
The Exact Nuclear Overhauser Enhancement: Recent Advances

 , ,
, ,
Abstract
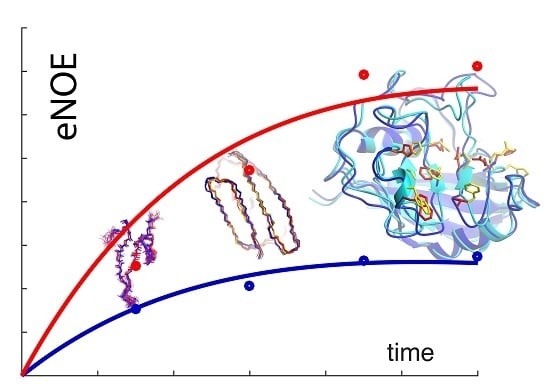
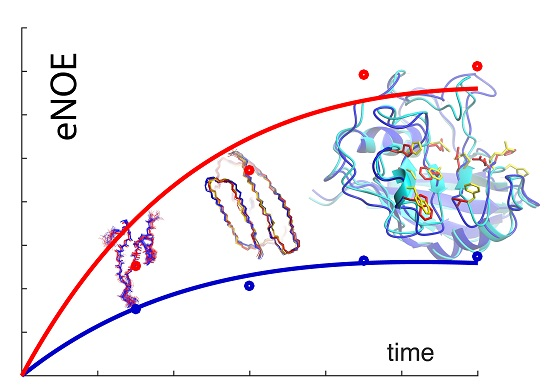
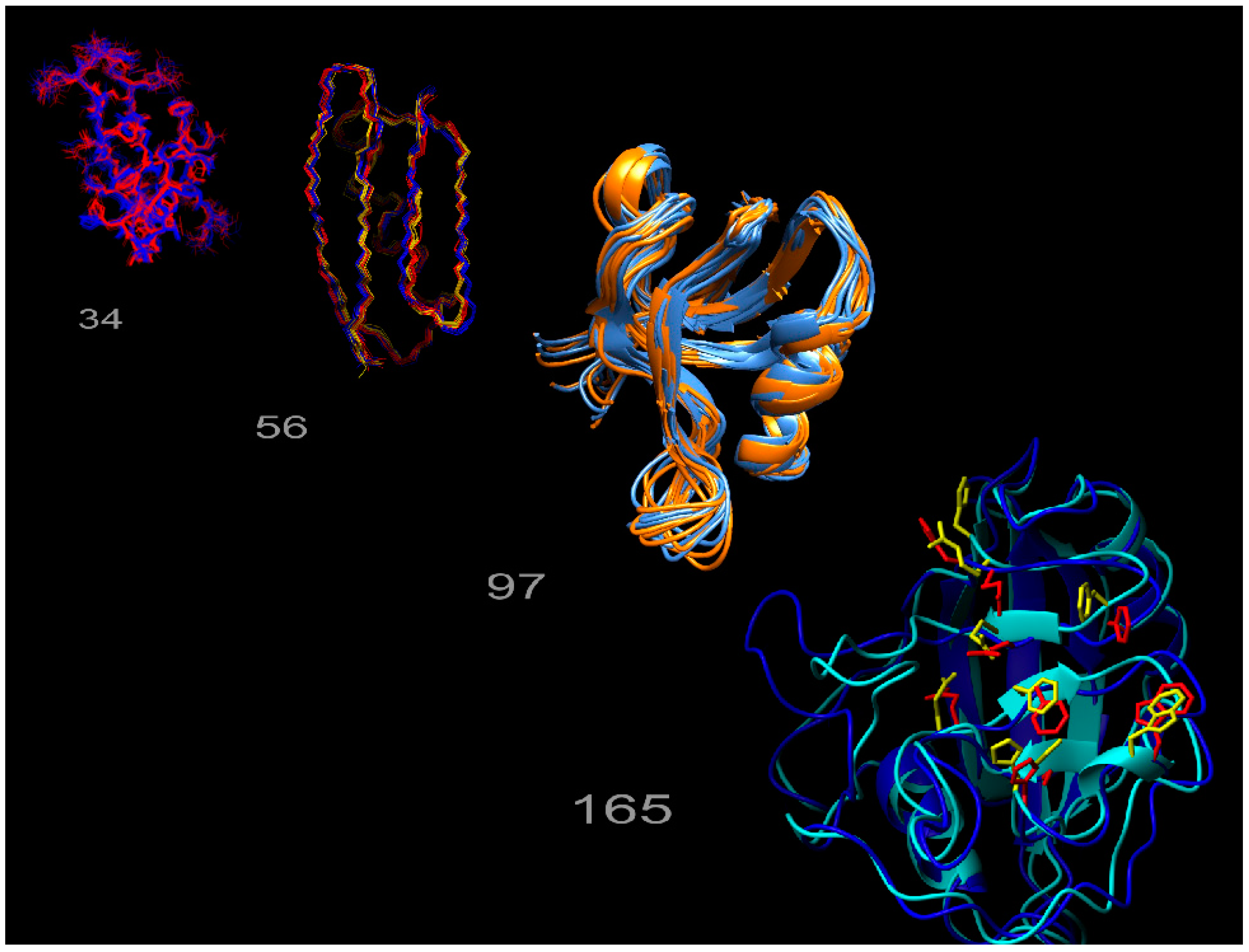
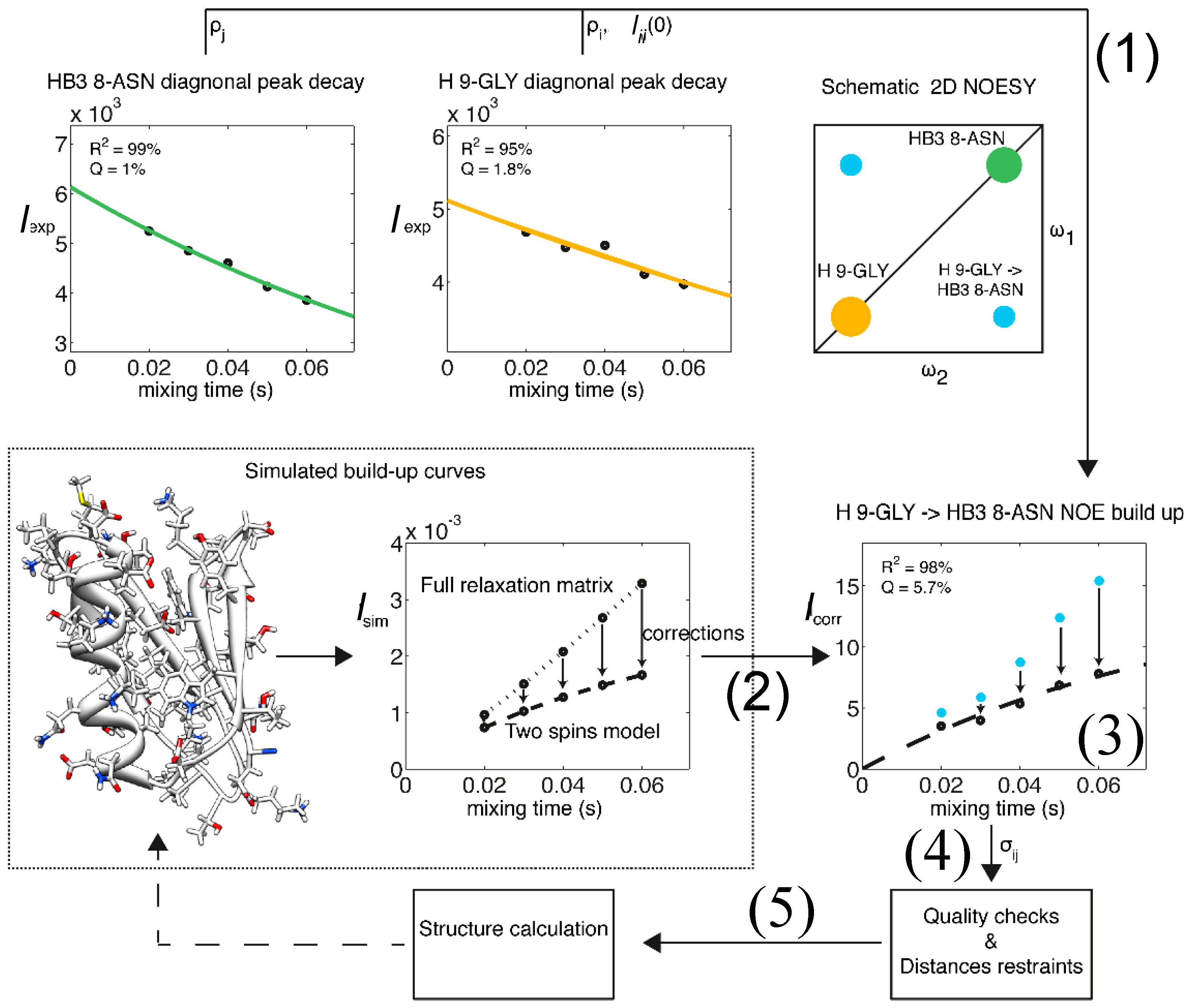
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
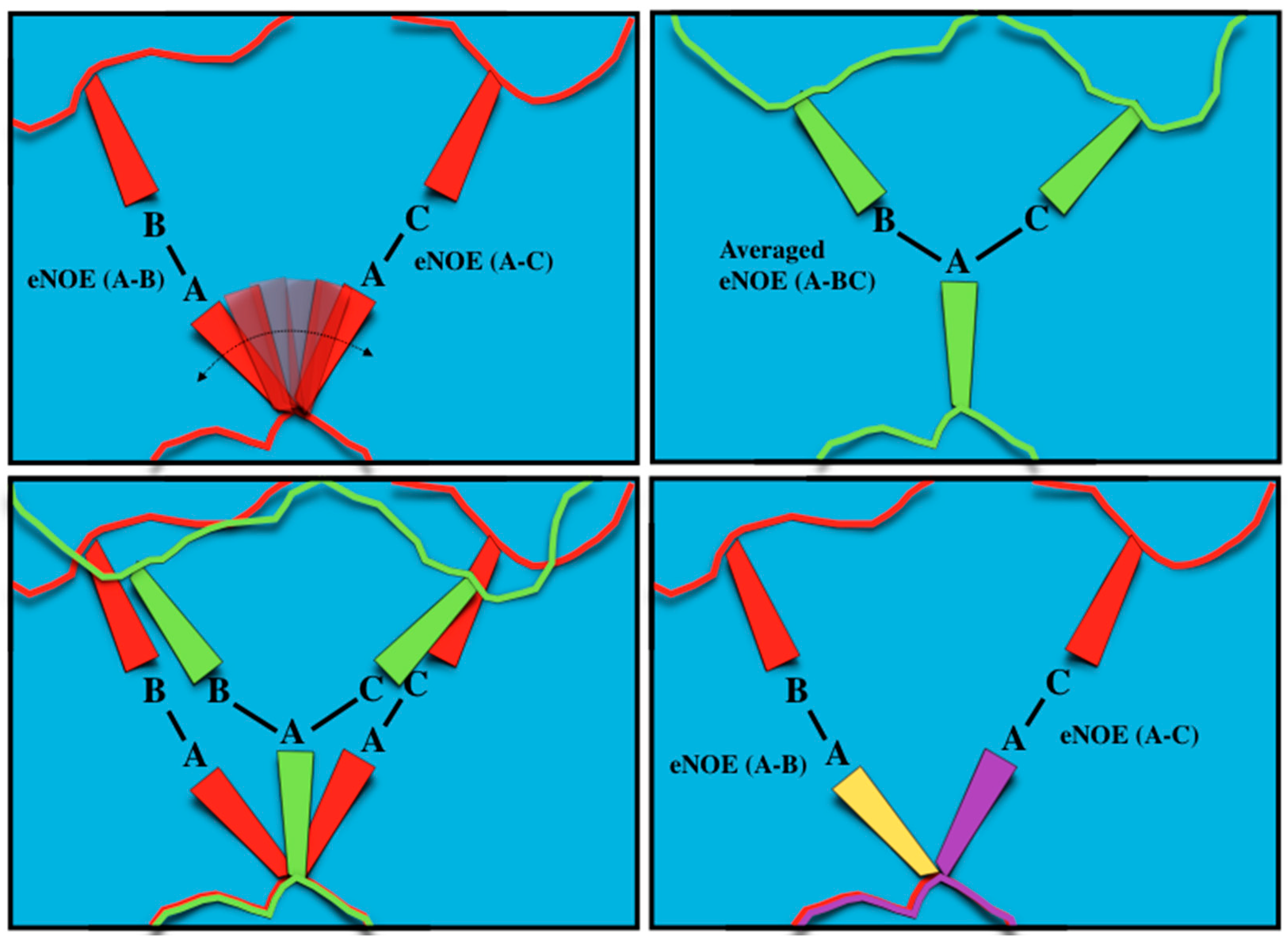
1.1. The Exact Nuclear Overhauser Enhancement Extraction Protocol
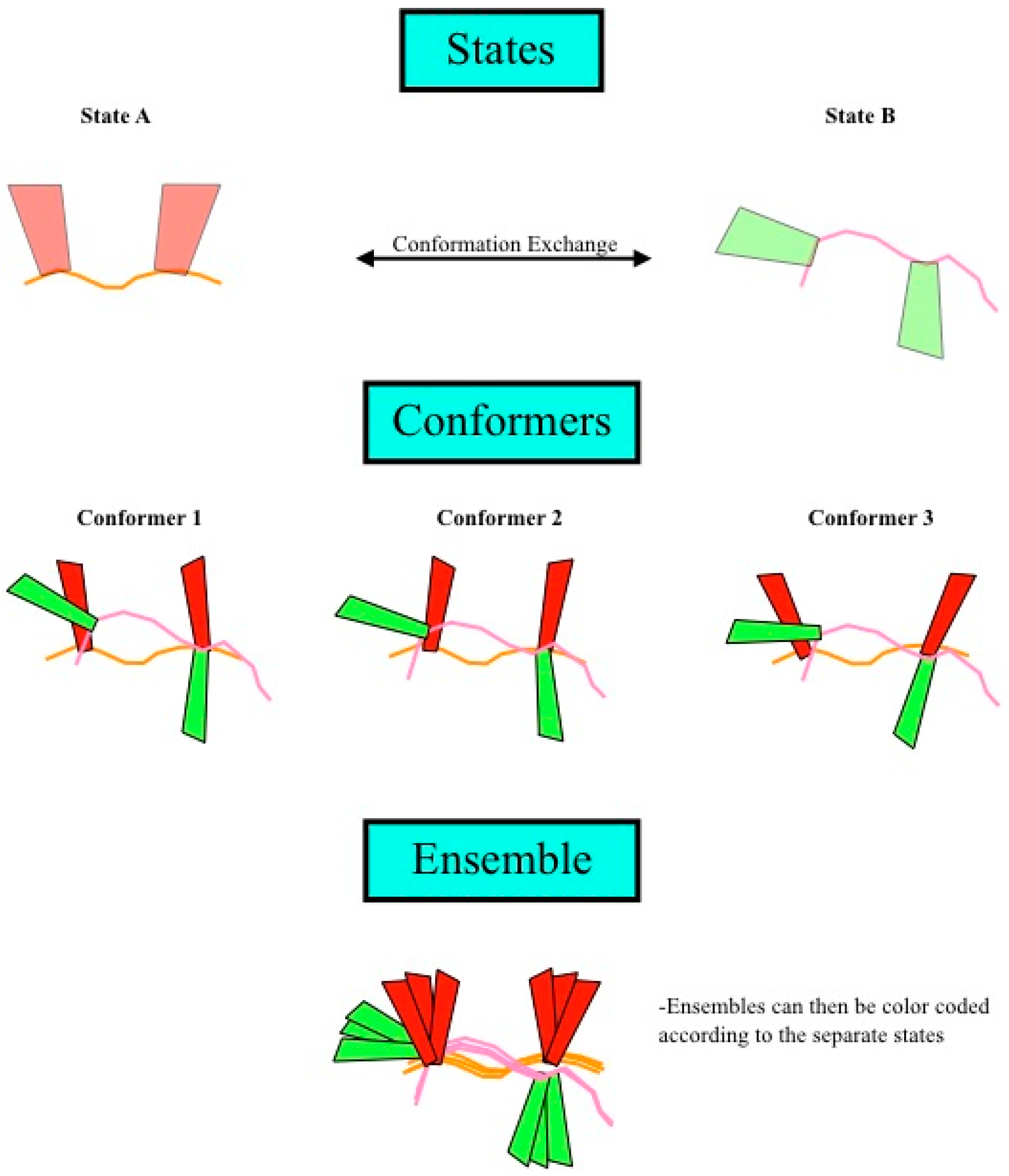
1.2. Exact Nuclear Overhauser Enhancements to Determine Dynamics through Multiple-State Ensembles
1.3. Definitions of States, Conformers and Ensembles
2. Exact Nuclear Overhauser Enhancement Recent Advances
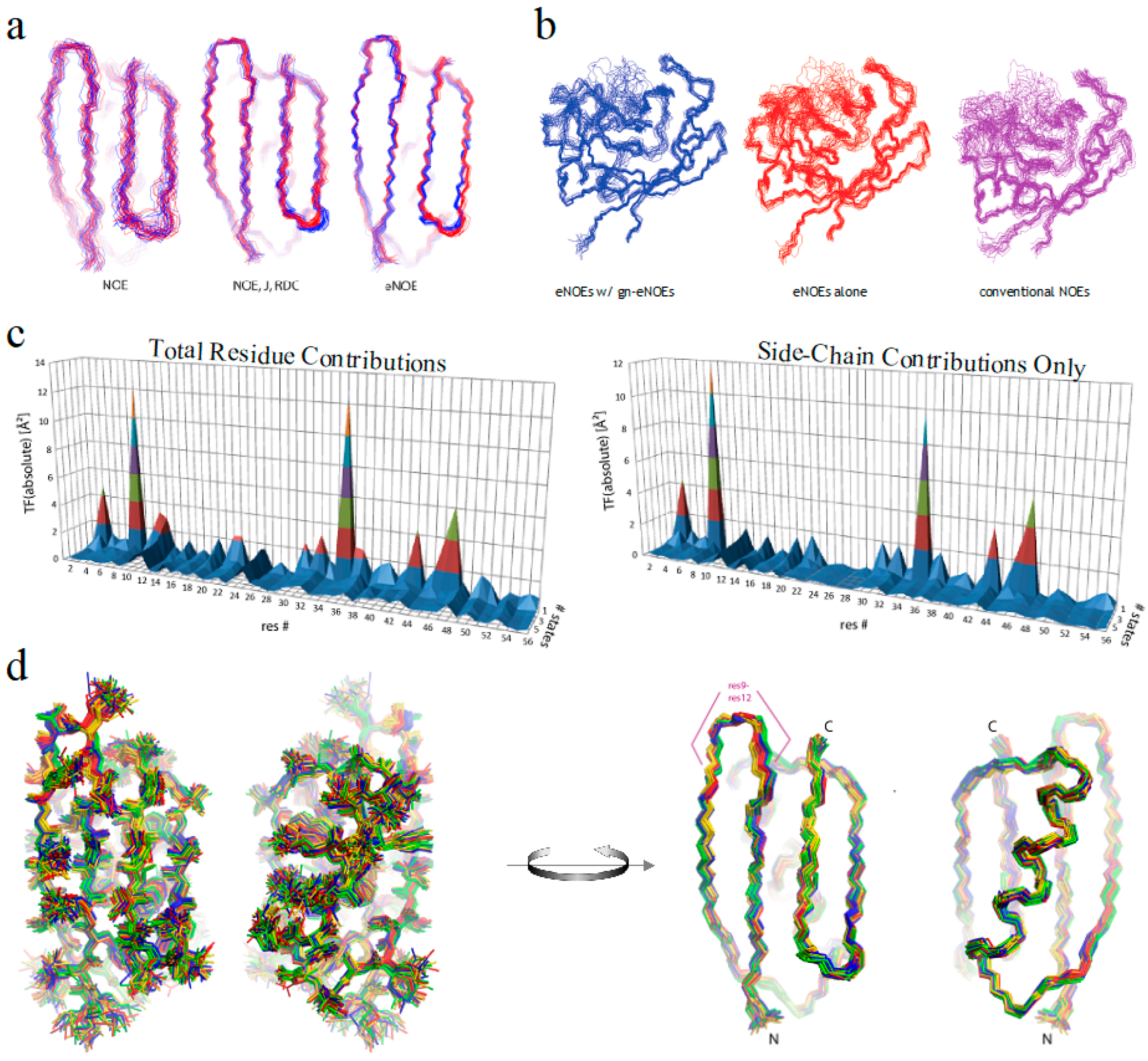
2.1. Exact Nuclear Overhauser Enhancement Methodology
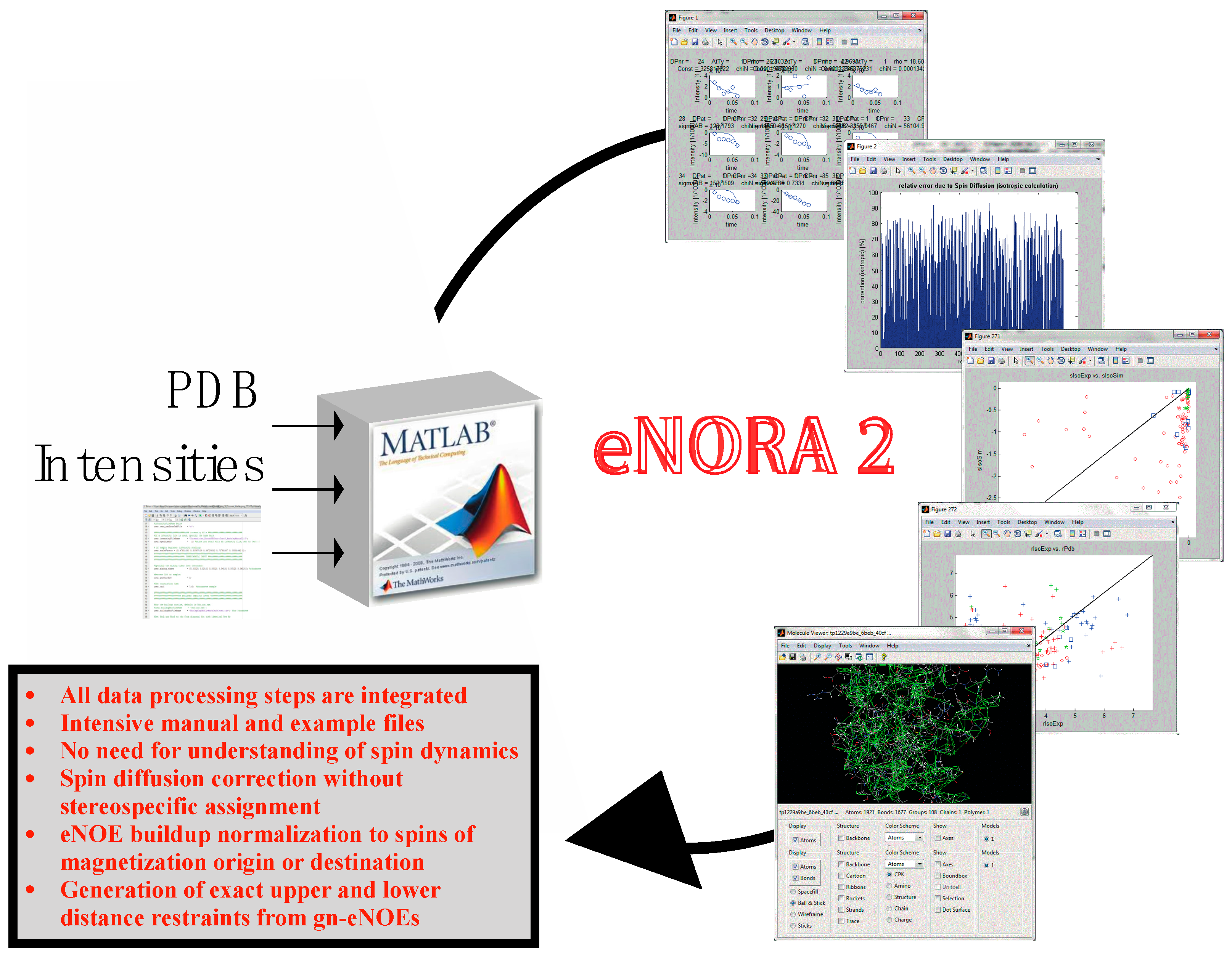
2.2. The eNORA2 Program
2.3. Exact Nuclear Overhauser Enhancement Analysis of Pin1 WW domain
2.4. Exact Nuclear Overhauser Enhancement Analysis of Cyclophilin A
3. Current Research and Development
3.1. Applying Exact Nuclear Overhauser Enhancements to Pin1
3.2. Extension of Exact Nuclear Overhauser Enhancements to the Proteasome
3.3. Applying F1F2-Selective NMR Spectroscopy to Exact Nuclear Overhauser Enhancements
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Van den Bedem, H.; Fraser, J.S. Integrative, dynamic structural biology at atomic resolution—It’s about time. Nat. Methods 2015, 12, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Barends, T.R.M.; Foucar, L.; Ardevol, A.; Nass, K.; Aquila, A.; Botha, S.; Doak, R.B.; Falahati, K.; Hartmann, E.; Hilpert, M.; et al. Direct observation of ultrafast collective motions in CO myoglobin upon ligand dissociation. Science 2015, 350, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Hekstra, D.R.; White, K.I.; Socolich, M.A.; Henning, R.W.; Ranganathan, R.; Biology, C. Electric field-stimulated protein mechanics. Nature 2016, 540, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Law, A.B.; Sapienza, P.J.; Zhang, J.; Zuo, X.; Petit, C.M. Native State Volume Fluctuations in Proteins as a Mechanism for Dynamic Allostery. J. Am. Chem. Soc. 2017, 139, 3599–3602. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Mehrabi, P.; Ren, Z.; Sljoka, A.; Ing, C.; Bezginov, A.; Ye, L.; Pomès, R.; Prosser, R.S.; Pai, E.F. The role of dimer asymmetry and protomer dynamics in enzyme catalysis. Science 2017, 355, eaag2355. [Google Scholar] [CrossRef] [PubMed]
- Capdevila, D.A.; Braymer, J.J.; Edmonds, K.A.; Wu, H.; Giedroc, D.P. Entropy redistribution controls allostery in a metalloregulatory protein. Proc. Natl. Acad. Sci. USA 2017, 114, 4424–4429. [Google Scholar] [CrossRef] [PubMed]
- Berlow, R.B.; Dyson, H.J.; Wright, P.E. Hypersensitive termination of the hypoxic response by a disordered protein switch. Nature 2017. [Google Scholar] [CrossRef] [PubMed]
- Papaleo, E.; Saladino, G.; Lambrughi, M.; Lindorff-Larsen, K.; Gervasio, F.L.; Nussinov, R. The Role of Protein Loops and Linkers in Conformational Dynamics and Allostery. Chem. Rev. 2016, 116, 6391–6423. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.N.; Bach, A.; Engström, Å.; Wang, H.; Strømgaard, K.; Gianni, S.; Jemth, P. A Sequential Binding Mechanism in a PDZ Domain. Biochemistry 2009, 48, 7089–7097. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.E.; Maragakis, P.; Lindorff-Larsen, K.; Piana, S.; Dror, R.O. Atomic-level characterization of the structural dynamics of proteins. Science 2011, 330, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Shaw, D.E. How Fast-Folding Proteins Fold. Science 2011, 334, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Eichmann, C.; Preissler, S.; Riek, R.; Deuerling, E. Cotranslational structure acquisition of nascent polypeptides monitored by NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2010, 107, 9111–9116. [Google Scholar] [CrossRef] [PubMed]
- Fersht, A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding; W.H. Freeman and Co.: New York, NY, USA, 1999; Volume 13409, ISBN 0716732688. [Google Scholar]
- Zhuravlev, P.I.; Papoian, G.A. Protein functional landscapes, dynamics, allostery: A tortuous path towards a universal theoretical framework. Q. Rev. Biophys. 2010, 43, 295–332. [Google Scholar] [CrossRef] [PubMed]
- Chill, J.H.; Naider, F. A solution NMR view of protein dynamics in the biological membrane. Curr. Opin. Struct. Biol. 2011, 21, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Ward, J.M.; Yuwen, T.; Podkorytov, I.S.; Skrynnikov, N.R. Microsecond time-scale conformational exchange in proteins: Using long molecular dynamics trajectory to simulate NMR relaxation dispersion data. J. Am. Chem. Soc. 2012, 134, 2555–2562. [Google Scholar] [CrossRef] [PubMed]
- Olsson, S.; Noé, F. Mechanistic Models of Chemical Exchange Induced Relaxation in Protein NMR. J. Am. Chem. Soc. 2016, 139, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.G.; Massi, F. Characterization of the dynamics of biomacromolecules using rotating-frame spin relaxation NMR spectroscopy. Chem. Rev. 2006, 106, 1700–1719. [Google Scholar] [CrossRef] [PubMed]
- Jardetzky, O. On the nature of molecular conformations inferred from high-resolution NMR. BBA Protein Struct. 1980, 621, 227–232. [Google Scholar] [CrossRef]
- Wüthrich, K. NMR of Proteins and Nucleic Acids; Wiley: Hoboken, NJ, USA, 1986; Volume 32, ISBN 0471828939. [Google Scholar]
- Solomon, I. Relaxation processes in a system of two spins. Phys. Rev. 1955, 99, 559–565. [Google Scholar] [CrossRef]
- Strotz, D.; Orts, J.; Minges, M.; Vögeli, B. The experimental accuracy of the uni-directional exact NOE. J. Magn. Reson. 2015, 259, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Keepers, J.W. Validation of the CHARMM27 force field for nucleic acids using 2D nuclear Overhauser effect spectroscopy. J. Magn. Reson. 1984, 57, 404. [Google Scholar]
- Vögeli, B.; Kazemi, S.; Güntert, P.; Riek, R. Spatial elucidation of motion in proteins by ensemble-based structure calculation using exact NOEs. Nat. Struct. Mol. Biol. 2012, 19, 1053–1057. [Google Scholar] [CrossRef] [PubMed]
- Vögeli, B.; Güntert, P.; Riek, R. Multiple-state ensemble structure determination from eNOE spectroscopy. Mol. Phys. 2013, 111, 437–454. [Google Scholar] [CrossRef]
- Vögeli, B. The nuclear Overhauser effect from a quantitative perspective. Prog. Nuclear Magn. Reson. Spectrosc. 2014, 78, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Wagner, G.; Ernst, R.R.; Wüthrich, K. Buildup rates of the nuclear overhauser effect measured by two-dimensional proton magnetic resonance spectroscopy: Implications for studies of protein conformation. J. Am. Chem. Soc. 1981, 103, 3654–3658. [Google Scholar] [CrossRef]
- Fesik, S.W.; Zuiderweg, E.R. Heteronuclear three-dimensional nmr spectroscopy. A strategy for the simplification of homonuclear two-dimensional NMR spectra. J. Magn. Reson. 1988, 78, 588–593. [Google Scholar] [CrossRef]
- Vögeli, B.; Friedmann, M.; Leitz, D.; Sobol, A.; Riek, R. Quantitative determination of NOE rates in perdeuterated and protonated proteins: Practical and theoretical aspects. J. Magn. Reson. 2010, 204, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Vögeli, B.; Segawa, T.F.; Leitz, D.; Sobol, A.; Choutko, A.; Trzesniak, D.; Van Gunsteren, W.; Riek, R. Exact distances and internal dynamics of perdeuterated ubiquitin from NOE buildups. J. Am. Chem. Soc. 2009, 131, 17215–17225. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.N.; Strotz, D.; Riek, R.; Vögeli, B. Extending the eNOE data set of large proteins by evaluation of NOEs with unresolved diagonals. J. Biomol. NMR 2015, 62, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Vögeli, B.; Olsson, S.; Riek, R.; Güntert, P. Compiled data set of exact NOE distance limits, residual dipolar couplings and scalar couplings for the protein GB3. Data Brief 2015, 5, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Vögeli, B.; Olsson, S.; Riek, R.; Güntert, P. Complementarity and congruence between exact NOEs and traditional NMR probes for spatial decoding of protein dynamics. J. Struct. Biol. 2015, 191, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Vögeli, B.; Olsson, S.; Güntert, P.; Riek, R. The Exact NOE as an Alternative in Ensemble Structure Determination. Biophys. J. 2016, 110, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Olsson, S.; Strotz, D.; Vögeli, B.; Riek, R.; Cavalli, A. The Dynamic Basis for Signal Propagation in Human Pin1-WW. Structure 2016, 24, 1464–1475. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.N.; Vögeli, B.; Bibow, S.; Strotz, D.; Orts, J.; Güntert, P.; Riek, R. A Structural Ensemble for the Enzyme Cyclophilin Reveals an Orchestrated Mode of Action at Atomic Resolution. Angew. Chemie Int. Ed. 2015, 54, 11657–11661. [Google Scholar] [CrossRef] [PubMed]
- Kalk, A.; Berendsen, H.J. Proton magnetic relaxation and spin diffusion in proteins. J. Magn. Reson. 1976, 24, 343–366. [Google Scholar] [CrossRef]
- Orts, J.; Vögeli, B.; Riek, R. Relaxation matrix analysis of spin diffusion for the NMR structure calculation with eNOEs. J. Chem. Theory Comput. 2012, 8, 3483–3492. [Google Scholar] [CrossRef] [PubMed]
- Güntert, P.; Mumenthaler, C.; Wüthrich, K. Torsion angle dynamics for NMR structure calculation with the new program Dyana. J. Mol. Biol. 1997, 273, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Güntert, P.; Buchner, L. Combined automated NOE assignment and structure calculation with CYANA. J. Biomol. NMR 2015, 62, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Leitz, D.; Vögeli, B.; Greenwald, J.; Riek, R. Temperature Dependence of 1HN-1HN Distances in Ubiquitin As Studied by Exact Measurements of NOEs. J. Phys. Chem. 2011, 115, 7648–7660. [Google Scholar] [CrossRef] [PubMed]
- Orts, J.; Vögeli, B.; Riek, R.; Güntert, P. Stereospecific assignments in proteins using exact NOEs. J. Biomol. NMR 2013, 57, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Strotz, D.; Orts, J.; Chi, C.N.; Riek, R.; Vögeli, B. The eNORA2 exact NOE analysis program. J. Chem. Theory Comput. Accepted.
- Lu, K.P.; Hanes, S.D.; Hunter, T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 1996, 380, 544–547. [Google Scholar] [PubMed]
- Ranganathan, R.; Lu, K.P.; Hunter, T.; Noel, J.P. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell 1997, 89, 875–886. [Google Scholar] [CrossRef]
- Bayer, E.; Goettsch, S.; Mueller, J.W.; Griewel, B.; Guiberman, E.; Mayr, L.M.; Bayer, P. Structural analysis of the mitotic regulator hPin1 in solution: Insights into domain architecture and substrate binding. J. Biol. Chem. 2003, 278, 26183–26193. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.A.; Bouchard, J.J.; Peng, J.W. Interdomain interactions support interdomain communication in human pin1. Biochemistry 2013, 52, 6968–6981. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhou, H.X. Dynamically Driven Protein Allostery Exhibits Disparate Responses for Fast and Slow Motions. Biophys. J. 2015, 108, 2771–2774. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Mahoney, B.J.; Zhang, M.; Zintsmaster, J.S.; Peng, J.W. Negative Regulation of Peptidyl-Prolyl Isomerase Activity by Interdomain Contact in Human Pin1. Structure 2015, 23, 2224–2233. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.W. Investigating dynamic interdomain allostery in Pin1. Biophys. Rev. 2015, 7, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Olsson, S.; Frellsen, J.; Boomsma, W.; Mardia, K.V.; Hamelryck, T. Inference of Structure Ensembles of Flexible Biomolecules from Sparse, Averaged Data. PLoS ONE 2013, 8, e79439. [Google Scholar] [CrossRef] [PubMed]
- Olsson, S.; Vögeli, B.R.; Cavalli, A.; Boomsma, W.; Ferkinghoff-Borg, J.; Lindorff-Larsen, K.; Hamelryck, T. Probabilistic determination of native state ensembles of proteins. J. Chem. Theory Comput. 2014, 10, 3484–3491. [Google Scholar] [CrossRef] [PubMed]
- Henzler-Wildman, K.A.; Lei, M.; Thai, V.; Kerns, S.J.; Karplus, M.; Kern, D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 2007, 450, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Kern, D.; Eisenmesser, E.Z.; Wolf-Watz, M. Enzyme Dynamics during Catalysis Measured by NMR Spectroscopy. Meth. Enzym. 2005, 394, 507–524. [Google Scholar] [PubMed]
- Eisenmesser, E.Z.; Millet, O.; Labeikovsky, W.; Korzhnev, D.M.; Wolf-Watz, M.; Bosco, D.A.; Skalicky, J.J.; Kay, L.E.; Kern, D. Intrinsic dynamics of an enzyme underlies catalysis. Nature 2005, 438, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.S.; Clarkson, M.W.; Degnan, S.C.; Erion, R.; Kern, D.; Alber, T. Hidden alternative structures of proline isomerase essential for catalysis. Nature 2009, 462, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Zydowsky, L.D.; Etzkorn, F.A.; Chang, H.Y.; Ferguson, S.B.; Stolz, L.A.; Ho, S.I.; Walsh, C.T. Active site mutants of human cyclophilin A separate peptidyl-prolyl isomerase activity from cyclosporin A binding and calcineurin inhibition. Protein Sci. 1992, 1, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Camilloni, C.; Sahakyan, A.B.; Holliday, M.J.; Isern, N.G.; Zhang, F.; Eisenmesser, E.Z.; Vendruscolo, M. Cyclophilin A catalyzes proline isomerization by an electrostatic handle mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, 10203–10208. [Google Scholar] [CrossRef] [PubMed]
- Gamble, T.R.; Vajdos, F.F.; Yoo, S.; Worthylake, D.K.; Houseweart, M.; Sundquist, W.I.; Hill, C.P. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 1996, 87, 1285–1294. [Google Scholar] [CrossRef]
- Sprangers, R.; Kay, L.E. Quantitative dynamics and binding studies of the 20S proteasome by NMR. Nature 2007, 445, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Religa, T.L.; Sprangers, R.; Kay, L.E. Dynamic Regulation of Archaeal Proteasome Gate Opening As Studied by TROSY NMR. Science 2010, 328, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Ruschak, A.M.; Kay, L.E. Proteasome allostery as a population shift between interchanging conformers. Proc. Natl. Acad. Sci. USA 2012, 109, E3454–E3462. [Google Scholar] [CrossRef] [PubMed]
- Chiarparin, E.; Pelupessy, P.; Bodenhausen, G. Selective cross-polarization in solution state NMR. Mol. Phys. 1998, 95, 759–767. [Google Scholar] [CrossRef]
- Pelupessy, P.; Chiarparin, E.; Bodenhausen, G. Excitation of selected proton signals in NMR of isotopically labeled macromolecules. J. Magn. Reson. 1999, 138, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Walinda, E.; Morimoto, D.; Shirakawa, M.; Sugase, K. F 1 F 2-selective NMR spectroscopy. J. Biomol. NMR 2017, 68, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Vögeli, B.; Orts, J.; Strotz, D.; Güntert, P.; Riek, R. Discrete three-dimensional representation of macromolecular motion from eNOE-based ensemble calculation. Chimia (Aarau) 2012, 66, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Vögeli, B.; Orts, J.; Strotz, D.; Chi, C.; Minges, M.; Wälti, M.A.; Güntert, P.; Riek, R. Towards a true protein movie: A perspective on the potential impact of the ensemble-based structure determination using exact NOEs. J. Magn. Reson. 2014, 241, 53–59. [Google Scholar] [CrossRef] [PubMed]









© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nichols, P.J.; Born, A.; Henen, M.A.; Strotz, D.; Orts, J.; Olsson, S.; Güntert, P.; Chi, C.N.; Vögeli, B. The Exact Nuclear Overhauser Enhancement: Recent Advances. Molecules 2017, 22, 1176. https://doi.org/10.3390/molecules22071176
Nichols PJ, Born A, Henen MA, Strotz D, Orts J, Olsson S, Güntert P, Chi CN, Vögeli B. The Exact Nuclear Overhauser Enhancement: Recent Advances. Molecules. 2017; 22(7):1176. https://doi.org/10.3390/molecules22071176
Chicago/Turabian StyleNichols, Parker J., Alexandra Born, Morkos A. Henen, Dean Strotz, Julien Orts, Simon Olsson, Peter Güntert, Celestine N. Chi, and Beat Vögeli. 2017. "The Exact Nuclear Overhauser Enhancement: Recent Advances" Molecules 22, no. 7: 1176. https://doi.org/10.3390/molecules22071176
APA StyleNichols, P. J., Born, A., Henen, M. A., Strotz, D., Orts, J., Olsson, S., Güntert, P., Chi, C. N., & Vögeli, B. (2017). The Exact Nuclear Overhauser Enhancement: Recent Advances. Molecules, 22(7), 1176. https://doi.org/10.3390/molecules22071176