Nortriterpenoids from the Fruiting Bodies of the Mushroom Ganoderma resinaceum


Abstract
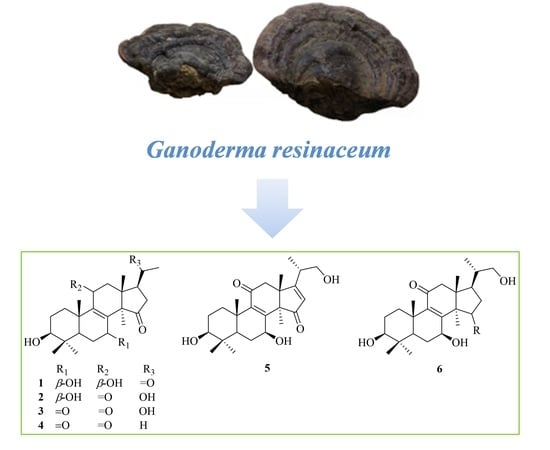
:
1. Introduction
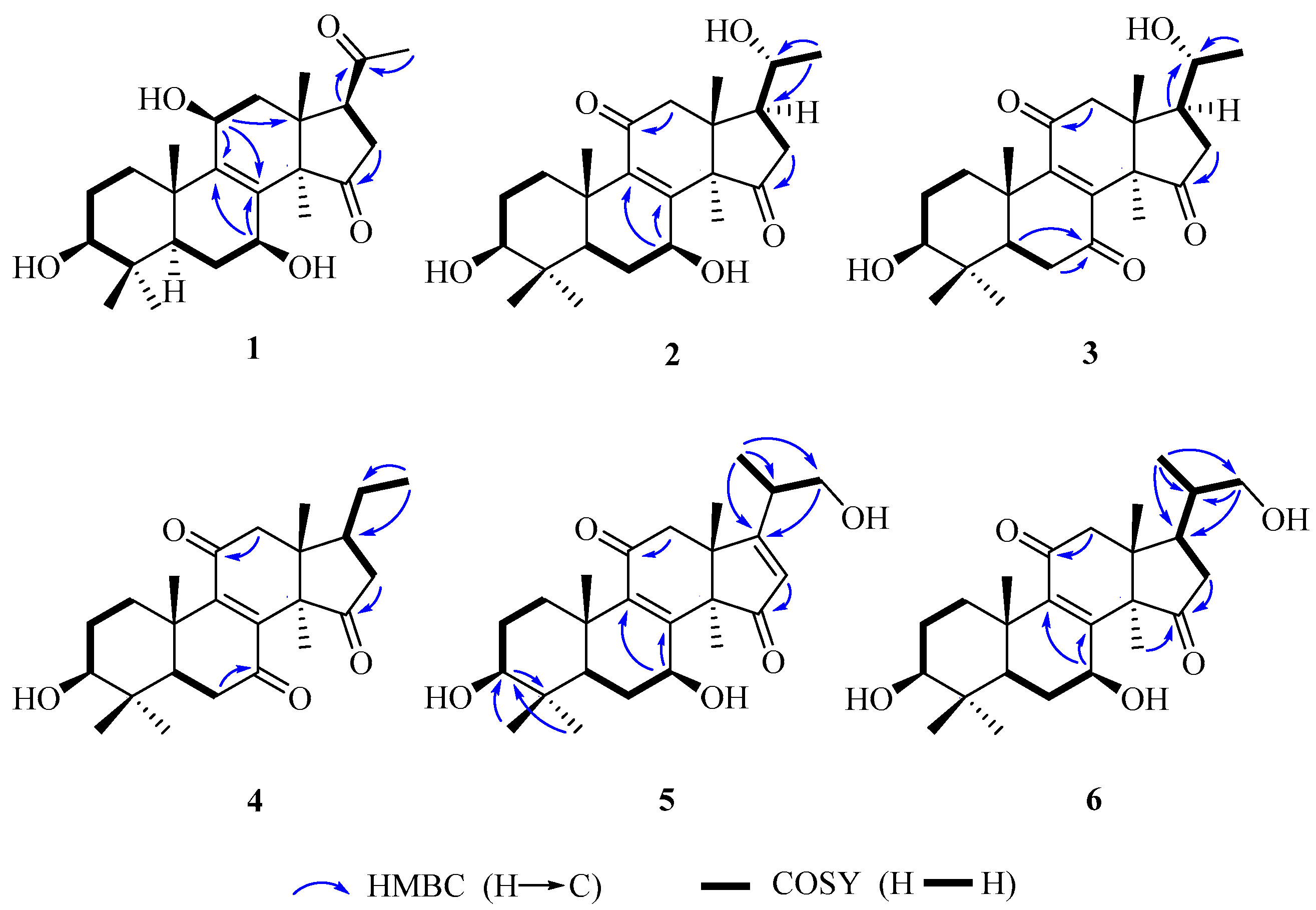
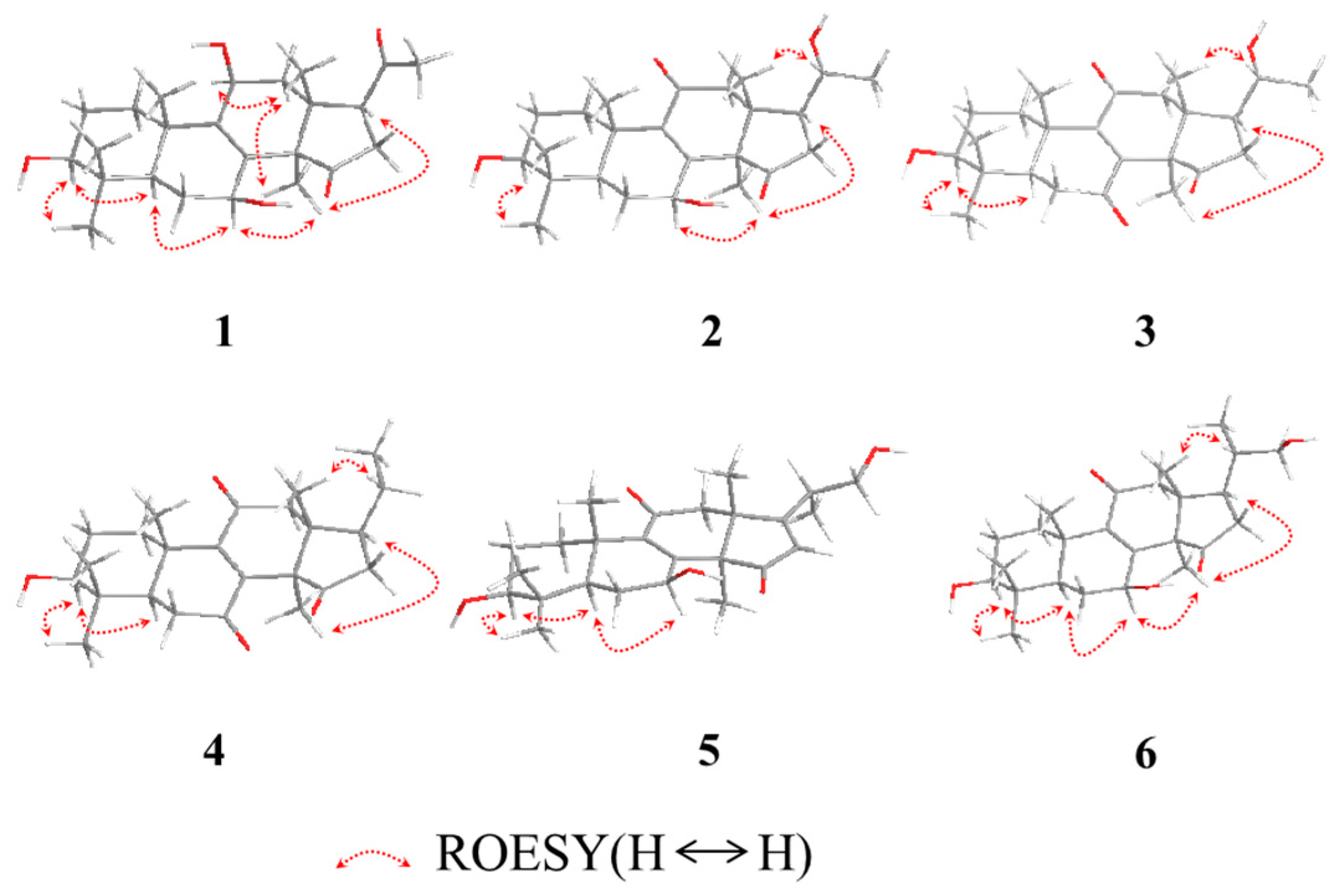
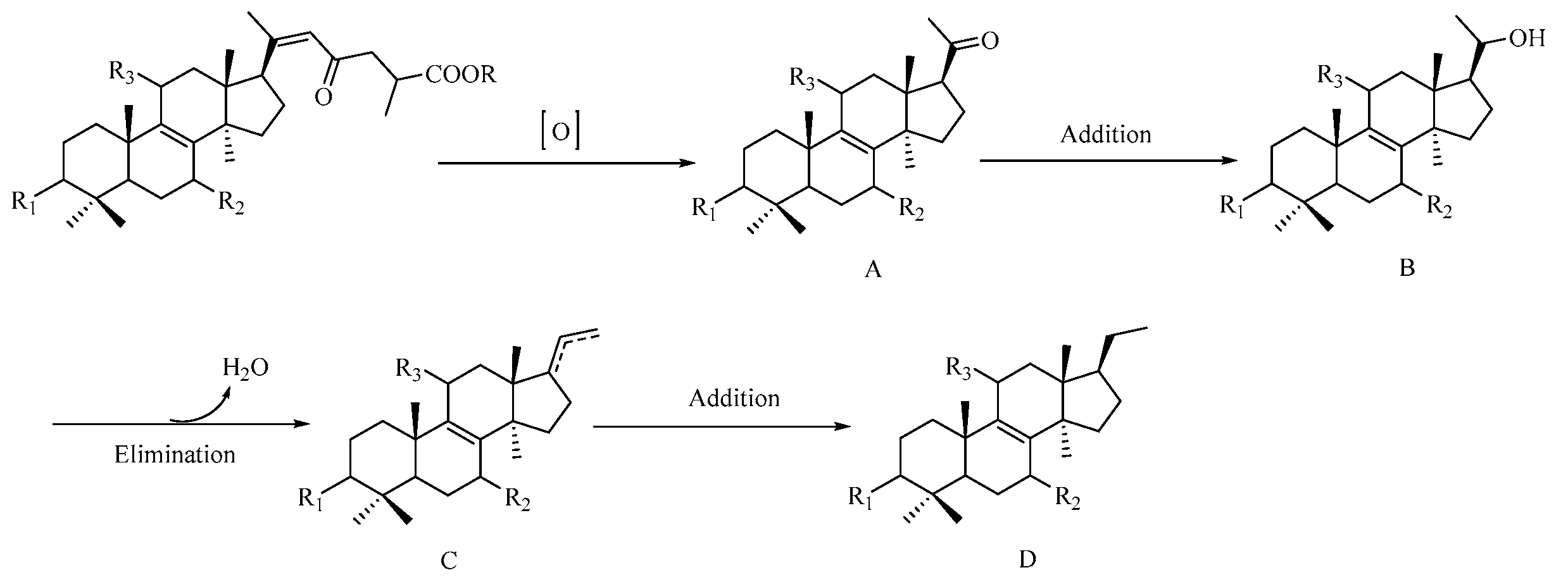
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Extraction and Isolation
3.3.1. Lucidone I (1)
3.3.2. Lucidone J (2)
3.3.3. Lucidone K (3)
3.3.4. Lucidone L (4)
3.3.5. Ganosineniol B (5)
3.3.6. Ganosineniol C (6)
3.4. Activity Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhao, J.; Zhang, X.-Q.; Li, S.-P.; Yang, F.-Q.; Wang, Y.-T.; Ye, W.-C. Quality evaluation of Ganoderma through simultaneous determination of nine triterpenes and sterols using pressurized liquid extraction and high performance liquid chromatography. J. Sep. Sci. 2006, 29, 2609–2615. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-W.; Gao, J.-L.; Guan, J.; Qian, Z.-M.; Feng, K.; Li, S.-P. Evaluation of Antiproliferative Activities and Action Mechanisms of Extracts from Two Species of Ganoderma on Tumor Cell Lines. J. Agric. Food Chem. 2009, 57, 3087–3093. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-F.; Liu, J.-Q.; Yan, Y.-X.; Chen, J.-C.; Lu, Y.; Guo, Y.-H.; Qiu, M.-H. Three new triterpenoids containing four-membered ring from the fruiting body of Ganoderma sinense. Org. Lett. 2010, 12, 1656–1659. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-L.; Yu, Y.-S.; Yen, G.-C. Lucidenic acid B induces apoptosis in human leukemia cells via a mitochondria-mediated pathway. J. Agric. Food Chem. 2008, 56, 3973–3980. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki, K.; Akihisa, T.; Tokuda, H.; Ukiya, M.; Oshikubo, M.; Kimura, Y.; Asano, T.; Nomura, A.; Nishino, H. Lucidenic acids P and Q, methyl lucidenate P, and other triterpenoids from the fungus Ganoderma lucidum and their inhibitory effects on Epstein-Barr virus activation. J. Nat. Prod. 2003, 66, 1582–1585. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.S.; Shi, L.S.; Kuo, S.C. Cytotoxicity of Ganoderma lucidum triterpenes. J. Nat. Prod. 2001, 64, 1121–1122. [Google Scholar] [CrossRef] [PubMed]
- Akihisa, T.; Nakamura, Y.; Tagata, M.; Tokuda, H.; Yasukawa, K.; Uchiyama, E.; Suzuki, T.; Kimura, Y. Anti-inflammatory and anti-tumor-promoting effects of triterpene acids and sterols from the fungus Ganoderma lucidum. Chem. Biodivers. 2007, 4, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-Q.; Ip, F.C.F.; Zhang, D.-M.; Chen, L.-X.; Zhang, W.; Li, Y.-L.; Ip, N.Y.; Ye, W.-C. Triterpenoids with neurotrophic activity from Ganoderma lucidum. Nat. Prod. Res. 2011, 25, 1607–1613. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.-R.; Liu, J.-Q.; Han, Z.-H.; Yuan, X.-X.; Luo, H.-R.; Qiu, M.-H. Protective effects of triterpenoids from Ganoderma resinaceum on H2O2-induced toxicity in HepG2 cells. Food Chem. 2013, 141, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Zhang, Q.; Ma, C.-M.; Hattori, M. Anti-human immunodeficiency virus-1 protease activity of new lanostane-type triterpenoids from Ganoderma sinense. Chem. Pharm. Bull. 2009, 57, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Ayissi, B.M.K.; Mossebo, D.C. Some noteworthy taxonomic variations in the complex wood-decayer Ganoderma resinaceum (Basidiomycota) with reference to collections from tropical Africa. Kew Bull. 2014, 69, 9542. [Google Scholar] [CrossRef]
- Zengin, G.; Sarikurkcu, C.; Gunes, E.; Uysal, A.; Ceylan, R.; Uysal, S.; Gungor, H.; Aktumsek, A. Two Ganoderma species: Profiling of phenolic compounds by HPLC-DAD, antioxidant, antimicrobial and inhibitory activities on key enzymes linked to diabetes mellitus, Alzheimer’s disease and skin disorders. Food Funct. 2015, 6, 2794–2802. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.-M.; Li, S.-H.; Xiao, W.-L.; Sun, H.-D.; Che, C.-T. Two new lanostanoids from Ganoderma resinaceum. J. Asian Nat. Prod. Res. 2007, 9, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Nishitoba, T.; Sato, H.; Sakamura, S. New terpenoids from Ganoderma lucidum and their bitterness. Agric. Biol. Chem. 1985, 49, 1547–1549. [Google Scholar] [CrossRef]
- Nishitoba, T.; Sato, H.; Sakamura, S. New terpenoids, ganolucidic acid D, ganoderic acid L, lucidone C and lucidenic acid G, from the fungus Ganoderma lucidum. Agric. Biol. Chem. 1986, 50, 809–811. [Google Scholar] [CrossRef]
- Nishitoba, T.; Sato, H.; Sakamura, S. Triterpenoids from the fungus Ganoderma lucidum. Phytochemistry 1987, 26, 1777–1784. [Google Scholar] [CrossRef]
- Li, W.; Lou, L.-L.; Zhu, J.-Y.; Zhang, J.-S.; Liang, A.-A.; Bao, J.-M.; Tang, G.-H.; Yin, S. New lanostane-type triterpenoids from the fruiting body of Ganoderma hainanense. Fitoterapia 2016, 115, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-Q.; Wang, C.-F.; Li, Y.; Luo, H.-R.; Qiu, M.-H. Isolation and bioactivity evaluation of terpenoids from the medicinal fungus Ganoderma sinense. Planta Med. 2012, 78, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Oyetayo, O.V. Medicinal Uses of Mushrooms in Nigeria: Towards Full and Sustainable Exploitation. Afr. J. Tradit. Complement. Altern. Med. 2011, 8, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-R.; Huo, X.-K.; Dong, P.-P.; Wang, C.; Huang, S.-S.; Zhang, B.-J.; Zhang, H.-L.; Deng, S.; Liu, K.-X.; Ma, X.-C.; et al. Inhibitory Effects of Highly Oxygenated Lanostane Derivatives from the Fungus Ganoderma lucidum on P-Glycoprotein and α-Glucosidase. J. Nat. Prod. 2015, 78, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Bao, L.; Xiong, W.; Ma, K.; Han, J.; Wang, W.; Yin, W.; Liu, H. Lanostane triterpenes from the Tibetan medicinal mushroom Ganoderma leucocontextum and their inhibitory effects on HMG-CoA reductase and α-glucosidase. J. Nat. Prod. 2015, 78, 1977–1989. [Google Scholar] [CrossRef] [PubMed]
- Fatmawati, S.; Kondo, R.; Shimizu, K. Structure-activity relationships of lanostane-type triterpenoids from Ganoderma lingzhi as α-glucosidase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 5900–5903. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-Q.; Qian, Z.-M.; Li, S.-P. Inhibition of Three Selected Beverage Extracts on α-Glucosidase and Rapid Identification of Their Active Compounds Using HPLC-DAD-MS/MS and Biochemical Detection. J. Agric. Food Chem. 2010, 58, 6608–6613. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–15 are available from corresponding authors. |





{kind=link}
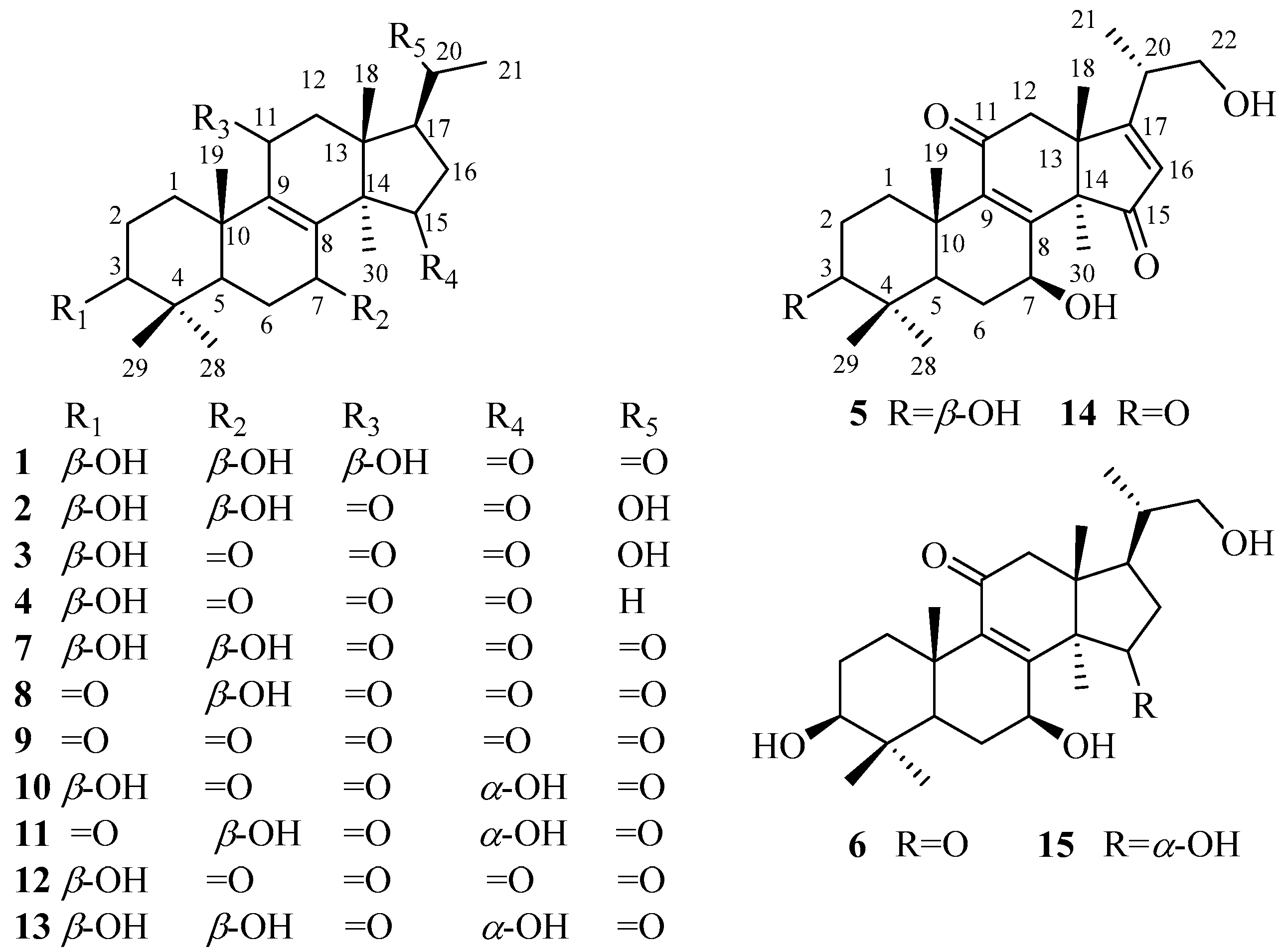{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|
| δC mult. | δH mult. (J in Hz) | δC mult. | δH mult. (J in Hz) | δC mult. | δH mult. (J in Hz) | δC mult. | δH mult. (J in Hz) | |
| 1 | 36.0, CH2 | 2.20, dt (12.6, 3.0); 1.36, m | 36.0, CH2 | 2.78, dt (13.8, 3.0); 1.02, m | 34.9, CH2 | 2.75, dt (13.8, 3.6); 1.16, dt (13.8, 3.6) | 34.9, CH2 | 2.80, m; 1.26, dt (13.5, 3.0) |
| 2 | 28.3, CH2 | 1.60, m; 1.53, m | 28.4, CH2 | 1.60, m; 1.68, m | 28.5, CH2 | 1.66, m; 1.60, m | 28.2, CH2 | 1.72, brd (13.2); 1.66, m |
| 3 | 79.5, CH | 3.08, dd (12.0, 4.2) | 79.1, CH | 3.17, dd (12.0, 4.8) | 78.3, CH | 3.14, dd (11.4, 4.8) | 78.3, CH | 3.21, dd (12.0, 4.2) |
| 4 | 40.1, C | 39.4, C | 40.4, C | 40.3, C | ||||
| 5 | 51.7, CH | 0.93, m | 50.4, CH | 0.96, d (13.2) | 52.8, CH | 1.50, d(1.8) | 52.6, CH | 1.60, d (14.4) |
| 6 | 28.94, CH2 | 2.10, m; 1.61, m | 28.0, CH2 | 2.20, dd (13.2, 9.0); 1.59, m | 37.5, CH2 | 2.60, d (14.4); 2.40, dd (14.4, 1.8) | 37.5, CH2 | 2.68, t (14.4); 2.49, d (14.4) |
| 7 | 66.9, CH | 4.54, t (9.6) | 68.2, CH | 4.85, t (9.0) | 202.3, C | 202.0, C | ||
| 8 | 136.8, C | 158.2, C | 147.3, C | 147.8, C | ||||
| 9 | 146.2, C | 143.3, C | 154.1, C | 153.6, C | ||||
| 10 | 40.2, C | 39.8, C | 41.9, C | 41.9, C | ||||
| 11 | 65.3, CH | 4.48, d (7.2) | 201.5, C | 202.3, C | 201.5, C | |||
| 12 | 42.0, CH2 | 2.51, dd (14.4, 7.2); 2.00, d (14.4) | 51.3, CH2 | 2.89, d (16.2); 2.82, d (16.2) | 50.2, CH2 | 2.90, d (15.6); 2.78, d (15.6) | 50.5, CH2 | 3.05, d (15.6); 2.65, d (15.6) |
| 13 | 43.8 C | 46.6, C | 45.3, C | 45.7, C | ||||
| 14 | 61.0, C | 60.3, C | 58.1, C | 58.7, C | ||||
| 15 | 220.2, C | 218.6, C | 210.2, C | 210.8, C | ||||
| 16 | 37.3, CH2 | 2.82, dd (19.8, 8.4); 2.45, dd (19.8, 9.0) | 40.0, CH2 | 2.63, dd (19.8, 9.0); 2.06, dd (19.8, 9.6) | 38.5, CH2 | 2.60, dd (18.6, 9.0); 1.69, dd (18.6, 8.4) | 41.3, CH2 | 2.82, m; 1.87, dd (18.0, 7.8) |
| 17 | 54.8, CH | 3.22, overlapped | 48.9, CH | 2.28, dd (18.6, 9.0) | 47.9, CH | 2.28, dd (18.6, 9.6) | 46.2, CH | 2.34, m |
| 18 | 19.7, CH3 | 0.79, s | 18.1, CH3 | 1.03, s | 16.9, CH3 | 0.80, s | 16.7, CH3 | 0.83, s |
| 19 | 22.2, CH3 | 1.21, s | 19.0, CH3 | 1.25, s | 18.2, CH3 | 1.26, s | 18.3, CH3 | 1.30, s |
| 20 | 209.3, C | 69.9, CH | 3.79, m | 69.9, CH | 3.65, m | 32.9, CH2 | 2.13, m | |
| 21 | 31.8, CH3 | 2.13, s | 23.8, CH3 | 1.20, d (6.0) | 23.7, CH3 | 1.10, d (6.0) | 20.5, CH3 | 0.99, t (6.0) |
| 28 | 28.89, CH3 | 0.90, s | 28.8, CH3 | 1.04, s | 28.2, CH3 | 0.94, s | 28.5, CH3 | 1.00, s |
| 29 | 16.2, CH3 | 0.77, s | 16.4, CH3 | 0.86, s | 16.8, CH3 | 1.13, s | 16.3, CH3 | 0.88, s |
| 30 | 23.5, CH3 | 1.08, s | 24.6, CH3 | 1.42, s | 16.3, CH3 | 0.82, s | 22.0, CH3 | 1.57, s |
| No. | 5a | 6b | ||
|---|---|---|---|---|
| δC mult. | δH mult. (J in Hz) | δC mult. | δH mult. (J in Hz) | |
| 1 | 34.7, CH2 | 2.94, dt (13.2, 3.6); 1.07, m | 36.0, CH2 | 2.71, dt (13.2, 3.6); 0.95, m |
| 2 | 27.7, CH2 | 1.70, m | 28.4, CH2 | 1.58, m; 1.53, m |
| 3 | 78.2, CH | 3.26, dd (12.0, 4.8) | 79.0, CH | 3.08, dd (12.0, 4.8) |
| 4 | 38.7, C | 39.7, C | ||
| 5 | 49.3, CH | 0.98, d (13.2) | 50.4, CH | 0.87, d (13.8) |
| 6 | 26.1, CH2 | 2.20, dd (13.2, 7.2); 1.64, m | 28.1, CH2 | 2.11, m; 1.49, m |
| 7 | 67.1, CH | 4.84, d (4.2) | 68.1, CH | 4.78, t (3.0) |
| 8 | 158.4, C | 159.0, C | ||
| 9 | 142.2, C | 144.2, C | ||
| 10 | 39.2, C | 40.0, C | ||
| 11 | 197.6, C | 200.6, C | ||
| 12 | 44.2, CH2 | 2.98, d (16.2); 2.58, d (16.2) | 51.6, CH2 | 2.85, d (16.8); 2.55, d (16.8) |
| 13 | 51.3, C | 46.7, C | ||
| 14 | 58.2, C | 60.2, C | ||
| 15 | 211.0, C | 218.9, C | ||
| 16 | 123.5, CH | 5.84, s | 41.6, CH2 | 2.68, d (13.2); 2.10, m |
| 17 | 185.6, C | 43.7, CH | 2.15, m | |
| 18 | 30.6, CH3 | 1.24, s | 16.4, CH3 | 0.77, s |
| 19 | 18.6, CH3 | 1.23, s | 19.0, CH3 | 1.15, s |
| 20 | 36.3, CH | 2.65, m | 39.8, CH | 1.58, m |
| 21 | 16.5, CH3 | 1.18, d (7.2) | 17.5, CH3 | 1.00, d (6.6) |
| 22 | 65.6, CH2 | 3.79, m | 67.6, CH2 | 3.45, dd (11.4, 3.3); 3.32, dd (11.4, 6.0) |
| 28 | 28.2, CH3 | 1.07, s | 28.8, CH3 | 0.95, s |
| 29 | 15.6, CH3 | 0.88, s | 18.0, CH3 | 0.91, s |
| 30 | 33.5, CH3 | 1.58, s | 25.0, CH3 | 1.30, s |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.-Q.; Chen, L.-X.; Zhao, J.; Tang, Y.-P.; Li, S.-P. Nortriterpenoids from the Fruiting Bodies of the Mushroom Ganoderma resinaceum. Molecules 2017, 22, 1073. https://doi.org/10.3390/molecules22071073
Chen X-Q, Chen L-X, Zhao J, Tang Y-P, Li S-P. Nortriterpenoids from the Fruiting Bodies of the Mushroom Ganoderma resinaceum. Molecules. 2017; 22(7):1073. https://doi.org/10.3390/molecules22071073
Chicago/Turabian StyleChen, Xian-Qiang, Ling-Xiao Chen, Jing Zhao, Yu-Ping Tang, and Shao-Ping Li. 2017. "Nortriterpenoids from the Fruiting Bodies of the Mushroom Ganoderma resinaceum" Molecules 22, no. 7: 1073. https://doi.org/10.3390/molecules22071073
APA StyleChen, X. -Q., Chen, L. -X., Zhao, J., Tang, Y. -P., & Li, S. -P. (2017). Nortriterpenoids from the Fruiting Bodies of the Mushroom Ganoderma resinaceum. Molecules, 22(7), 1073. https://doi.org/10.3390/molecules22071073