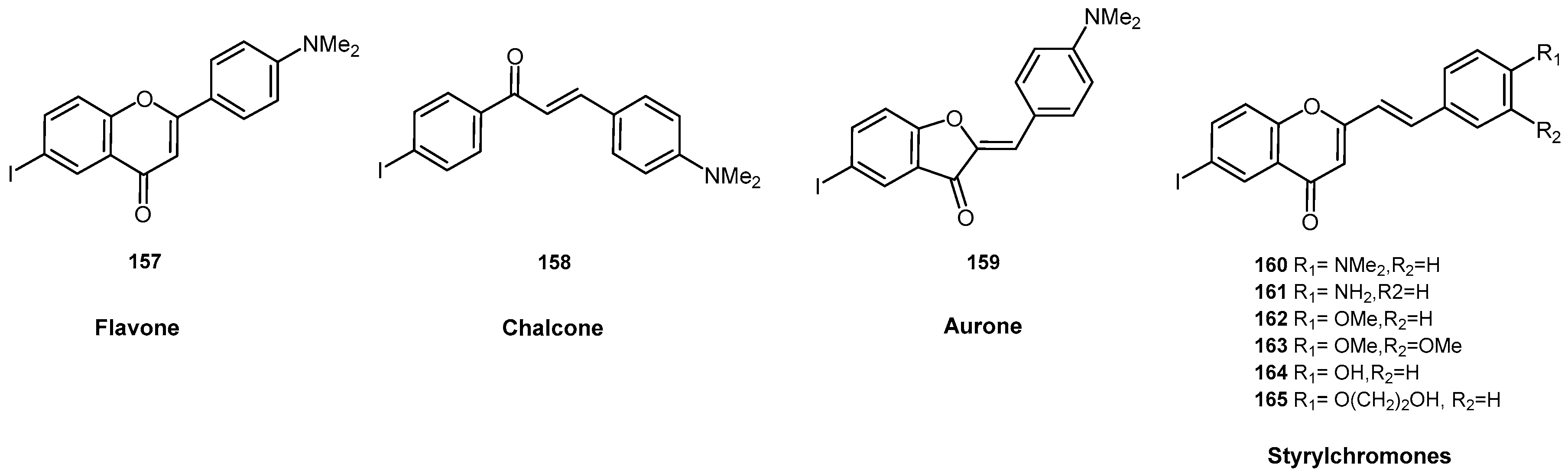
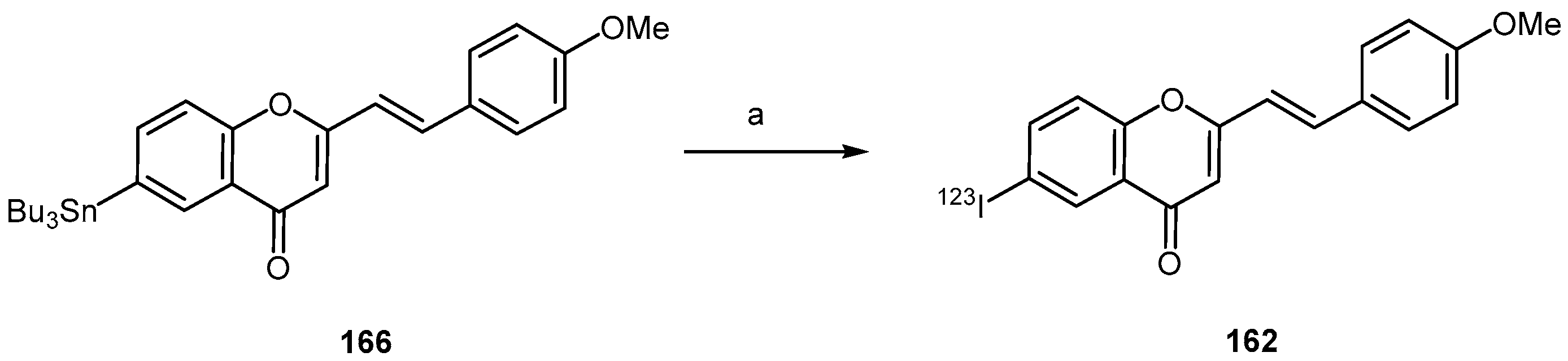
- Fuchigami, T.; Yamashita, Y.; Kawasaki, M.; Ogawa, A.; Haratake, M.; Atarashi, R.; Sano, K.; Nakagaki, T.; Ubagai, K.; Ono, M.; et al. Characterisation of radioiodinated flavonoid derivatives for SPECT imaging of cerebral prion deposits. Sci. Rep. 2015, 5, 18440. [Google Scholar] [CrossRef] [PubMed]
- Walle, T. Bioavailability of resveratrol. Ann. N. Y. Acad. Sci. 2011, 1215, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.L.; Kosmeder, J.W.; Pezzuto, J.M. Biological effects of resveratrol. Antioxid. Redox Signal. 2001, 3, 1041–1064. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.-K.; Moon, M.-H.; Bae, B.-C.; Lee, Y.-J.; Seol, J.-W.; Kang, H.-S.; Kim, J.-S.; Kang, S.-J.; Park, S.-Y. Autophagy induced by resveratrol prevents human prion protein-mediated neurotoxicity. Neurosci. Res. 2012, 73, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.S.; Moon, M.-H.; Jeong, J.K.; Seol, J.W.; Lee, Y.J.; Park, B.H.; Park, S.Y. SIRT1, a histone deacetylase, regulates prion proteininduced neuronal cell death. Neurobiol. Aging 2012, 33, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.K.; Moon, M.H.; Lee, Y.J.; Seol, J.W.; Park, S.Y. Autophagy induced by the class III histone deacetylase Sirt1 prevents prion peptide neurotoxicity. Neurobiol. Aging 2013, 34, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, J.; Shi, Q.; Zhang, B.Y.; Chen, C.; Chen, L.N.; Sun, J.; Wang, H. Scrapie infection in experimental rodents and SMBS15 cells decreased the brain endogenous levels and activities of Sirt1. J. Mol. Neurosci. 2015, 55, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, B.-Y.; Zhang, J.; Xiao, K.; Chen, L.-N.; Wang, H.; Sun, J.; Shi1, Q.; Dong, X.-P. Treatment of SMB-S15 Cells with Resveratrol Efficiently Removes the PrPSc Accumulation In Vitro and Prion Infectivity In Vivo. Mol. Neurobiol. 2016, 53, 5367–5376. [Google Scholar] [CrossRef] [PubMed]
- Ismail, T.; Shafi, S.; Srinivas, J.; Sarkar, D.; Qurishi, Y.; Khazir, J.; Alam, M.S.; Kumar, H.M.S. Synthesis and tyrosinase inhibition activity of trans-stilbene derivatives. Bioorg. Chem. 2016, 64, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Birar, V.C.; Sheerin, A.N.; Milkovicova, J.; Faragher, R.G.A.; Ostler, E.L. A facile, stereoselective, one-pot synthesis of resveratrol derivatives. Chem. Cent. J. 2015, 9, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.K.; Kumar, V.; Sharma, A.; Sharma, A.; Kumar, R. An unusual, mild and convenient one-pot two-step access to (E)-stilbenes from hydroxy-substituted benzaldehydes and phenylacetic acids under microwave activation: A new facet of the classical Perkin reaction. Tetrahedron 2007, 63, 11070–11077. [Google Scholar] [CrossRef]
- Ferré-Filmon, K.; Delaude, L.; Demonceau, A.; Noels, A.F. Stereoselective Synthesis O (E)-Hydroxystilbenoids by Ruthenium-Catalyzed Cross-Metathesis. Eur. J. Org. Chem. 2005, 3319–3325. [Google Scholar] [CrossRef]
- Schmidt, B.; Elizarov, N.; Berger, R.; Hölter, F. Scope and limitations of the Heck–Matsuda-coupling of phenol diazonium salts and styrenes: A protecting group economic synthesis of phenolic stilbenes. Org. Biomol. Chem. 2013, 11, 3674–3691. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.V.; García, J.I.; Mayoral, J.A. An expedient synthesis of resveratrol through a highly recoverable. Tetrahedron 2016, 1–4. (in press). [Google Scholar]
- El-Deeb, I.Y.; Tatsuya Funakoshi, T.; Shimomoto, Y.; Matsubara, R.; Hayashi, M. Dehydrogenative Formation of Resorcinol Derivatives Using Pd/C−Ethylene Catalytic System. J. Org. Chem. 2017, 82, 2630–2640. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Raymond, L.D.; Raymond, G.J.; Maxson, L.; Silveir, J.; Baron, G.S. Inhibition of protease-resistant prion protein accumulation in vitro by curcumin. J. Virol. 2003, 77, 5499–5502. [Google Scholar] [CrossRef] [PubMed]
- Hafner-Bratkovic, I.; Gaspersic, J.; Smid, L.M.; Bresjanac, M.; Jerala, R. Curcumin binds to the alpha-helical intermediate and to the amyloid form of prion protein—A new mechanism for the inhibition of PrPSc accumulation. J. Neurochem. 2008, 104, 1553–1564. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-F.; Yu, K.-H.; Jheng, C.-P.; Chung, R.; Lee, C.-I. Curcumin Reduces Amyloid Fibrillation of Prion Protein and Decreases Reactive Oxidative Stress. Pathogens 2013, 2, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Pabon, H.J.J. A synthesis of curcumin and related compounds. Rec. Trav. Chim. 1964, 83, 379–386. [Google Scholar] [CrossRef]
- Khan, N.; Afaq, F.; Saleem, M.; Ahmad, N.; Mukhtar, H. Targeting multiple signaling pathways by green tea polyphenol (−)-epigallocatechin-3-gallate. Cancer Res. 2006, 66, 2500–2505. [Google Scholar] [CrossRef] [PubMed]
- Ramassamy, C. Emerging role of polyphenolic compounds in the treatment of neurodegenerative diseases: A review of their intracellular targets. Eur. J. Pharmacol. 2006, 545, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Chacko, S.M.; Thambi, P.T.; Kuttan, R.; Nishigaki, I. Beneficial effects of green tea: A literature review. Chin. Med. 2010, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Ingolfsson, H.I.; Thakur, P.; Herold, K.F.; Hobart, E.A.; Ramsey, N.B.; Periole, X.; de Jong, D.H.; Zwama, M.; Yilmaz, D.; Hall, K.; et al. Phytochemicals Perturb Membranes and Promiscuously Alter Protein Function. ACS Chem. Biol. 2014, 9, 1788–1798. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, H.; Koga, K.; Fujimura, Y.; Yamada, K. A receptor for green tea polyphenol EGCG. Nat. Struct. Mol. Biol. 2004, 11, 380–381. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Gordon, O.N.; Edwards, R.L.; Luis, P.B.J. Degradation of Curcumin: From Mechanism to Biological Implications. Agric. Food Chem. 2015, 63, 7606–7614. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B. Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Tu, T.; D’Avignon, D.A.; Gross, M.L. Balance of Beneficial and Deleterious Health Effects of Quinones: A Case Study of the Chemical Properties of Genistein and Estrone Quinones. J. Am. Chem. Soc. 2009, 131, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Dohler, F.; Sepulveda-Falla, D.; Krasemann, S.; Altmeppen, H.; Schlüter, H.; Hildebrand, D.; Zerr, I.; Matschke, J.; Glatzel, M. High molecular mass assemblies of amyloid-β oligomers bind prion protein in patients with Alzheimer’s disease. Brain 2014, 137, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Gimbel, D.A.; Nygaard, H.B.; Coffey, E.E.; Gunther, E.C.; Laurén, J.; Gimbel, Z.A.; Strittmatter, S.M. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 2010, 30, 6367–6374. [Google Scholar] [CrossRef] [PubMed]




























































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | EC50 (μM) ScN2a | EC50 (μM) N167 | EC50 (μM) F3 | Binding to hPrP121–231 (%RUmax) | Permeability PAMPA-BBB Assay (logD7.4) | Efflux Ratio * MDCK-MDR1 |
|---|---|---|---|---|---|---|
| Quinacrine | 0.23 | 0.59 | 1.88 | 87.3 | 1.89 | 5 |
| 37 | 0.10 | 0.42 | 0.68 | 120.3 | 3.36 | 2.4 |
| 38 | 0.42 | 0.49 | 0.80 | 67.5 | 3.75 | - |
| 39 | 0.13 | 0.23 | 0.19 | 169.5 | 3.41 | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blázquez-Sánchez, M.T.; De Matos, A.M.; Rauter, A.P. Exploring Anti-Prion Glyco-Based and Aromatic Scaffolds: A Chemical Strategy for the Quality of Life. Molecules 2017, 22, 864. https://doi.org/10.3390/molecules22060864
Blázquez-Sánchez MT, De Matos AM, Rauter AP. Exploring Anti-Prion Glyco-Based and Aromatic Scaffolds: A Chemical Strategy for the Quality of Life. Molecules. 2017; 22(6):864. https://doi.org/10.3390/molecules22060864
Chicago/Turabian StyleBlázquez-Sánchez, María Teresa, Ana M. De Matos, and Amélia P. Rauter. 2017. "Exploring Anti-Prion Glyco-Based and Aromatic Scaffolds: A Chemical Strategy for the Quality of Life" Molecules 22, no. 6: 864. https://doi.org/10.3390/molecules22060864
APA StyleBlázquez-Sánchez, M. T., De Matos, A. M., & Rauter, A. P. (2017). Exploring Anti-Prion Glyco-Based and Aromatic Scaffolds: A Chemical Strategy for the Quality of Life. Molecules, 22(6), 864. https://doi.org/10.3390/molecules22060864