Review of Ligand Specificity Factors for CYP1A Subfamily Enzymes from Molecular Modeling Studies Reported to-Date


Abstract
:
1. Introduction
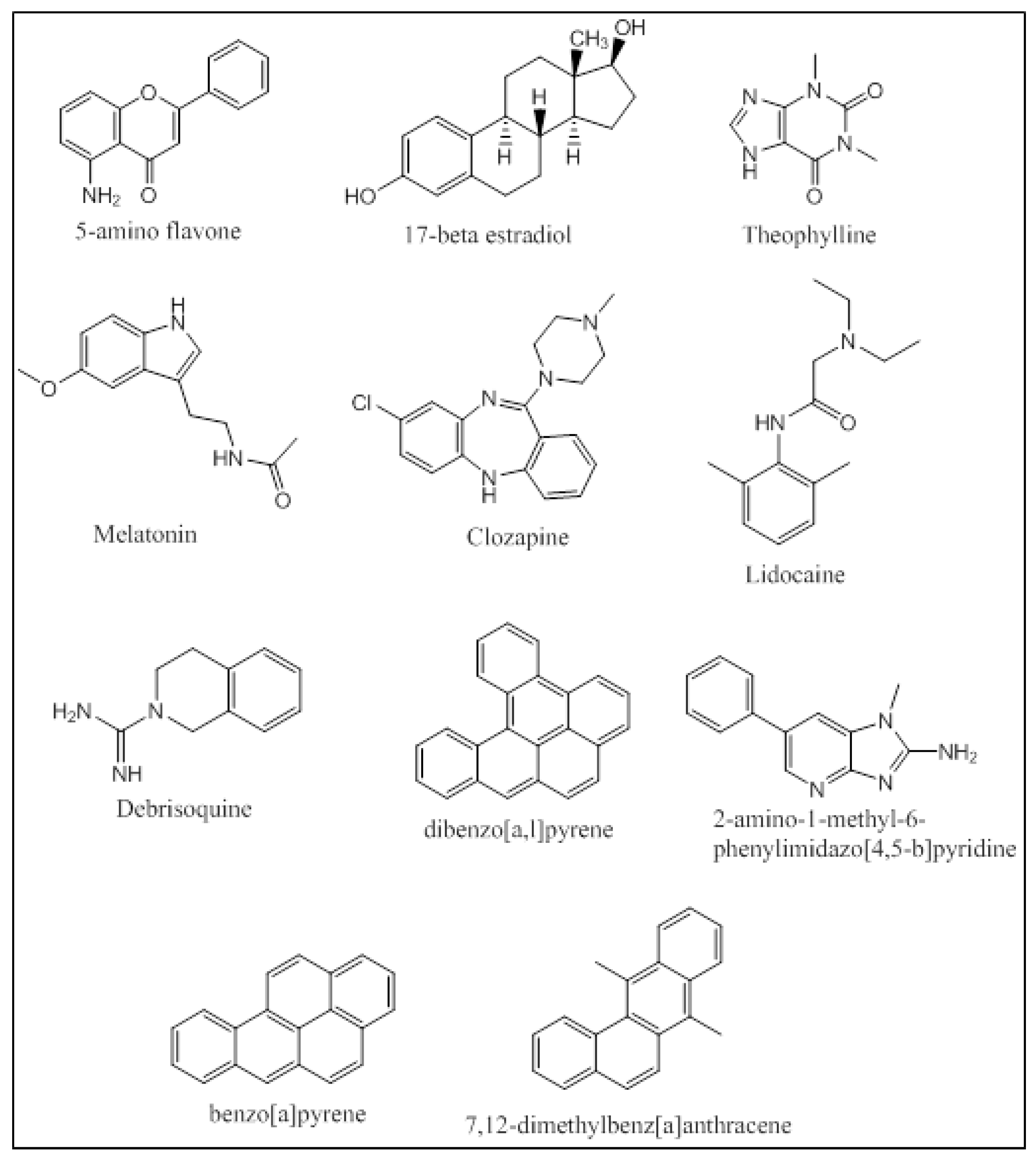
2. CYP1A Substrates
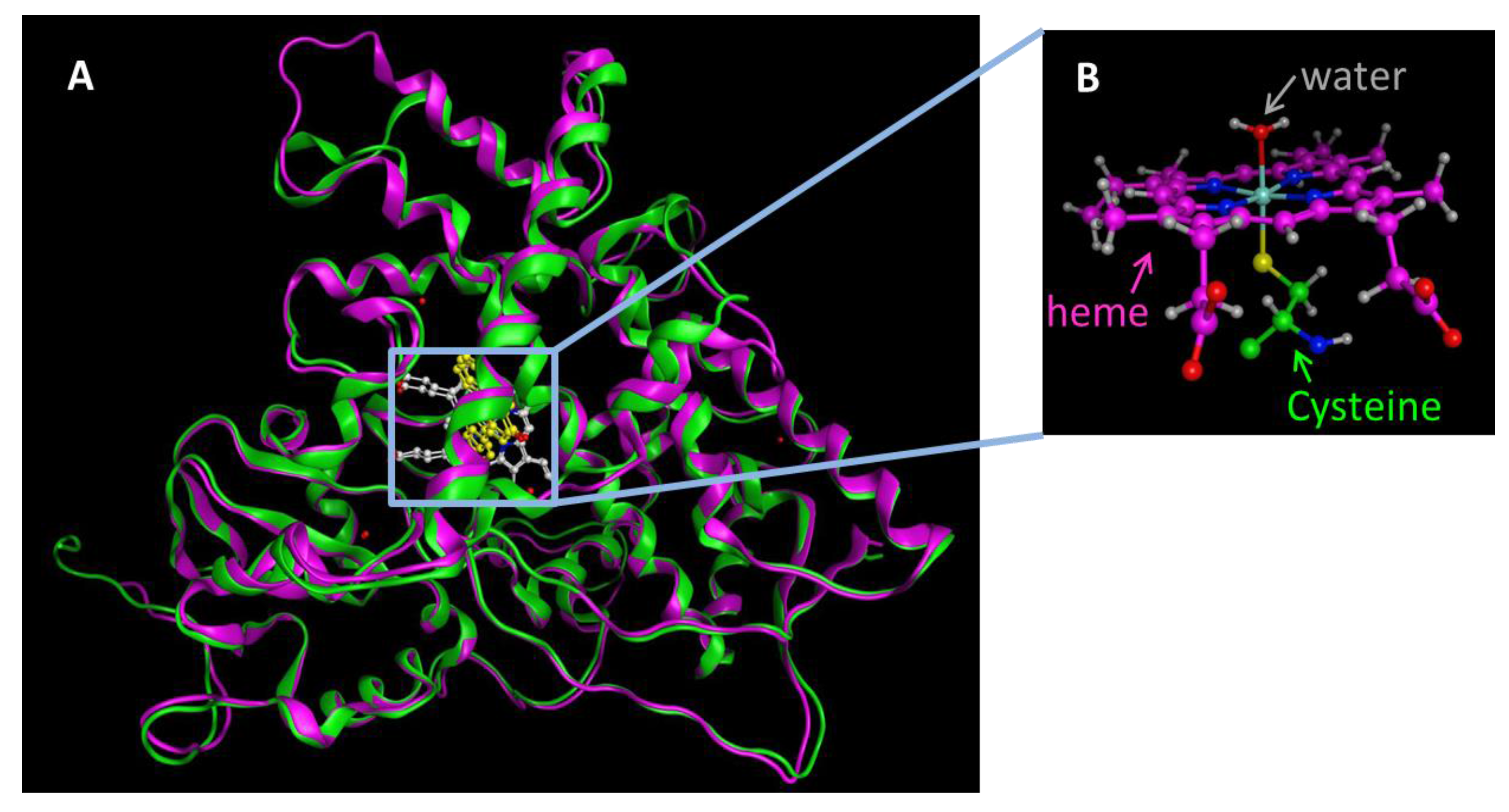
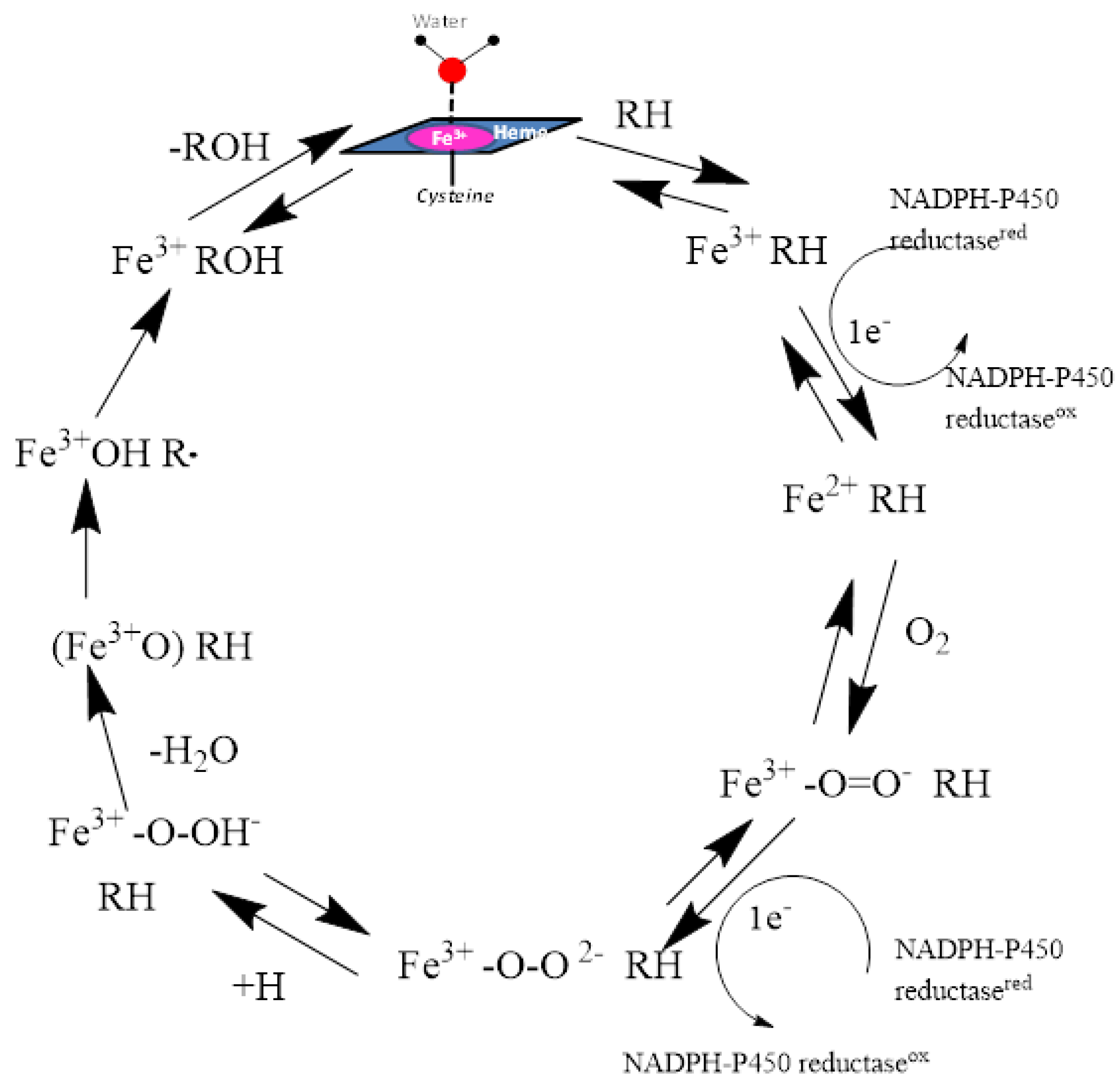
3. Structure and Catalytic Cycle
4. Inhibition of CYP1A Enzymes
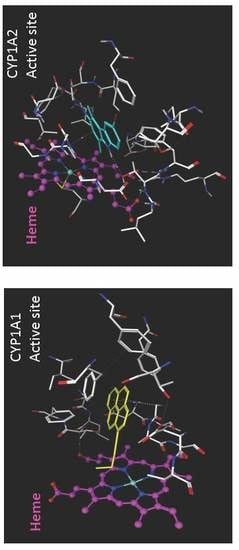
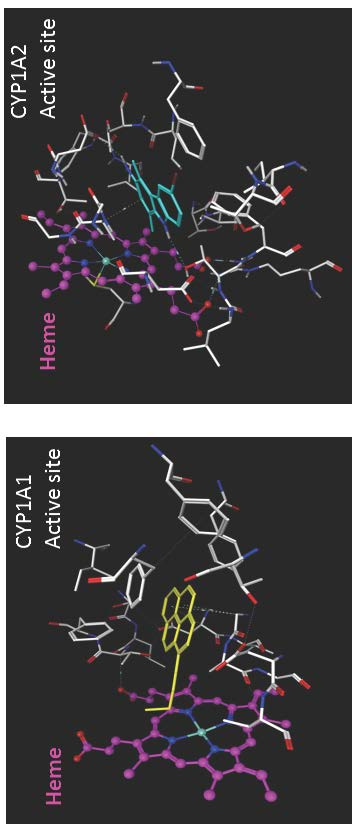
5. Substrate Binding Site Characteristics
6. Ligand-Based Studies on Isoform Selectivity
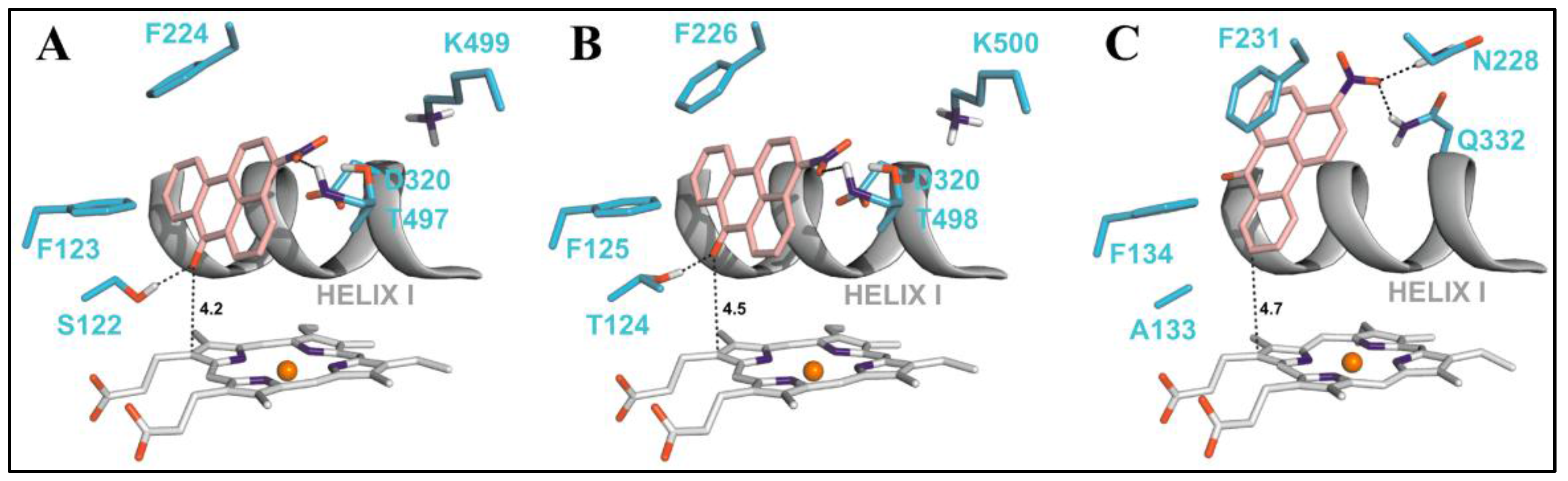
7. Protein-Based Studies Using Molecular Docking and Molecular Dynamics
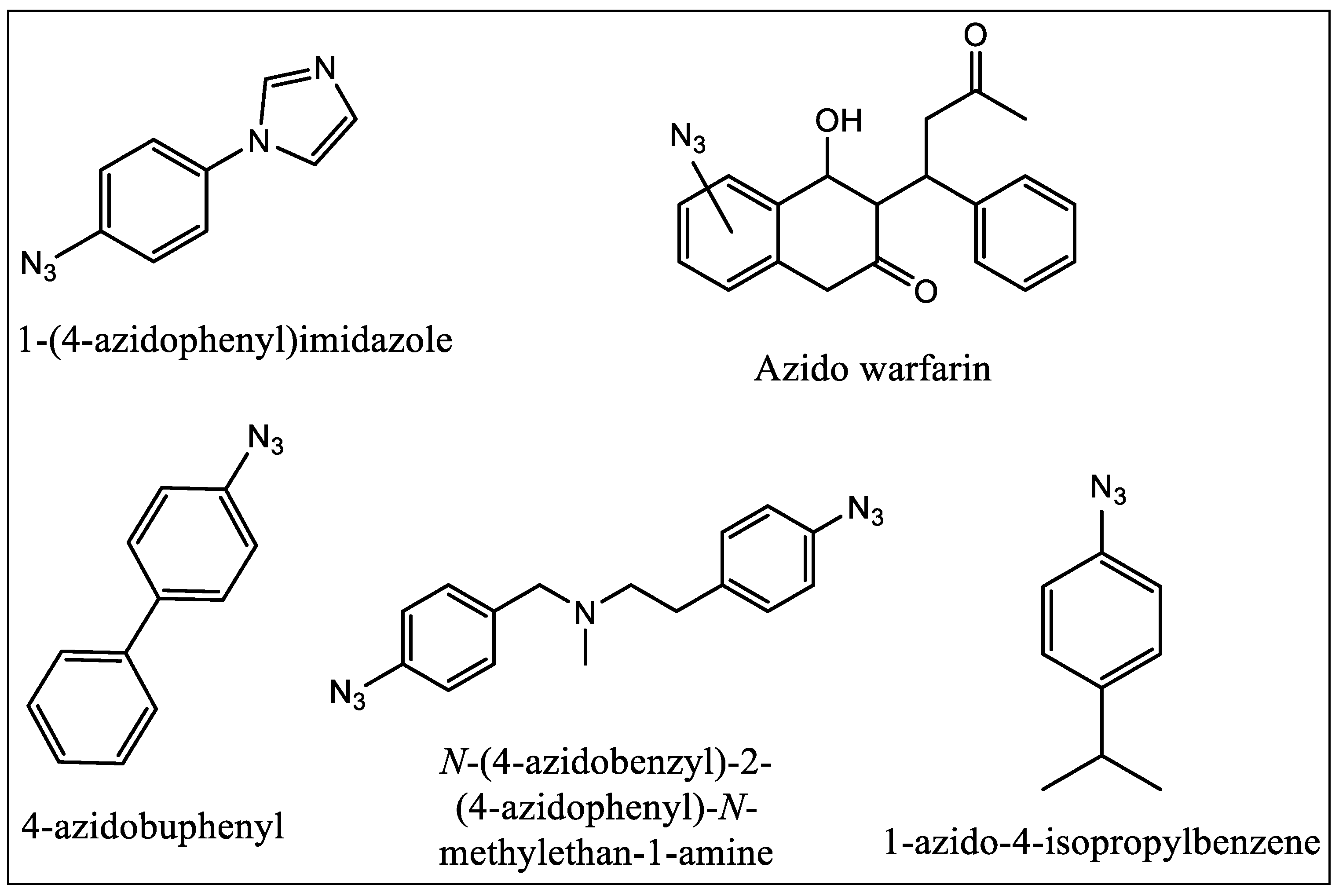
8. Photo Affinity Ligands (PALs) as Binding Site Probes
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Nelson, D.R. Progress in tracing the evolutionary paths of cytochrome P450. Biochim. Biophys. Acta 2011, 1814, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Corchero, J.; Pimprale, S.; Kimura, S.; Gonzalez, F.J. Organization of the CYP1A cluster on human chromosome 15: Implications for gene regulation. Pharmacogenetics 2001, 11, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ikeya, K.; Jaiswal, A.K.; Owens, R.A.; Jones, J.E.; Nebert, D.W.; Kimura, S. Human CYP1A2: Sequence, gene structure, comparison with the mouse and rat orthologous gene, and differences in liver 1A2 mRNA expression. Mol. Endocrinol. 1989, 3, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Kawajiri, K.; Watanabe, J.; Gotoh, O.; Tagashira, Y.; Sogawa, K.; Fujii-Kuriyama, Y. Structure and drug inducibility of the human cytochrome P-450c gene. Eur. J. Biochem. 1986, 159, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Yamazaki, H.; Mimura, M.; Inui, Y.; Guengerich, F.P. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: Studies with liver microsomes of 30 Japanese and 30 Caucasians. J. Pharmacol. Exp. Ther. 1994, 270, 414–423. [Google Scholar] [PubMed]
- Stiborova, M.; Martinek, V.; Rydlova, H.; Koblas, T.; Hodek, P. Expression of cytochrome P450 1A1 and its contribution to oxidation of a potential human carcinogen 1-phenylazo-2-naphthol (Sudan I) in human livers. Cancer Lett. 2005, 220, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Smith, G.; Wolf, C.R. Genetic polymorphisms in xenobiotic metabolism. Eur. J. Cancer 1994, 30, 1921–1935. [Google Scholar] [CrossRef]
- Lee, A.J.; Cai, M.X.; Thomas, P.E.; Conney, A.H.; Zhu, B.T. Characterization of the oxidative metabolites of 17beta-estradiol and estrone formed by 15 selectively expressed human cytochrome p450 isoforms. Endocrinology 2003, 144, 3382–3398. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Idle, J.R.; Krausz, K.W.; Gonzalez, F.J. Metabolism of melatonin by human cytochromes p450. Drug Metab. Dispos. 2005, 33, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.; Kisselev, P.; Ericksen, S.S.; Szklarz, G.D.; Chernogolov, A.; Honeck, H.; Schunck, W.H.; Roots, I. Arachidonic and eicosapentaenoic acid metabolism by human CYP1A1: Highly stereoselective formation of 17(R),18(S)-epoxyeicosatetraenoic acid. Biochem. Pharmacol. 2004, 67, 1445–1457. [Google Scholar] [CrossRef] [PubMed]
- Conney, A.H. Induction of microsomal enzymes by foreign chemicals and carcinogenesis by polycyclic aromatic hydrocarbons: G. H. A. Clowes Memorial Lecture. Cancer Res. 1982, 42, 4875–4917. [Google Scholar] [PubMed]
- Shimada, T.; Fujii-Kuriyama, Y. Metabolic activation of polycyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and 1B1. Cancer Sci. 2004, 95, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Guengerich, F.P. Cytochrome P450 activation of arylamines and heterocyclic amines. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 27–49. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S. Summary of information on human CYP enzymes: Human P450 metabolism data. Drug Metab. Rev. 2002, 34, 83–448. [Google Scholar] [CrossRef] [PubMed]
- Stiborova, M.; Frei, E.; Wiessler, M.; Schmeiser, H.H. Human enzymes involved in the metabolic activation of carcinogenic aristolochic acids: Evidence for reductive activation by cytochromes P450 1A1 and 1A2. Chem. Res. Toxicol. 2001, 14, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Ueng, Y.F.; Hsieh, C.H.; Don, M.J.; Chi, C.W.; Ho, L.K. Identification of the main human cytochrome P450 enzymes involved in safrole 1′-hydroxylation. Chem. Res. Toxicol. 2004, 17, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Granfors, M.T.; Backman, J.T.; Laitila, J.; Neuvonen, P.J. Tizanidine is mainly metabolized by cytochrome p450 1A2 in vitro. Br. J. Clin. Pharmacol. 2004, 57, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Gunes, A.; Dahl, M.L. Variation in CYP1A2 activity and its clinical implications: Influence of environmental factors and genetic polymorphisms. Pharmacogenomics 2008, 9, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Lobo, E.D.; Bergstrom, R.F.; Reddy, S.; Quinlan, T.; Chappell, J.; Hong, Q.; Ring, B.; Knadler, M.P. In vitro and in vivo evaluations of cytochrome P450 1A2 interactions with duloxetine. Clin. Pharmacokinet. 2008, 47, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Turpeinen, M.; Hofmann, U.; Klein, K.; Murdter, T.; Schwab, M.; Zanger, U.M. A predominate role of CYP1A2 for the metabolism of nabumetone to the active metabolite, 6-methoxy-2-naphthylacetic acid, in human liver microsomes. Drug Metab. Dispos. 2009, 37, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Sansen, S.; Yano, J.K.; Reynald, R.L.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J. Biol. Chem. 2007, 282, 14348–14355. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.A.; Szklarz, G.D.; Scott, E.E. Human cytochrome P450 1A1 structure and utility in understanding drug and xenobiotic metabolism. J. Biol. Chem. 2013, 288, 12932–12943. [Google Scholar] [CrossRef] [PubMed]
- Androutsopoulos, V.P.; Tsatsakis, A.M.; Spandidos, D.A. Cytochrome P450 CYP1A1: Wider roles in cancer progression and prevention. BMC Cancer 2009, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Liehr, J.G.; Ricci, M.J. 4-Hydroxylation of estrogens as marker of human mammary tumors. Proc. Natl. Acad. Sci. USA 1996, 93, 3294–3296. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S.; Guengerich, F.P. Contributions of human enzymes in carcinogen metabolism. Chem. Res. Toxicol. 2012, 25, 1316–1383. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F.; Chan, E.; Zhou, Z.W.; Xue, C.C.; Lai, X.; Duan, W. Insights into the structure, function, and regulation of human cytochrome P450 1A2. Curr. Drug Metab. 2009, 10, 713–729. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F.; Wang, B.; Yang, L.P.; Liu, J.P. Structure, function, regulation and polymorphism and the clinical significance of human cytochrome P450 1A2. Drug Metab. Rev. 2010, 42, 268–354. [Google Scholar] [CrossRef] [PubMed]
- Bruno, R.D.; Njar, V.C. Targeting cytochrome P450 enzymes: A new approach in anti-cancer drug development. Bioorg. Med. Chem. 2007, 15, 5047–5060. [Google Scholar] [CrossRef] [PubMed]
- Ueng, Y.F.; Jan, W.C.; Lin, L.C.; Chen, T.L.; Guengerich, F.P.; Chen, C.F. The alkaloid rutaecarpine is a selective inhibitor of cytochrome P450 1A in mouse and human liver microsomes. Drug Metab. Dispos. 2002, 30, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.W.; Britt, S.G.; Pohl, L.R. Inactivation of cytochrome P-450 by 2-isopropyl-4-pentenamide and other xenobiotics leads to heme-derived protein adducts. Chem. Biol. Interact. 1986, 58, 345–352. [Google Scholar] [CrossRef]
- Halpert, J. Further studies of the suicide inactivation of purified rat liver cytochrome P-450 by chloramphenicol. Mol. Pharmacol. 1982, 21, 166–172. [Google Scholar] [PubMed]
- Kesharwani, S.S.; Nandekar, P.P.; Pragyan, P.; Rathod, V.; Sangamwar, A.T. Characterization of differences in substrate specificity among CYP1A1, CYP1A2 and CYP1B1: An integrated approach employing molecular docking and molecular dynamics simulations. J. Mol. Recognit. 2016, 29, 370–390. [Google Scholar] [CrossRef] [PubMed]
- Kesharwani, S.S.; Nandekar, P.P.; Pragyan, P.; Sangamwar, A.T. Comparative proteomics among cytochrome p450 family 1 for differential substrate specificity. Protein J. 2014, 33, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Taylor, S.F.; Dupart, P.S.; Arnold, C.L.; Sridhar, J.; Jiang, Q.; Wang, Y.; Skripnikova, E.V.; Zhao, M.; Foroozesh, M. Pyranoflavones: A group of small-molecule probes for exploring the active site cavities of cytochrome P450 enzymes 1A1, 1A2, and 1B1. J. Med. Chem. 2013, 56, 4082–4092. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sridhar, J.; Foroozesh, M. Cytochrome P450 family 1 inhibitors and structure-activity relationships. Molecules 2013, 18, 14470–14495. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, J.; Liu, J.; Foroozesh, M.; Klein Stevens, C.L. Inhibition of cytochrome p450 enzymes by quinones and anthraquinones. Chem. Res. Toxicol. 2012, 25, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, J.; Liu, J.; Komati, R.; Schroeder, R.; Jiang, Q.; Tram, P.; Riley, K.; Foroozesh, M. Ortho-Methylarylamines as Time-Dependent Inhibitors of Cytochrome P450 1A1 Enzyme. Drug Metab. Lett. 2016. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, J.; Liu, J.; Foroozesh, M.; Stevens, C.L. Insights on cytochrome p450 enzymes and inhibitors obtained through QSAR studies. Molecules 2012, 17, 9283–9305. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.F.; Lake, B.G.; Dickins, M. Quantitative structure-activity relationships (QSARs) in inhibitors of various cytochromes P450: The importance of compound lipophilicity. J. Enzym. Inhib. Med. Chem. 2007, 22, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.F.; Modi, S.; Dickins, M. Structure-activity relationship for human cytochrome P450 substrates and inhibitors. Drug Metab. Rev. 2002, 34, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.; Marchand-Geneste, N.; Giraudel, J.L.; Shimada, T. Docking and QSAR comparative studies of polycyclic aromatic hydrocarbons and other procarcinogen interactions with cytochromes P450 1A1 and 1B1. SAR QSAR Environ. Res. 2012, 23, 87–109. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.F.; Dickins, M. Substrate SARs in human P450s. Drug Discov. Today 2002, 7, 918–925. [Google Scholar] [CrossRef]
- Lewis, D.F.; Jacobs, M.N.; Dickins, M.; Lake, B.G. Quantitative structure--activity relationships for inducers of cytochromes P450 and nuclear receptor ligands involved in P450 regulation within the CYP1, CYP2, CYP3 and CYP4 families. Toxicology 2002, 176, 51–57. [Google Scholar] [CrossRef]
- Shimada, T.; Oda, Y.; Gillam, E.M.; Guengerich, F.P.; Inoue, K. Metabolic activation of polycyclic aromatic hydrocarbons and other procarcinogens by cytochromes P450 1A1 and P450 1B1 allelic variants and other human cytochromes P450 in Salmonella typhimurium NM2009. Drug Metab. Dispos. 2001, 29, 1176–1182. [Google Scholar] [PubMed]
- Sridhar, J.; Ellis, J.; Dupart, P.; Liu, J.; Stevens, C.L.; Foroozesh, M. Development of flavone propargyl ethers as potent and selective inhibitors of cytochrome P450 enzymes 1A1 and 1A2. Drug Metab. Lett. 2012, 6, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Iori, F.; da Fonseca, R.; Ramos, M.J.; Menziani, M.C. Theoretical quantitative structure-activity relationships of flavone ligands interacting with cytochrome P450 1A1 and 1A2 isozymes. Bioorg. Med. Chem. 2005, 13, 4366–4374. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, J.; Foroozesh, M.; Stevens, C.L. QSAR models of cytochrome P450 enzyme 1A2 inhibitors using CoMFA, CoMSIA and HQSAR. SAR QSAR Environ. Res. 2011, 22, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Novotarskyi, S.; Sushko, I.; Korner, R.; Pandey, A.K.; Tetko, I.V. A comparison of different QSAR approaches to modeling CYP450 1A2 inhibition. J. Chem. Inf. Model. 2011, 51, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Roy, P.P. Comparative QSAR studies of CYP1A2 inhibitor flavonoids using 2D and 3D descriptors. Chem. Biol. Drug Des. 2008, 72, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Vasanthanathan, P.; Olsen, L.; Jorgensen, F.S.; Vermeulen, N.P.; Oostenbrink, C. Computational prediction of binding affinity for CYP1A2-ligand complexes using empirical free energy calculations. Drug Metab. Dispos. 2010, 38, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Vasanthanathan, P.; Taboureau, O.; Oostenbrink, C.; Vermeulen, N.P.; Olsen, L.; Jorgensen, F.S. Classification of cytochrome P450 1A2 inhibitors and noninhibitors by machine learning techniques. Drug Metab. Dispos. 2009, 37, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, J.; Jin, P.; Liu, J.; Foroozesh, M.; Stevens, C.L. In silico studies of polyaromatic hydrocarbon inhibitors of cytochrome P450 enzymes 1A1, 1A2, 2A6, and 2B1. Chem. Res. Toxicol. 2010, 23, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Liu, G.Y.; Cheng, J.; Chen, X.J.; Ju, X.L. CoMFA and molecular docking studies of benzoxazoles and benzothiazoles as CYP450 1A1 inhibitors. Eur. J. Med. Chem. 2010, 45, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Tanaka, K.; Takenaka, S.; Murayama, N.; Martin, M.V.; Foroozesh, M.K.; Yamazaki, H.; Guengerich, F.P.; Komori, M. Structure-function relationships of inhibition of human cytochromes P450 1A1, 1A2, 1B1, 2C9, and 3A4 by 33 flavonoid derivatives. Chem. Res. Toxicol. 2010, 23, 1921–1935. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Gonzalez, I.; Mojica, R.; Madrigal-Bujaidar, E.; Camacho-Carranza, R.; Escobar-Garcia, D.; Espinosa-Aguirre, J.J. The antigenotoxic effects of grapefruit juice on the damage induced by benzo(a)pyrene and evaluation of its interaction with hepatic and intestinal Cytochrome P450 (Cyp) 1A1. Food Chem. Toxicol. 2011, 49, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Platt, K.L.; Edenharder, R.; Aderhold, S.; Muckel, E.; Glatt, H. Fruits and vegetables protect against the genotoxicity of heterocyclic aromatic amines activated by human xenobiotic-metabolizing enzymes expressed in immortal mammalian cells. Mutat. Res. 2010, 703, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Tassaneeyakul, W.; Guo, L.Q.; Fukuda, K.; Ohta, T.; Yamazoe, Y. Inhibition selectivity of grapefruit juice components on human cytochromes P450. Arch. Biochem. Biophys. 2000, 378, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Santes-Palacios, R.; Romo-Mancillas, A.; Camacho-Carranza, R.; Espinosa-Aguirre, J.J. Inhibition of human and rat CYP1A1 enzyme by grapefruit juice compounds. Toxicol. Lett. 2016, 258, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Nguyen, T.T.; Dupart, P.S.; Sridhar, J.; Zhang, X.; Zhu, N.; Stevens, C.L.; Foroozesh, M. 7-Ethynylcoumarins: Selective inhibitors of human cytochrome P450s 1A1 and 1A2. Chem. Res. Toxicol. 2012, 25, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Rogan, E.G.; Badawi, A.F.; Devanesan, P.D.; Meza, J.L.; Edney, J.A.; West, W.W.; Higginbotham, S.M.; Cavalieri, E.L. Relative imbalances in estrogen metabolism and conjugation in breast tissue of women with carcinoma: Potential biomarkers of susceptibility to cancer. Carcinogenesis 2003, 24, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Parl, F.F.; Dawling, S.; Roodi, N.; Crooke, P.S. Estrogen metabolism and breast cancer: A risk model. Ann. N. Y. Acad. Sci. 2009, 1155, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Nakajima, M.; Yokoi, T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005, 227, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.T.; Lee, A.J. NADPH-dependent metabolism of 17beta-estradiol and estrone to polar and nonpolar metabolites by human tissues and cytochrome P450 isoforms. Steroids 2005, 70, 225–244. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Takemura, H.; Shimoi, K.; Yamamoto, K. A 3D model of CYP1B1 explains the dominant 4-hydroxylation of estradiol. J. Chem. Inf. Model. 2010, 50, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Labas, A.; Kramos, B.; Olah, J. Combined Docking and Quantum Chemical Study on CYP-Mediated Metabolism of Estrogens in Man. Chem. Res. Toxicol. 2017, 30, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Kunze, K.L.; Trager, W.F. Isoform-selective mechanism-based inhibition of human cytochrome P450 1A2 by furafylline. Chem. Res. Toxicol. 1993, 6, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Sesardic, D.; Boobis, A.R.; Murray, B.P.; Murray, S.; Segura, J.; de la Torre, R.; Davies, D.S. Furafylline is a potent and selective inhibitor of cytochrome P450IA2 in man. Br. J. Clin. Pharmacol. 1990, 29, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.F.V.; Lake, B.G. Molecular modelling of CYP1A subfamily members based on an alignment with CYP102: Rationalization of CYP1A substrate specificity in terms of active site amino acid residues. Xenobiotica 1996, 26, 723–753. [Google Scholar] [CrossRef] [PubMed]
- Stiborova, M.; Frei, E.; Schmeiser, H.H.; Arlt, V.M.; Martinek, V. Mechanisms of enzyme-catalyzed reduction of two carcinogenic nitro-aromatics, 3-nitrobenzanthrone and aristolochic acid I: Experimental and theoretical approaches. Int. J. Mol. Sci. 2014, 15, 10271–10295. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.K.A.; Akhtar, S.; Arif, J.M. Development of In Silico Protocols to Predict Structural Insights into the Metabolic Activation Pathways of Xenobiotics. Interdiscip. Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.X.; Zhou, Z.W.; He, Z.X.; Zhang, X.; Zhou, S.F.; Zhu, S. Estimation of the binding modes with important human cytochrome P450 enzymes, drug interaction potential, pharmacokinetics, and hepatotoxicity of ginger components using molecular docking, computational, and pharmacokinetic modeling studies. Drug Des. Dev. Ther. 2015, 9, 841–866. [Google Scholar]
- Singh, A.; Thornton, E.R.; Westheimer, F.H. The photolysis of diazoacetylchymotrypsin. J. Biol. Chem. 1962, 237, 3006–3008. [Google Scholar] [PubMed]
- Swanson, R.A.; Dus, K.M. Specific covalent labeling of cytochrome P-450CAM with 1-(4-azidophenyl)imidazole, an inhibitor-derived photoaffinity probe for P-450 heme proteins. J. Biol. Chem. 1979, 254, 7238–7246. [Google Scholar] [PubMed]
- Obach, R.S.; Spink, D.C.; Chen, N.; Kaminsky, L.S. Azidowarfarin photoaffinity probes of purified rat liver cytochrome P4501A1. Arch. Biochem. Biophys. 1992, 294, 215–222. [Google Scholar] [CrossRef]
- Yun, C.H.; Hammons, G.J.; Jones, G.; Martin, M.V.; Hopkins, N.E.; Alworth, W.L.; Guengerich, F.P. Modification of cytochrome P450 1A2 enzymes by the mechanism-based inactivator 2-ethynylnaphthalene and the photoaffinity label 4-azidobiphenyl. Biochemistry 1992, 31, 10556–10563. [Google Scholar] [CrossRef] [PubMed]
- Cvrk, T.; Hodek, P.; Strobel, H.W. Identification and characterization of cytochrome P4501A1 amino acid residues interacting with a radiolabeled photoaffinity diazido-benzphetamine analogue. Arch. Biochem. Biophys. 1996, 330, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Larroque, C.; van Lier, J.E. Hydroperoxysterols as a probe for the mechanism of cytochrome P-450scc-mediated hydroxylation. Homolytic versus heterolytic oxygen-oxygen bond scission. J. Biol. Chem. 1986, 261, 1083–1087. [Google Scholar] [PubMed]
- White, R.E.; Sligar, S.G.; Coon, M.J. Evidence for a homolytic mechanism of peroxide oxygen--oxygen bond cleavage during substrate hydroxylation by cytochrome P-450. J. Biol. Chem. 1980, 255, 11108–11111. [Google Scholar] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sridhar, J.; Goyal, N.; Liu, J.; Foroozesh, M. Review of Ligand Specificity Factors for CYP1A Subfamily Enzymes from Molecular Modeling Studies Reported to-Date. Molecules 2017, 22, 1143. https://doi.org/10.3390/molecules22071143
Sridhar J, Goyal N, Liu J, Foroozesh M. Review of Ligand Specificity Factors for CYP1A Subfamily Enzymes from Molecular Modeling Studies Reported to-Date. Molecules. 2017; 22(7):1143. https://doi.org/10.3390/molecules22071143
Chicago/Turabian StyleSridhar, Jayalakshmi, Navneet Goyal, Jiawang Liu, and Maryam Foroozesh. 2017. "Review of Ligand Specificity Factors for CYP1A Subfamily Enzymes from Molecular Modeling Studies Reported to-Date" Molecules 22, no. 7: 1143. https://doi.org/10.3390/molecules22071143
APA StyleSridhar, J., Goyal, N., Liu, J., & Foroozesh, M. (2017). Review of Ligand Specificity Factors for CYP1A Subfamily Enzymes from Molecular Modeling Studies Reported to-Date. Molecules, 22(7), 1143. https://doi.org/10.3390/molecules22071143